

Abstract
Purpose
Enzastaurin is a selective inhibitor of protein kinase C β. Prior phase I studies did not show increased drug exposures with escalating once daily administration. Limits from gastrointestinal absorption may be overcome by twice daily dosing, potentially improving antitumor effects.
Experimental Design
We conducted a phase I dose escalation study in 26 patients with recurrent malignant glioma, stratified by use of enzyme-inducing antiepileptic drugs, to investigate whether divided twice daily dosing results in higher exposures compared with once daily dosing. Phosphorylated glycogen synthase 3 β was analyzed as a potential biomarker of enzastaurin activity.
Results
Enzastaurin was poorly tolerated at all dose levels evaluated (500, 800, and 1,000 mg total daily), with thrombocytopenia and prolonged QTc as dose-limiting toxicities. The average drug concentration of enzastaurin under steady-state conditions was doubled by twice daily dosing compared with daily dosing [1.990; 90% confidence interval (CI), 1.450-2.730]. Additionally, geometric mean ratios doubled with 800 versus 500 mg dosing for both daily (2.687; 90% CI, 1.232-5.860) and twice daily regimens (1.852; 90% CI, 0.799-4.292). Two patients achieved long-term benefit (over 150 weeks progression free).
Conclusions
Higher and more frequent dosing of enzastaurin resulted in improved drug exposure but with unacceptable toxicity at the doses tested. Phosphorylated glycogen synthase 3 β may be a useful biomarker of the biological activity of enzastaurin. Enzastaurin has activity in a subset of malignant glioma patients and warrants continued study in combination with other agents using a maximal once daily dose of 500 mg.
Malignant gliomas are uncommon but lethal tumors. Once the primary treatment modalities of surgery, chemotherapy, and radiation have failed, the prognosis for patients with recurrent disease is poor. When patients have tumor progression, conventional chemotherapy has not been shown to prolong survival (1). Gliomas are relatively resistant to standard chemotherapy because of blood-brain barrier impedance to drug delivery, tumor hypoxia, and their relatively low growth fraction. Most importantly, gliomas tend to have significant intrinsic resistance to most standard cytotoxic agents. New agents directed against novel cellular targets may improve patient outcomes.
The protein kinase C (PKC) enzymes are a family of serine/threonine kinase signaling molecules involved in numerous cellular functions including cell growth, proliferation, and programmed cell death. The β isoform of PKC lies in the signal cascade of vascular endothelial growth factor, and inhibition of this pathway leads to a block in tumor angiogenesis and growth in vitro (2). Enzastaurin is a potent selective inhibitor of PKCβ and has been shown to have antiangiogenic and antitumor effects in various preclinical models (3-6). Enzastaurin and its metabolites (LY326020 and LY485912) prevent substrate phosphorylation by competing with ATP for the ATP binding site of the enzyme, thereby inhibiting PKC activation and its target proteins such as AKT (7, 8). Enzastaurin treatment has also been shown to suppress glycogen synthase 3 β (GSK3β) phosphorylation in a variety of human cancer cell lines, including HCT116 colon carcinoma cells, glioblastomas, and B-cell lymphomas (9-11). Additionally, we have recently shown that enzastaurin has direct inhibitory activity against GSK3 activity, associated with overall inhibition of GSK3 phosporylation (12). Thus, phosphorylated GSK3β may be a useful biomarker for monitoring the activity of enzastaurin.
A prior phase I trial of enzastaurin established a phase II dose of 525 mg/day based on achieving a biologically active target plasma concentration, and showed an inability to significantly increase plasma levels of enzastaurin and its metabolites with increasing doses of the drug (13). A subsequent phase I/II trial in recurrent high-grade gliomas suggested activity but raised the question of whether suboptimal concentrations of the drug were being achieved in the brain, especially for patients on enzyme-inducing antiepileptic drugs (EIAED; ref. 14). We hypothesized that the lack of increase in plasma exposures after higher doses could be due to dose-limiting saturation in absorption of enzastaurin from the gastrointestinal tract. We designed this trial to determine if the dose schedule, in addition to the total dose, influences exposure to enzastaurin and its metabolites in patients on EIAEDs and those who are on non-EIAED.
Patients and Methods
Inclusion/exclusion criteria
Adult patients with histologically proven malignant glioma with progression of disease after prior radiation therapy were eligible for this National Cancer Institute Institutional Review Board–approved trial. Patients were required to have a Karnofsky Performance Status of ≥60, and adequate organ function as measured by WBC of ≥3,000/μL, absolute neutrophil count of ≥1,500/mm3, platelet count of ≥100,000/mm3, hemoglobin of ≥10 gm/dL, aspartate aminotransferase and bilirubin of less than or equal to twice the upper limit of normal, and creatinine of ≤1.5 mg/dL. They must have recovered from toxic effects of prior therapy and not received nitrosureas within 6 wk, procarbazine within 3 wk, other cytotoxic therapy within 4 wk, noncytotoxic investigational agent within 2 wk, or other noncytotoxic agents within 1 wk of study entry. Any prior treatment with EIAEDs was discontinued for at least 2 wk before enrollment for patients in group A (non-EIAEDs). Patients in group B were taking one of the following EIAEDs: carbamazapine, oxcarbazapine, or phenytoin. Other concurrent tumor therapy, active malignancy, active infection, pregnancy, breast feeding, QTc of >460 ms, and clinically significant dysrrhythmia were exclusion criteria for participation.
Study design
This was a phase I trial designed to explore drug exposure as a function of total dose and dosing schedule, and to establish the maximum tolerated dose for once daily and twice daily dosing stratified by use of EIAEDs. Twelve patients per dose level were planned for group A, and six patients per dose level were planned for group B. Dose limiting toxicity (DLT) was defined as any CTC v3.0 hematologic or nonhematologic grade of ≥3 toxicity thought to be due to or possibly due to enzastaurin during the first 6 wk of treatment. For group A, the maximum tolerated dose was defined as the dose level below that one third of patients enrolled experience a DLT. Likewise, for group B, the maximum tolerated dose was defined as the dose level below that two or more of six patients experience DLT during the first cycle of therapy.
Treatment
Due to expected interpatient variability, each patient served as their own control in a crossover design where patients alternated between daily and twice daily divided dosing midway through the first 6-wk cycle. Half of patients at each dose level started with once daily dosing, and the other half with twice daily dosing. All patients proceeding to cycle two and beyond were treated with twice daily dosing. Enzastaurin was supplied as 100 and 125 mg tablets, self-administered within 30 min of a high-fat meal (at least 40 grams). Blood chemistry and hematology was monitored weekly through cycle one, then biweekly thereafter. Physical and neurologic exams were done before initiating every cycle and midway through cycle one. Serial electrocardiograms were obtained at steady-state midcycle and end-cycle one for correlation with pharmacokinetic parameters.
Pharmacokinetics
Pharmacokinetic sampling was done for comparison between once daily and twice daily dosing in weeks 2 to 3 and 5 to 6. If a patient was on the once daily regimen, blood samples for drug concentration measurement were collected on day 19 to 21 from the start of the once daily dosing, at predose, and 1, 2, 4, 6, and 24 h post-dose (before next dose administration). If a patient was on twice daily dosing, then samples were taken on day 19 to 21 from the start of the twice daily regimen at predose (before the morning dose), 1, 2, 4, 6, 12 h postdose (before evening dose), and 12 h after the evening dose (before next dose administration). Thus, each patient had 2 d of sampling on approximately day 21 and 42 of cycle 1. In addition, a 4-h sample on day 1 was obtained for enzastaurin concentration measurement coincident with the sample collected for measuring GSK3β activity. High-performance liquid chromatography with tandem mass spectrometry was used to analyze concentrations of enzastaurin, and its active metabolite, LY326020 (Advion BioSciences, Inc.).
Pharmacokinetic parameters for enzastaurin, its active metabolite (LY326020), and total analytes (sum of enzastaurin and its metabolite) were determined from the plasma concentration versus time data using standard noncompartmental methods (WinNonlin Enterprise, Version 5.0.1; Pharsight Corp.). The lower limit of quantification for enzastaurin was 0.970 nmol/L (0.5 ng/mL), and 1.178 nmol/L (0.5 ng/mL) for LY326020. Pharmacokinetic parameters of interest for enzastaurin were steady-state AUC, maximum plasma concentration (Cmax,ss), and apparent clearance. For the active metabolite (LY326020) and for total analytes (sum of enzastaurin and its metabolites), Cmax,ss and steady-state AUC were estimated. Log-transformed pharmacokinetic parameters (AUCτ,ss, Cmax,ss) were analyzed using a mixed-effects model allowing for the fixed effect of dose, regimen, and the random effect of patient to assess the potential effect of dose and regimen on enzastaurin pharmacokinetics. The geometric mean ratio of the enzastaurin parameters (twice daily versus once daily; 800 versus 500 mg) were reported along with their 90% confidence interval (CI).
Biomarker assays
Concurrent samples were also collected to assay GSK3β activity as measured by its phosphorylated form (pGSK3β). These studies were done as an exploratory analysis of whether pGSK3β may be a useful biomarker of PKCβ pathway inhibition. For each sample, 5 μg of protein from peripheral blood mononuclear cells were assayed for the presence of pGSK3β. Reactions were done in duplicate using the Assay Designs phospho-GSK 3β Enzyme Immunometric Assay. A recombinant phosphorylated GSK3β standard of known concentration, included in the kit, was serially diluted and assayed along with the samples. Reactions were done per manufacturer’s protocol. At the final step of the assay, samples were read at an absorbance of 450 nm using a plate reader. The mean absorbance reading of the blank wells are subtracted from sample absorbance readings. The quantity of pGSK3β for each sample was calculated based on a standard curve constructed from the average net absorbance for each standard versus pGSK3β concentration in each standard.
Response and survival analysis
Tumor response and survival were secondary end points. Contrast-enhanced magnetic resonance imaging was evaluated every 6 wk where tumor response was assessed with standard Macdonald and Levin criteria using largest cross-sectional diameters of measurable lesions, or scored evaluations of nonmeasurable but evaluable disease (15, 16). The designation of stable disease required a minimum of 6-wk duration. Progression-free survival was estimated using Kaplan Meier methodology using the earlier of either date of documented disease progression or date of death.
Results
Treatment and toxicity
Twenty-two of 26 patients who enrolled into the study between April 2005 and December 2006 were evaluable. Before completing the first cycle, two patients had documented disease progression and two withdrew for clinical deterioration. Median age was 56 years (range, 24-71); median Karnofsky Performance Status was 90 (range, 70-100); 17 (77%) patients had glioblastoma. Patients received an average of 2 chemotherapy regimens for their tumor before study participation (range, 1-3) and all patients received prior radiation therapy.
Twelve group A patients were treated at the first dose level of enzastaurin 800 mg given either once daily, or as 400 mg twice daily. Three of the first 9 patients experienced DLT; 2 grade 3 thrombocytopenia (1 also with asymptomatic grade 3 QTc prolongation) and 1 grade 3 elevated alanine aminotransferase. This was considered unacceptable toxicity and subsequently, the next cohort of patients was treated with enzastaurin 500 mg given daily or in divided doses. Two of 5 patients treated at this dose level experienced DLT; 1 patient died from intracerebral hemorrhage in the setting of grade 3 thrombocytopenia, and another patient had grade 3 asymptomatic QTc prolongation. Because greater than one third of patients in this lower dose level had DLT, enrollment to this dose level then closed. Only 5 of 6 planned patients were accrued to the group B arm of the study and treated with enzastaurin 1,000 mg/day, when 2 patients had dose limiting grade 3 thrombocytopenia. All DLTs occurred while taking enzastaurin on the twice daily schedule or at ≥800 mg once daily. The only exception was one case of grade 3 prolonged QTc in a patient being treated at 500 mg once daily who had a history of QTc prolongation before study entry. Serial electrocardiogram evaluations at the middle and end of cycle one revealed transient asymptomatic prolonged QTc as described in Table 1 along with the complete toxicity data. There was no significant relationship between QTcB changes from baseline and concentrations of enzastaurin LY326020.
Table 1.
Toxicity possibly or probably related to treatment with enzastaurin (N = 22)
| Toxicity | Grade 1-2 | Grade 3-4 | Grade 5 |
|---|---|---|---|
| Thrombocytopenia | 6 (27%) | 5 (23%) | 0 |
| QTc prolongation | 5 (23%) | 2 (9%)* | 0 |
| Elevated AST | 9 (41%) | 0 | 0 |
| Elevated ALT | 5 (23%) | 1 | 0 |
| Intracranial hemorrhage | 0 | 0 | 1† |
| Hypermagnesemia | 1 | 0 | 0 |
| Proteinuria | 1 | 0 | 0 |
| Hematuria | 1 | 0 | 0 |
| Nausea | 1 | 0 | 0 |
Abbreviations: AST, aspartate aminotransferase; ALT, alanine aminotransferase.
Cases were transient, asymptomatic, and required no intervention or therapy.
Occurred in the setting of grade 3 thrombocytopenia.
Pharmacokinetics
Of 21 patients entered in group A, 17 were evaluable for toxicity and had data available for pharmacokinetic analysis. Summary of the pharmacokinetic parameters for enzastaurin, its metabolite LY326020, total analytes (sum of enzastaurin and its metabolites), and the geometric mean ratio (exponentiating LS means) are presented in Table 2. The average drug concentration of enzastaurin under steady-state conditions during multiple dosing was doubled by twice daily dosing compared with daily dosing (1.990; 90% CI, 1.450-2.730), whereas the metabolite was ~44% higher (1.439; 90% CI, 1.248-1.658). Total analyte was ~52% higher for divided dosing at 800 mg/day (1.519; 90% CI, 1.189-1.941), and 97% higher at 500 mg/day (1.971; 90% CI, 1.286, 3.020). Supplementary Table S1 presents the geometric mean ratios for comparisons of DL1 (800 mg) and DL-1 (500 mg). Exposures approximately doubled with 800 mg versus 500 mg dosing for both daily (2.687; 90% CI, 1.232-5.860) and twice daily regimens (1.852; 90% CI, 0.799-4.292). Overall, increased drug exposure was associated with both higher and twice daily dosing (Fig. 1).
Table 2.
Summary of plasma pharmacokinetic parameters of enzastaurin and LSN326020 for group A (non-EIAED)
| Parameter | Geometric Mean (%CV)
|
||||||
|---|---|---|---|---|---|---|---|
| DL 1 (800 mg daily or 400 mg twice daily; 12 patients)
|
DL-1 (500 mg daily or 250 mg twice daily; 5 patients)
|
DL 1 + DL-1
|
|||||
| 800 mg daily n = 12 | 400 mg twice daily n = 12 | Ratio of regimen* twice daily/daily (90%CI) | 500 mg daily n = 5 | 250 mg twice daily n = 3† | Ratio of regimen* twice daily/daily (90% CI) | Ratio of regimen twice daily/daily (90% CI) | |
| Enzastaurin | |||||||
| Cmax,ss (nmol/L) | 2,180 (75) | 2,710 (86) | 1.226 (0.958-1.569) | 979 (97) | 1,390 (106) | 1.590 (1.014-2.494) | 1.396 (1.080-1.805) |
| Tmax (h)‡ | 4.00 (1.00-6.00) | 4.03 (2.00-6.08) | — | 2.08 (2.00-4.75) | 6.03 (4.00-6.17) | — | — |
| Cav,ss (nmol/L) | 1,130 (89) | 1,950 (106) | 1.652 (1.219-2.238) | 472 (109) | 1,040 (124) | 2.397 (1.377-4.173) | 1.990 (1.450-2.730) |
| CL/F (L/h) | 57.3 (89) | 33.2 (106) | — | 85.6 (109) | 38.9 (124) | — | — |
| LY326020 | |||||||
| Cmax (nmol/L) | 1,130 (43) | 1,360 (47) | 1.257 (1.091-1.448) | 611 (48) | 730 (55) | 1.424 (1.099-1.845) | 1.338 (1.154-1.551) |
| Tmax (h)‡ | 5.96 (4.00-23.92) | 6.00 (0.00-11.73) | — | 4.08 (2.00-4.75) | 6.03 (4.00-6.17) | — | — |
| Cav,ss (nmol/L) | 955 (47) | 1210 (51)§ | 1.353 (1.175-1.559) | 522 (46) | 676 (55) | 1.529 (1.195-1.956) | 1.439 (1.248-1.658) |
| MR (ratio) | 0.846 (.57) | 0.643 (.60)§ | — | 1.11 (.63) | 0.651 (.66) | — | |
| Total analyte | |||||||
| Cmax (nmol/L) | 3,620 (59) | 4,570 (72) | 1.269 (1.008-1.599) | 1,740 (76) | 2,420 (88) | 1.559 (1.043-2.329) | 1.407 (1.116-1.773) |
| Tmax (h)‡ | 4.00 (2.00-6.00) | 4.03 (2.00-6.08) | — | 3.87 (2.00-4.75) | 6.03 (4.00-6.17) | — | — |
| Cav,ss (nmol/L) | 2,370 (66) | 3,610 (76)§ | 1.519 (1.189-1.941) | 1,120 (78) | 2,000 (95) | 1.971 (1.286-3.020) | 1.730 (1.353-2.213) |
Abbreviations: Cmax, maximum plasma concentration; Tmax, time of maximum observed drug concentration; Cav,ss, average drug concentration under steady-state conditions during multiple dosing; CL/F, apparent total body clearance of drug calculated after extravascular administration; CV, coefficient of variation; MR, metabolic ratio; n, number of subjects in pharmacokinetic analysis.
Treatment comparisons are based on least-square geometric mean values of the pharmacokinetic parameters.
Two pvatients did not have pharmacokinetic data available for the twice daily portion of their treatment course.
The median and range are presented.
n = 11.
Fig. 1.
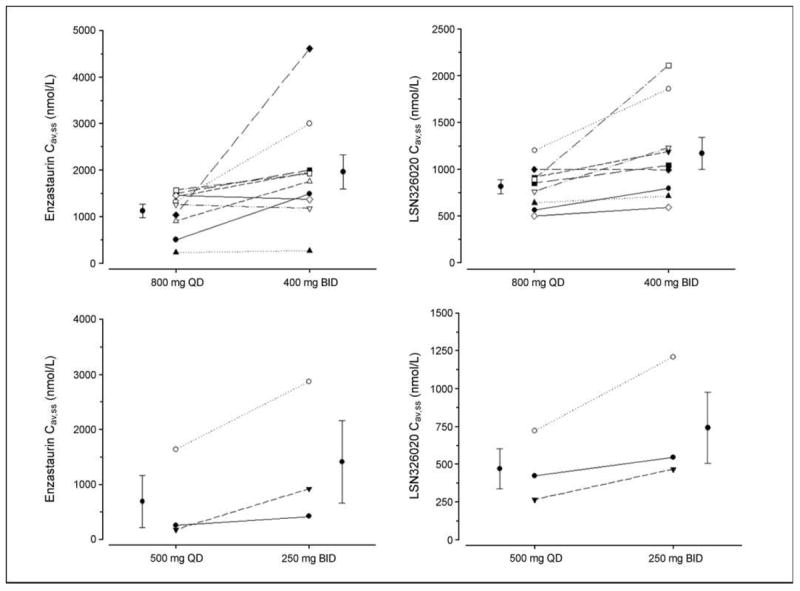
Exposures of enzastaurin and its major metabolite LSN326020 are increased with twice daily versus once daily dosing at all dose levels for patients on non-EIAEDs. QD, once daily; BID, twice daily.
Among the 5 patients enrolled to group B, only 2 had sufficient data available for pharmacokinetic evaluation; 2 patients progressed within 3 weeks of enrollment, and 1 patient did not have adequate vascular access for pharmacokinetic sampling. Only one patient in group B had pharmacokinetic data available to compare once daily versus twice daily dosing, and had pharmacokinetic results consistent with the patients described for group A where higher exposures occurred with twice daily versus once daily oral dosing (Table 3).
Table 3.
Plasma pharmacokinetic parameters of enzastaurin and LY326020 for Group B patients (non-EIAED)
| Parameter | 1,000 mg daily
|
500 mg twice daily
|
|
|---|---|---|---|
| Patient 1 | Patient 2 | Patient 2 | |
| Enzastaurin | |||
| Cmax,ss (nmol/L) | 3,012 | 2,730 | 4,093 |
| Tmax (h) | 4.00 | 2.00 | 4.00 |
| Cav,ss (nmol/L) | 1,340 | 1,350 | 3,050 |
| CL/F (L/h) | 60 | 60 | 27 |
| LY326020 | |||
| Cmax,ss (nmol/L) | 1,867 | 808 | 870 |
| Tmax (h) | 6.00 | 4.00 | 6.00 |
| Cav,ss (nmol/L) | 1,640 | 697 | 819 |
| MR (ratio) | 1.22 | 0.516 | 0.269 |
| Total Analyte | |||
| Cmax,ss (nmol/L) | 4,832 | 3,873 | 5,860 |
| Tmax (h) | 4.00 | 2.00 | 4.00 |
| Cav,ss (nmol/L) | 3,150 | 2,380 | 4,730 |
NOTE: Of five patients enrolled on this arm of the study, pharmacokinetic data are partially available for two patients.
Exposures in group B patients in this trial were >10-fold higher (Table 3) than those seen in our earlier once daily dosing phase I/II trial of enzastaurin in glioma patients where mean enzastaurin average drug concentration under steady-state conditions during multiple dosing was ~72 to 99 nmol/L in the 3 dose groups of 525, 700, or 900 mg daily (14). In that phase I/II trial, patients on enzyme inducers were seen to have a much higher proportionate exposure of LY326020 compared with the parent enzastaurin compound in line with an induction effect; something not observed in the patients in this trial.4 A review of concomitant medications showed that group B patients on the present trial who were evaluable for pharmacokinetic parameters were treated with oxcarbazepine, which is not a very potent inducer of CYP3A (17). By contrast, the pharmacokinetic samples in the JCAJ trial came from patients treated with phenytoin or carbamazepine, drugs known to be very potent inducers of CYP3A (18).
Response and survival outcomes
Nineteen patients were evaluable for radiographic response. Two patients (11%) had objective responses (1 complete and 1 partial response), whereas 8 patients (32%) achieved stable disease (Fig. 2). Two patients (1 anaplastic astrocytoma, 1 glioblastoma) have had long-term disease control and remain alive progression free over 150 weeks from treatment initiation. Both patients were young men (ages 34 and 39 years) treated for their first recurrence. Nevertheless, the median progression-free survival for the entire study group was only 1.4 months, and the median overall survival was 5.7 months.
Fig. 2.
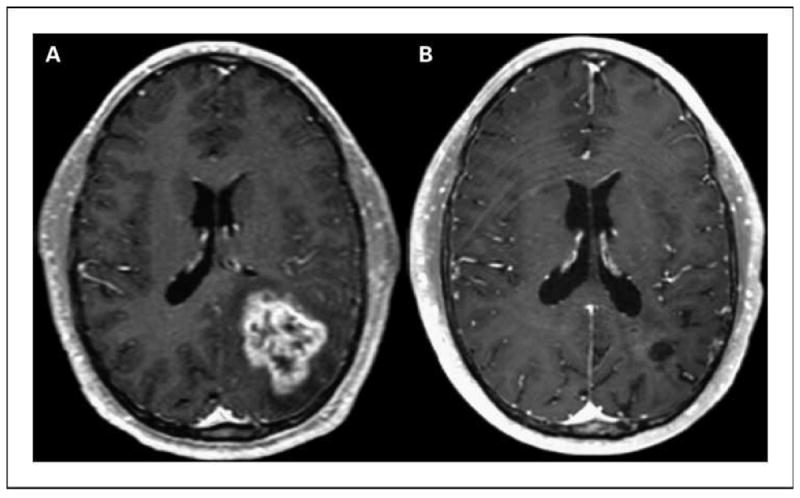
A, baseline study. B, complete response maintained on >3 y of therapy with enzastaurin.
Pharmacodynamics
Because enzastaurin inhibits the phosphorylation and activity of GSK3β in glioma cells in vitro (Supplementary Fig. S1; ref. 12), we analyzed pGSK3β in patient peripheral blood mononuclear cells as a potential biomarker of enzastaurin activity. Due to difficulties with both sample processing and collection, only six patients had pGSK3β assays successfully collected and done through steady-state. Five of these patients achieved disease control (one complete response, two partial response, and two stable disease) and had pGSK3β levels that were all decreased from baseline. The 6th patient had progressive disease and had increased pGSK3β at steady state after the first 3 weeks of therapy (Fig. 3A). Figure 3B shows the kinetics of pGSK3β for two patients after administration of enzastaurin, where levels in peripheral blood mononuclear cells decrease in a time dependent manner.
Fig. 3.
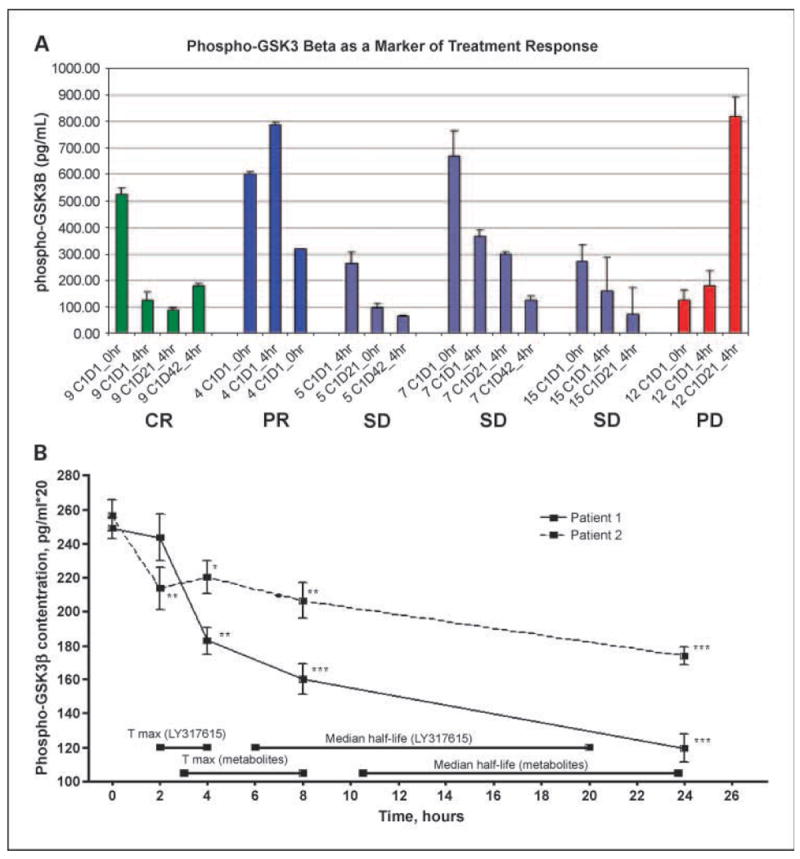
Phospho-GSK3b as a marker of treatment response. A, patients who achieved disease control [complete response (CR), partial response (PR), or stable disease] all had decreased levels of pGSK3β, in contrast to the patient who had disease progression and did not have suppression of pGSK3β with enzastaurin treatment. Error bars, one-SD of technical variation. B, time course of pGSK3β inhibition in peripheral blood mononuclear cells after enzastaurin administration. Peripheral blood mononuclear cells were harvested from patient blood samples obtained serially, and pGSK3β levels measured in triplicate through 96-well plate ELISA assays as described in Patients and Methods section. Points, mean; bars, SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
Enzastaurin is potentially a promising drug in the treatment of malignant glioma through its ability to inhibit PKCβ and thereby influence vascular endothelial growth factor–mediated signaling, as well as its ability to inhibit GSK3 (12). This trial investigated higher dosing and an alternate dose schedule in an effort to improve drug exposures of enzastaurin and its metabolites above the previously established phase II dose of 500 mg/day. We also explored pGSK3β as a potential biomarker of enzastaurin activity in vivo.
There were several reasons for wanting to evaluate higher serum levels of enzastaurin and its active metabolite in the treatment of gliomas. First, the phase II dose established by a prior phase I/II study was not a traditional toxicity-based maximum tolerated dose (14). Rather, it was determined by the pharmacokinetic end point of achieving a drug concentration that inhibits PKCβ in vitro. Whether this drug concentration maximally inhibits PKCβ in tumor-associated endothelial cells of human subjects is speculative. Given the wide variability in intrapatient pharmacokinetics, it is certainly possible that the current phase II dose does not optimally inhibit the target enzyme in all patients. Thus, higher serum enzastaurin levels may translate to higher concentrations of the drug in tumor-associated endothelium with clinical benefit.
Additionally, we observed that enzastaurin has direct cytotoxic activity against glioma cells in vitro, mediated at least in part through its ability to directly inhibit GSK3 (Supplementary Fig. S1). Both the GSK3 inhibitory effects and the cytotoxic effects of enzastaurin seem to be concentration dependent with higher drug levels resulting in greater inhibition of GSK3 and greater glioma cell cytotoxicity (Supplementary Fig. S1). Because this cytotoxic activity requires enzastaurin to directly reach tumor cells in situ (many of which reside behind a partially or fully intact blood-brain barrier), higher concentrations of drug may increase the total amount of drug that passes through the barrier for this lipophillic compound.
We hypothesized that the inability in previous clinical trials to increase serum concentrations of enzastaurin were secondary to saturation absorption in the gastrointestinal tract because all prior dosing had been given once per day. Thus, we designed this study to ask whether we could increase serum concentrations by dividing the dose and administering it twice rather than once daily. Given the significant interpatient variability in enzastaurin pharmacokinetics, we designed this trial so that each patient served as their own comparator between the daily and twice a day dosing.
Patients in our study experienced unacceptable toxicity at all dose levels tested, in contrast to prior studies where enzastaurin was very well-tolerated. Toxicity seemed to be dose dependent because DLTs were largely observed with twice daily dosing, which was in turn associated with 50% to 90% higher drug exposures than was the daily dosing. Furthermore, in contrast to the prior phase I/II study, where increasing doses of enzastaurin from 500 to 900 mg/day did not result in increasing serum levels of the drug (14), our study did result in significant increases in total analyte exposure between the 500 and 800 mg once daily cohorts. The reasons for these discrepant results are not clear but could simply be a statistical phenomenon related to the relatively small numbers of patients in both studies confounded by large interpatient variability in pharmacokinetics. One additional possibility is that in our study, we mandated that patients ingest their enzastaurin with a high-fat meal (at least 40 grams of fat per day) to maximize absorption of this lipid-soluble drug. Thus, we may have been able to partially overcome some of the presumed saturable absorption kinetics of single high-dose enzastaurin administration by administration of the drug with a fat-rich diet. Thus, although drug exposure could be increased by both administration of higher single doses of enzastaurin (possibly by the coadministration of a fat-rich diet) and by twice daily dosing, dose-limiting thrombocytopenia seems to preclude further study of higher dosing of enzastaurin in glioma patients.
Only a few complete data sets were available for evaluating pGSK3β as a biomarker of PKCβ inhibition. Interestingly, the five patients studied who achieved disease control all had decreased pGSK3β levels at steady-state compared with baseline values, whereas the one patient who had progressive disease at first follow-up had increased levels. This pattern suggests that sustained GSK3β down-regulation may be an integral part of the clinical activity of enzastaurin, although clearly, a larger number of patients will be required to confirm these observations.
From a clinical perspective, enzastaurin shows limited but potentially encouraging efficacy results for a subset of patients with recurrent malignant glioma. Two patients in this small cohort achieved long-term disease control, implying treatment effects beyond antiangiogenesis, possibly related to proapoptotic activity of enzastaurin. Studies of enzastaurin in other solid tumor malignancies have not shown the same objective response rates as seen in this and other trials of brain tumor patients (11, 14, 19), possibly suggestive of a unique direct effect of enzastaurin on glioma cells.
In conclusion, higher and more frequent daily dosing of enzastaurin can increase systemic exposure to drug but with unacceptable toxicity comprised primarily of thrombocytopenia and prolonged QTc. Enzastaurin remains a potentially promising drug in the treatment of recurrent malignant glioma and is currently being evaluated by the 500 mg once daily dosing schedule in combination with other agents. pGSK3β may be a useful biomarker for enzastaurin antiglioma activity and warrants further study as a correlative end point in future trials.
Supplementary Material
Translational Relevance.
Malignant gliomas are intrinsically resistant to treatment and carry a generally poor prognosis. Standard therapy for initial management of malignant glioma provides significant benefit for the minority of patients and virtually all eventually suffer from disease progression. Standard cytotoxic chemotherapies have failed to provide effective salvage regimens. Greater understanding of the molecular biology of these tumors has led to the development of novel agents specifically targeted against oncogenic pathways, with the potential to improve patient outcomes. The PKCβ inhibitor, enzastaurin, has shown clinical activity in patients with malignant glioma. This phase I trial improves our understanding of the pharmacokinetics and pharmacodynamics of this potentially useful new compound, and identifies a possible new biomarker of enzastaurin activity. Enzastaurin continues to be investigated in combination with other agents for this patient population.
Acknowledgments
Grant support: Eli Lilly and Company Cooperative Research and Development Agreement (CRADA #01364).
Footnotes
H. Fine, unpublished data.
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
H.A. Fine, Cooperative Research and Development Agreement, Eli Lilly.
References
- 1.Fine HA. Chemotherapy in the treatment of malignant astrocytomas in adults. In: Black PLL, editor. Astrocytomas. Boston: Blackwell; [Google Scholar]
- 2.Shen BQ, Lee DY, Zioncheck TF. Vascular endothelial growth factor governs endothelial nitricoxide synthase expression via a KDR/Flk-1 receptor and a protein kinase C signaling pathway. J Biol Chem. 1999;274:33057–63. doi: 10.1074/jbc.274.46.33057. [DOI] [PubMed] [Google Scholar]
- 3.Aiello LP, Bursell SE, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective β-isoform-selective inhibitor. Diabetes. 1997;46:1473–80. doi: 10.2337/diab.46.9.1473. [DOI] [PubMed] [Google Scholar]
- 4.Danis RP, Bingaman DP, Jirousek M, Yang Y. Inhibition of intraocular neovascularization caused by retinal ischemia in pigs by PKCβ inhibition with LY333531. Invest Ophthalmol Vis Sci. 1998;39:171–9. [PubMed] [Google Scholar]
- 5.Jirousek MR, Gillig JR, Gonzalez CM, et al. (S)-13-[(dimethylamino)methyl]-10,11,14,15-tetrahydro-4,9:16, 21-dimetheno-1H, 13H-dibenzo[e k] pyrrolo[3,4-h][1,4,13]oxadiazacyclohexadecene-1,3(2H)-d ione (LY333531) and related analogues: isozyme selective inhibitors of protein kinase C β. J Med Chem. 1996;39:2664–71. doi: 10.1021/jm950588y. [DOI] [PubMed] [Google Scholar]
- 6.Yoshiji H, Kuriyama S, Ways DK, et al. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res. 1999;59:4413–8. [PubMed] [Google Scholar]
- 7.Aeder SE, Martin PM, Soh JW, Hussaini IM. PKC-eta mediates glioblastoma cell proliferation through the Akt and mTOR signaling pathways. Oncogene. 2004;23:9062–9. doi: 10.1038/sj.onc.1208093. [DOI] [PubMed] [Google Scholar]
- 8.Kawakami Y, Nishimoto H, Kitaura J, et al. Protein kinase C βII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279:47720–5. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- 9.da Rocha AB, Mans DR, Regner A, Schwartsmann G. Targeting protein kinase C: new therapeutic opportunities against high-grade malignant gliomas? Oncologist. 2002;7:17–33. doi: 10.1634/theoncologist.7-1-17. [DOI] [PubMed] [Google Scholar]
- 10.Gokmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP. Elevated protein kinase C βII is an early promotive event in colon carcinogenesis. Cancer Res. 2001;61:1375–81. [PubMed] [Google Scholar]
- 11.Shipp MA, Ross KN, Tamayo P, et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat Med. 2002;8:68–74. doi: 10.1038/nm0102-68. [DOI] [PubMed] [Google Scholar]
- 12.Kotliarova S, Pastorino S, Kovell LC, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-κB, glucose regulation. Cancer Res. 2008;68:6643–51. doi: 10.1158/0008-5472.CAN-08-0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carducci MA, Musib L, Kies MS, et al. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C β inhibitor, in patients with advanced cancer. J Clin Oncol. 2006;24:4092–9. doi: 10.1200/JCO.2005.05.3447. [DOI] [PubMed] [Google Scholar]
- 14.Fine HA, Kim L, Royce C, et al. Results from phase II trial of enzastaurin (LY317615) in patients with recurrent high grade gliomas. ASCO Meeting Abstracts; 2005. p. 1504. [Google Scholar]
- 15.Levin VA, Crafts DC, Norman DM, Hoffer PB, Spire JP, Wilson CB. Criteria for evaluating patients undergoing chemotherapy for malignant brain tumors. J Neurosurg. 1977;47:329–35. doi: 10.3171/jns.1977.47.3.0329. [DOI] [PubMed] [Google Scholar]
- 16.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–80. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 17.Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol. 2006;61:246–55. doi: 10.1111/j.1365-2125.2005.02529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riva R, Albani F, Contin M, Baruzzi A. Pharmacokinetic interactions between antiepileptic drugs. Clinical considerations. Clin Pharmacokinet. 1996;31:470–93. doi: 10.2165/00003088-199631060-00005. [DOI] [PubMed] [Google Scholar]
- 19.Oh Y, Herbst RS, Burris H, et al. Enzastaurin, an oral serine/threonine kinase inhibitor, as second- or third-line therapy of non-small-cell lung cancer. J Clin Oncol. 2008;26:1135–41. doi: 10.1200/JCO.2007.14.3685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.