

Abstract
Single ventricle heart defects (SVDs) are congenital disorders that result in a variety of complications, including increased ventricular mechanical strain and mixing of oxygenated and deoxygenated blood, leading to heart failure without surgical intervention. Corrective surgery for SVDs are traditionally handled by the Fontan procedure, requiring a vascular conduit for completion. Although effective, current conduits are limited by their inability to aid in pumping blood into the pulmonary circulation. In this report, we propose an innovative and versatile design strategy for a tissue engineered pulsatile conduit (TEPC) to aid circulation through the pulmonary system by producing contractile force. Several design strategies were tested for production of a functional TEPC. Ultimately, we found that porcine extracellular matrix (ECM)-based engineered heart tissue (EHT) composed of human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) and primary cardiac fibroblasts (HCF) wrapped around decellularized human umbilical artery (HUA) made an efficacious basal TEPC. Importantly, the TEPCs showed effective electrical and mechanical function. Initial pressure readings from our TEPC in vitro (0.68 mmHg) displayed efficient electrical conductivity enabling them to follow electrical pacing up to a 2 Hz frequency. This work represents a proof of principle study for our current TEPC design strategy. Refinement and optimization of this promising TEPC design will lay the groundwork for testing the construct’s therapeutic potential in the future. Together this work represents a progressive step toward developing an improved treatment for SVD patients.
Keywords: Human induced pluripotent stem cells, Fontan Conduit, TEPC, Engineered heart tissue Tissue engineering
1. Introduction
Single ventricle defects (SVD) represent a class of congenital heart disorders that may be caused by a diverse group of structural anomalies that result in the formation of one functional ventricle, estimated to affect somewhere between 1 and 2 in 200 0 live births [1,2]. Children born with these defects have a 70% mortality rate if there is no appropriate surgical intervention [3]. Abnormal pulmonary blood flow, noted in SVD patients, has been linked to aberrant vascular remodeling that subsequently increases local resistance in the pulmonary circulation, which has been observed to increase the risk of clots and the hepatic blood stagnation that leads to chronic liver disease [4,5]. The Fontan procedure is the typical treatment for patients with single ventricle defects, where venous blood is drained directly to the pulmonary artery and passively travels through the pulmonary circulation [6]. Tissue-engineered vascular grafts have been used for the Fontan procedure because this strategy forgoes additional surgeries to harvest autologous vessels for these young patients, who are usually 2–3 years of age [3]. These grafts have been generated by seeding autologous bone marrow cells onto a synthetic, biodegradable tubular scaffold to induce infiltration of host cells to remodel the implant and ultimately lead to new vessel formation as the biomaterial is replaced with natural extracellular matrix (ECM) and cells [7,8]. This strategy has been used in clinical trials where these grafts displayed moderate safety and efficacy [6,9,10]. The main concept in this approach is that these grafts would become fully biologic and ideally be responsive to local injury and changing hemodynamics as well as grow with the child. This is in contrast to grafts that need to be replaced periodically throughout adolescence, as necessary with fully synthetic grafts [11,12]. Although this engineered tissue approach has shown some early success in clinical trials, the strategy does not address the need to provide pumping activity to assist the single ventricle. Decreased cardiac output is seen in SVD patients ultimately leads to insufficient blood perfusion to tissues, heart failure, and the development of pulmonary vascular diseases [2,4–6]. To decrease the risk of these cardiovascular pathologies, a contractile Fontan conduit is necessary that can act as a pumping assist device to improve blood flow and pressure in the pulmonary circulation. A pulsatile conduit may further improve overall heart function by alleviating excess stress of the single ventricle which is currently tasked with providing both systemic and pulmonary circulation pressure.
Previously, bioengineered cardiac tissues-derived from human induced pluripotent stem cells (hiPSCs) or neonatal rat cardiomyocytes (CMs) were developed to support circulation [13,14]. These tissues also used an in vivo development strategy by transplanting hiPSC-derived cardiac sheets around the inferior vena cava (IVC) and abdominal aorta. Although these designs were able to produce discernable pressure changes in vivo, they were not capable of producing recordable pressures without clamping the tissue and introducing luminal pre-load pressures well above the physiological value of the IVC, where Fontan conduits would be placed [13,14]. Additionally, beating frequency was sporadic in the absence of ectopic pacing of the hiPSC-based construct [13]. It is likely that in vivo mechanical training by the pulsatile environment of the aorta aided in the maturation of the CMs in these tissues, as noted in other engineered heart tissue applications [15–17]. However, the low-pressure mechanical environment of the vena cava, where Fontan conduits are implanted, may not provide the same benefits. A group performing a direct to in vivo strategy in the venous circulation did not report any robust contractility of their construct [18]. For this reason, that a standalone tissue engineered pulsatile conduit (TEPC) construct is needed for optimization in vitro, before moving to in vivo systems.
Here, we propose an innovative strategy for the production of hiPSC-derived CMs-based TEPCs to address shortcomings of previous attempts. First, three biomaterial scaffolds including polyglycolic acid (PGA), collagen type I, and decellularized porcine ventricle heart tissue used to generate engineered heart tissues (EHTs) with hiPSC-CMs and tested for contractile capacity. Second, the cell composition of the EHTs was tuned to enhance their contractile function. Finally, a robust TEPC was produced by wrapping compositionally optimized EHTs around decellularized human umbilical artery (HUA). Through the use of robust and scalable EHTs our group was able to produce a functioning TEPC with observable pressure generation in vitro. providing the basis for future studies to mature the tissue and test therapeutic potential in vivo. By optimizing each component of the TEPC, this work provides a solid foundation for the design of a robust contractile Fontan conduit.
2. Materials and methods
2.1. Animal rights
All experimental procedures involving animals were performed under the approval of the Institutional Animal Care and Use Committee (IACUC) of the Yale University. Animal care was performed in compliance with the NIH Guidelines for the Care and Use of Laboratory Animals.
2.2. Maintenance of hiPSCs and induction and enrichment of cardiomyocytes from hiPSC-CMs
hiPSCs were cultured in human embryonic stem cell (hESC) medium [20% KnockOut serum (KOSR) in Dulbecco’s modified eagle medium (DMEM/F12) medium supplemented with 10 ng/mL bFGF 1% non-essential amino acid (v/v), 1% Pen/Sep (v/v), 0.1 mM β-Mercaptoethanol (Sigma-Aldrich) and 2 mM L-Glutamine (all from ThermoFisher)]. During expansion, hiPSCs were cultured on irradiated mouse embryonic fibroblasts (MEFs) which are known to promote stem cell growth through secretion of soluble factors in the media [19]. 70–80% confluent (~4–5 day old) hiPSCs cultured on MEFs were treated with 1 mg/mL Dispase (ThermoFisher) for 7–9 min in 37 °C. The hiPSCs were mechanically dissociated using a 5 mL pipette, collected in a 15 mL falcon tube, and spun down at 200 RPM for 4 min to deplete the MEF cells. The hiPSCs were then plated on Growth Factor Reduced Matrigel (GFR-Matrigel; Corning,1:60 diluted) -coated wells for culture. The hiPSC were cultured for ~3–4 days in mTeSR medium (Stemcell Technologies) until ~90% confluency was reached. Cells were then treated with 20 μM CHIR99021(Selleckchem) on Day 0 for 24 h. The following day the media was changed to Roswell Park Memorial Institute (RPMI) 1640 (Gibco) supplemented with 1% B27 (Gibco) minus insulin and no additional growth factors. On Day 3, 5 μM IWP4 (Stemgent) was added to RPMI/B27(−) insulin media for 48 h. During the cardiac differentiation, the media was changed every other day with RPMI/B27(−) insulin media. After beating, iPSC-CMs were cultured in RPMI 1640 supplemented with 1% B27 complete (Gibco). hiPSC-CMs at Day 12 were treated with 4 mM lactate (Sigma-Aldrich) in glucose-free medium for two days to enrich hiPSC-CMs and then switched back to RPMI/B27(+) complete media.
2.3. Generation of beating PGA construct with hiPSC-CMs
Nonwoven-PGA polymer mesh (0.3 mm x 150 mg/cc, 20 cm x 30 cm sheet; BIOFELT) was cut into 5 mm x 5 mm squares and prepared for cell seeding as described previously [20,21]. Briefly, the PGA squares were immersed in 1.0 N sodium hydroxide (NaOH; Sigma-Aldrich) for 1 min to increase the fiber absorption of liquid. Meshes were then soaked in distilled water three times for 2 min each to remove the residual NaOH, sterilized with 70% ethanol (Sigma-Aldrich), and air-dried overnight under sterile conditions. Before seeding the cells, the PGA squares were coated with 0.1% gelatin (Sigma-Aldrich) at 37 °C for 1 h and then 0.7 × 106 hiPSC-CMs were seeded onto dried PGA and cultured for two weeks.
2.4. Generation of beating ring with hiPSC-CMs mixed with collagen gel
A polydimethylsiloxane (PDMS) mold (Dow Corning SYLGARD-184) was used to cast wells from 2% agarose solution in DMEM. The PDMS mold was prepared by mixing the elastomer base and curing reagent in a 9:1 ratio. The mixture was degassed in a vacuum chamber for 10 min to remove any air bubbles, and then cured at 37 °C overnight [22]. The agarose molds were equilibrated in RPMI 1640/B27(+) complete media with 10% fetal bovine serum (FBS; Gibco) overnight. To fabricate the ring-shaped tissue, 1.2 × 106 hiPSC-CMs on differentiation Day 14 were mixed in 33 μL of gel mixture and 6.4 μL of RPMI1640/B27(+) complete media. The cell suspension was then pipetted into a ring-shaped agarose mold. After 1 h, 5 mL of medium was added to each well. The medium was changed every other day and the rings were cultured for 14 days after seeding.
2.5. Generation of beating EHT with hiPSC-CMs
Engineered heart tissues were generated as previously described [23] with minor modifications. Briefly, laser-cut porcine ventricle heart tissues were mounted into a custom laser cut polytetrafluoroethylene (PTFE) culture device and decellularized with 0.5% sodium dodecyl sulfate (SDS; RPI) diluted in DPBS (Gibco) for 40 min. Decellularized tissues were washed with DPBS three times and incubated overnight at 37 °C with MEF media (DMEM with glucose, 10% FBS, 1% nonessential amino acids [NEAA], 1% L-glutamine, 1% penicillin streptomycin [P/S], 1% sodium pyruvate [all Gibco]; sterile filtered at 0.22 μm). For seeding, scaffolds were moistened with 50 μL of MEF media in the custom PDMS seeding wells. For standard sized EHTs, 75 μL of media containing 1 × 106 cells were seeded onto EHT evenly. After 2 h, 500 μL of MEF media was added, EHTs were cultured, undisturbed, for two days. For testing the effect of cellular composition on EHT contractility, we generated EHTs with a 7:3 ratio of enriched hiPSC-CMs and commercially available adult human cardiac fibroblasts (HCF; ScienCell, #6300). For TEPC generation, we scaled this tissue up to a scaffold size of 15 × 14.5 cm, and seeded 7 × 106 and 3 × 106 of enriched hiPSC-CMs and HCF, respectively. The medium was changed every other day with RPMI 1640/B27(+) complete and EHTs were cultured for 14 days after seeding the cells.
2.6. Umbilical artery decellularization
Umbilical cords were obtained from Yale New Haven Hospital with all patient identifying information removed prior to acquisition. The cords were dissected, and arteries were removed. Using tweezers, any remaining Wharton’s jelly was removed from the artery prior to decellularization as outlined in a previous study [24]. Cleaned arteries were placed in a CHAPS detergent buffer [8 mM CHAPS detergent (Dot Scientific), 25 mM ethylenediaminetetraacetic acid (EDTA; American Bioanalytical), 1 M sodium chloride (NaCl; American Bioanylitical)] for 24 h with vigorous shaking at 37 °C. The vessels were then washed with sterile DPBS and incubated in SDS buffer (1.8 mM SDS, 25 mM EDTA, 1 M NaCl) for 24 h with vigorous shaking at 37 °C. Arteries were then liberally washed with sterile DPBS to remove any remaining SDS. The now decellularized arteries were then incubated in DPBS supplemented with 20% FBS overnight to remove any remaining nuclei.
2.7. TEPC production
Collagenase treated and decellularized HUA were submerged in 0.1% gelatin and allowed to sit for 30 min to help fibroblasts initially adhere. After coating, 1.5 × 106 fibroblasts in 400 μL of cell suspension, were seeded on the HUA in a Petri dish for 1 h The HUA was rotated every 15 min during this one-hour period, and remaining fibroblast suspension was reapplied. An EHT, 4 days after seeding, was manually wrapped around the HUA to produce a TEPC. TEPCs were cultured on a mandrill in T75 flask for five days prior to testing.
2.8. Cytospin
Cells were detached from the culture plate using Accutase (Sigma-Aldrich) and washed with DPBS and resuspended in RPMI 1640/B27 (+) complete media at a concentration of 1 × 105 cells/100 μL. 100 μL of cell suspension was added to a slide chamber and spun at 400 RPM for 4 min. Slides were removed from cytocentrifuge, allowed to air dry and fixed with 4% Paraformaldehyde (PFA; Sigma-Aldrich) /DPBS prior to staining.
2.9. TEPC fixation, embedding, and sectioning
The TEPCs were washed with DPBS and fixed in 4% PFA for 2 h at room temperature (RT). The samples were then equilibrated in 30% sucrose (Sigma-Aldrich) and DPBS overnight until the sample sinks. With increasing ratios, the TEPCs were soaked in 1:5 optimal cutting temperature (OCT; Tissue-Tek) and then 1:1 of OCT. The TEPC was divided into three equal portions and position in a mold and embedded in 100% OCT with no bubbles until fully submerged. The three portions were sectioned using a cryostat (Leica Biosystems) and mounted on the slide for staining. The sections were washed with DPBS two times and incubated with primary antibodies: anti-cardiac troponin-T (cTNT, 1:500, Thermofisher, MS-295-P0 and Abcam, ab92546), anti-vimentin (1:400, Abcam, ab8069) diluted in blocking solution (5% normal goat serum [NGS; Thermofisher] in PBS) overnight at 4 °C in a humidified chamber. Sections were then washed three times with DPBS, incubated with appropriate secondary antibodies diluted 1:500 in blocking solution for 1 h at RT, washed again three times. The slides were mounted with mounting solution containing 4’ , 6-diamidino-2-phenylindole (DAPI; Invitrogen). Images were taken using a SP8 LIGHTNING Confocal Microscope (Leica Microsystems).
2.10. TUNEL assay
TUNEL assay was performed by In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s instructions. Three tissue sections were prepared for negative control, positive control, and sample. Tissue sections were washed with DPBS twice and permeabilized with 0.1% Triton X-100 (Fisher) for 20 min, then washed with DPBS. For positive control, tissue section was incubated with DNase I recombinant (1 mg/mL, Roche) for 10 min at RT. After treatment of DNase I for a positive control, all samples were incubated in 50 μL per samples (50 μL of Label solution for negative control and 45 μL of Label solution and 5μL of Enzyme solution for positive control and TUNEL sample) for 1 h in 37 °C. Images were acquired using a Nikon 80i microscope and captured using NIS-Elements AR software.
2.11. Tissue immunohistochemistry and histology
Tissue sections were boiled in 10 mM citrate buffer (pH 6) in 95 °C for 20 min and incubated with primary antibodies diluted in blocking solution (5% NGS in DPBS) overnight at 4 °C in a humidified chamber. Sections were washed three times with Tris-buffered saline, incubated with appropriate secondary antibodies diluted 1:500 in blocking solution for 1 h at RT, washed again three times. Images were acquired using a Nikon 80i microscope and captured using NIS-Elements AR software. For hematoxylin and eosin and Masson’s Trichrome staining, the tissue samples were paraffin embedded and cut into section of 5 μm by Yale Pathology Tissue Services based on standard protocol. Primary antibodies included: anti-cTNT (1:500, Thermo, 13-11), anti-vimentin (1:400, Abcam, ab8069), and anti-collagen I (1:300, Abcam, ab34710).
2.12. Alkaline phosphatase activity assay and immunostaining of cultured cells
Alkaline phosphatase activity was evaluated by using alkaline phosphatase staining kit II (Stemgent) according to the manufacturer’s instructions. To conduct immunostaining, cells were fixed with 4% paraformaldehyde (PFA, Electron Microscopy Sciences) and blocked by 10% goat serum (ThermoFisher) in PBST buffer (Dulbecco’s Phosphate-Buffered Saline (PBS, ThermoFisher) with 0.1% triton X-100 (Sigma–Aldrich)) for 30 min at room temperature. Cells were then incubated with primary antibody in PBST containing 1% goat serum at 4 °C overnight. Primary antibodies include: anti-NANOG (Abcam, ab218524), anti-SSEA-4 (Millipore, MAB4304), anti TRA-1–60 (Cell Signaling, 4746P), anti-Oct4 (Abcam, ab18976) and anti-cTnT (Thermo, 13-11). All dilutions were performed as directed by antibody manufacturers. Cells were then washed and incubated with secondary antibody (1:1000 in PBST with 1% goat serum) for 1 h at room temperature and washed with PBS. Nuclei were counterstained with DAPI (Thermo). Stained samples were analyzed using a fluorescent microscope (Leica Microsystems).
2.13. Karyotyping analysis
As previously described, hiPSC-CMs were cultured on GFR-Matrigel-coated cover slips and sent to the laboratory of Yale Cytogenetic Services [21]. Karyotyping analysis was completed via standard G-banding chromosome analysis according to the standard procedures. Ten cells were randomly selected and analyzed.
2.14. Teratoma formation
After lactate selection and two days of recovery, hiPSC-CMs were dissociated with 10 μm/mL collagenase A and B (Roche) for 30 min, followed by Accutase for 10 min, spun down at 1000 RPM for 5 min. 5 × 106 cells were then mixed with 100 μL of RPMI 1640/B27 (+) complete, 5 μM Y-27632 (ROCK Inhibitor; STEMCELL Technologies) and 70 μL of GFR-Matrigel on ice. hiPSCs were dissociated with dispase for 8 min and spun down at 200 RPM for 3 min. Then, 5 × 106 cells were mixed with 100 μL of media (hESC), 5 μM Y-27632 and 70 μL of GFR-Matrigel on ice, and then subcutaneously injected into 8- to 12-week-old nonobese diabetic SCID-γ mice (Taconic). Tumors were collected between 6 and 8 weeks after the cell injection.
2.15. Mechanical and functional testing of EHTs
As previously described [23], EHTs were mechanically tested in a temperature-controlled perfusion bath equipped with electrodes for field stimulation. Throughout the measurements, scaffolds were perfused with freshly oxygenated Tyrode’s solution (140 mM of sodium chloride [NaCl], 5.4 mM of potassium chloride [KCl], 1 mM of magnesium chloride [MgCl2 ], 25 mM of HEPES, 10 mM of glucose, and 1.8 mM of calcium chloride [CaCl2 ]; pH adjusted to 7.30 and all purchased from Sigma-Aldrich) in 37 °C. Peak active force measured at 5% stretch from culture length and during 1 Hz field stimulus was normalized by cross-sectional area as determined by optical coherence tomography in order to calculate peak stress.
2.16. Lumen pressure measurement and electrical pacing
The TEPC was tied onto the inner ends of two cannulas of a specialized glass dish filled with warm Tyrode’s buffer. The whole set up was then placed in a large, normoxic 37 °C incubator. One cannula was hooked up to a syringe pump for adjustment of preload lumen pressure, the other cannula was open for insertion of a Millar catheter pressure sensor (Size 1.4 F, Model SPR-671). The catheter was calibrated and pre-equilibrated in warm Tyrode’s buffer before being inserted in the lumen of the TEPC. Once set, carbon electrodes were placed on both sides of the TEPC and connected to a WPI A385 stimulus isolator box. Diligent Wave- Forms generator was used to control the electrical input from the stimulator box. A square wave of 10 ms impulses of 60 mA current was applied to the TEPC at 1 and 2 Hz frequencies. PowerLab software was used to record pressure changes in real time during measurements.
2.17. Statistics
All experimental samples were run in either duplicate or triplicate, and in at least three biological repeats. Differences between samples were considered significant if P-value < 0.05 as determined using unpaired two tailed Student’s T-test and One-Way ANOVA. All graph illustrations and statistical analyses were compiled using GraphPad Prism8 and the data were presented as means +/− standard error of the mean.
3. Results
3.1. Validation of safety and function of hiPSCs and hiPSC-CMs
The hiPSC line used in this work was derived from female neonatal skin tissue as described previously, and differentiated into CMs according to schematic diagram inFig. 1A [20,22,25]. First, validation of the expression of pluripotency markers including OCT4, NANOG, SSEA4, TRA-1–60 and alkaline phosphatase (AP) were confirmed through immunofluorescence staining in these hiPSCs (Fig. 1B). Cardiac differentiation was induced through temporal Wnt signaling modulation as previously reported [25]. hiPSC-CMs start to beat between 8–10 days after the addition of CHIR99021, a GSK3 small molecule inhibitor that activates Wnt signaling. Once these cells start to beat, a metabolic [lactate] selection method was used to enrich each differentiation batch for hiPSC-CMs [26]. Immunostaining of hiPSC-CM culture also confirmed the expression of CM marker, cTnT, and lack of pluripotency markers such as OCT4, NANOG, SSEA4 and TRA 1–60 (Fig. 1C). Additionally, a transient 2-day metabolic selection significantly enriched hiPSC-CMs (Fig. 1D). Karyotype analysis of hiPSC-CMs also revealed stable normal chromosomal integrity (Fig. 1E). To further examine the safety of using hiPSC-CMs in vivo, 5 × 106 hiPSCs were injected into left hind limb of immunocompromised (Rag2−/−;Il2rg−/−) mice as a positive control, and 5 × 106 of enriched hiPSC-CMs in the right hind limb of the same animal. We observed a teratoma from the left hind limb of mice injected with hiPSCs (Fig. 1F and G), but not from right hind limb injected with hiPSC-CMs (Fig. 1F). Representative tissues of endoderm, mesoderm and ectoderm were noted in the hiPSCs-derived teratoma tissue (Fig. 1H–J).
Fig. 1.
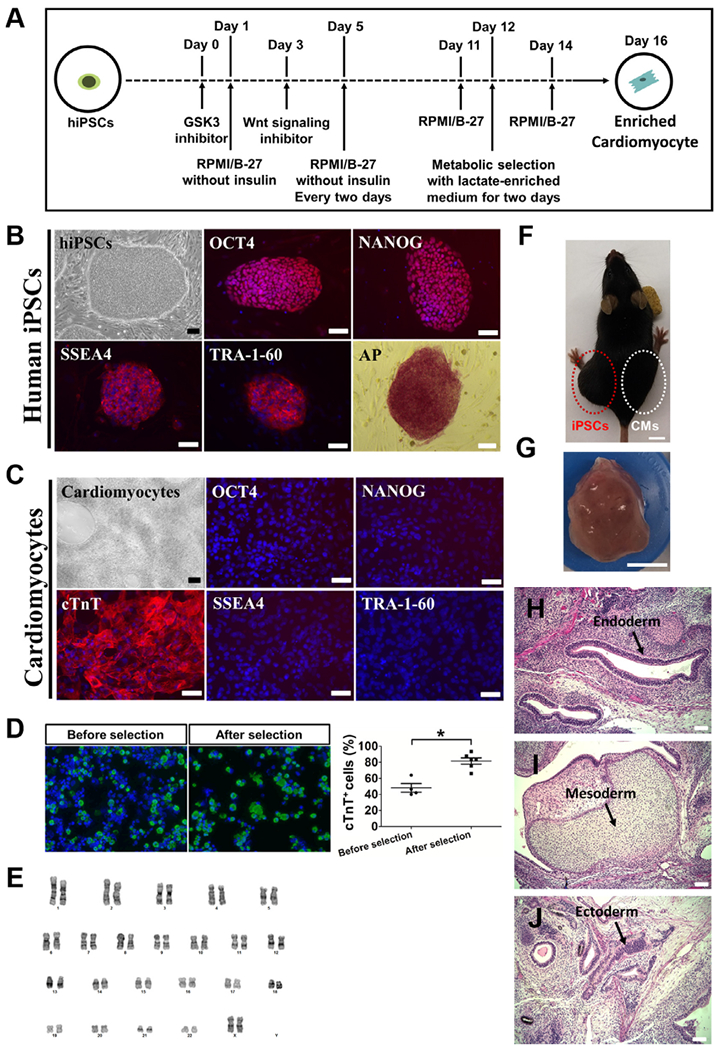
Validation of hiPSCs and the evaluation of the safety of using hiPSC-CMs. (A) Schematic diagram depicting design strategy of cardiac differentiation from hiPSC. (B) Typical hiPSC colony and immunostaining with pluripotency markers (OCT4, NANOG, SSEA-4 and TRA-1–60). hiPSCs were positive for all pluripotency markers and the activity of alkaline phosphatase (AP) was observed. Nucleus was stained by DAPI. Scale bars = 100 μm. (C) Typical iPSC-CMs and positive staining for cTnT (red). Nucleus was stained by DAPI. hiPSC-CMs were negative for pluripotency markers. Scale bars = 100 μm (D) Day 12 culture were then grown in glucose-depleted DMEM media containing 4 mM lactate for 2 days to enrich cardiomyocytes. Cells (before and after enrichment) were dissociated and cytospun onto the glass slides for hiPSC-CM quantification by cTnT (green). Nucleus was stained by DAPI. Scale bars=100 μm. Samples were analyzed by Student’s T-test (*P < 0.05). (E) Karyotyping of hiPSC-CMs demonstrated stable normal chromosomal integrity. (F) Examination of teratoma formation from hiPSCs (left hind limb) and hiPSC-CMs (right hind limb) using immunodeficient mice. Scale bars = 1 cm. (G) Teratoma generated from hiPSCs injected to left hind limb. Scale bars=1 cm. (H-J) Representative H&E stained sections derived from teratoma. Arrows indicate gut-like epithelium (endoderm) (H) , cartilage (mesoderm) (I) , and neuroepithelial rosettes (ectoderm) (J). Scale bars= 100 μm. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Three biomaterial scaffold approaches for EHT generation using hiPSC-CMs
Three biomaterial scaffolds (PGA, collagen type I, and decellularized porcine ventricle ECM) were used to establish preliminary EHT candidates that best suited the design of a functional hiPSC-CM-based TEPC according to the illustration shown in Fig. 2. Because our group has been previously successful in making engineered vascular tissues using PGA [20], initial attempts to make robust EHTs used this scaffold. 7 × 105 beating hiPSC-CMs (Supplementary Video S1) were seeded on a flat 5 × 5 mm PGA mesh, in a dropwise fashion to encourage even cell distribution, and cultured for two weeks (Fig. 2A, B). Contractility of hiPSC-CMs seeded on PGA was visually weak (Supplementary Video S2, Supplementary Table S1). This is likely due to poor alignment of the hiPSC-CMs and poor remodeling of the PGA scaffold, which is noted to occur over much longer timescales in other applications of this scaffold material [20,27,28]
Fig. 2.
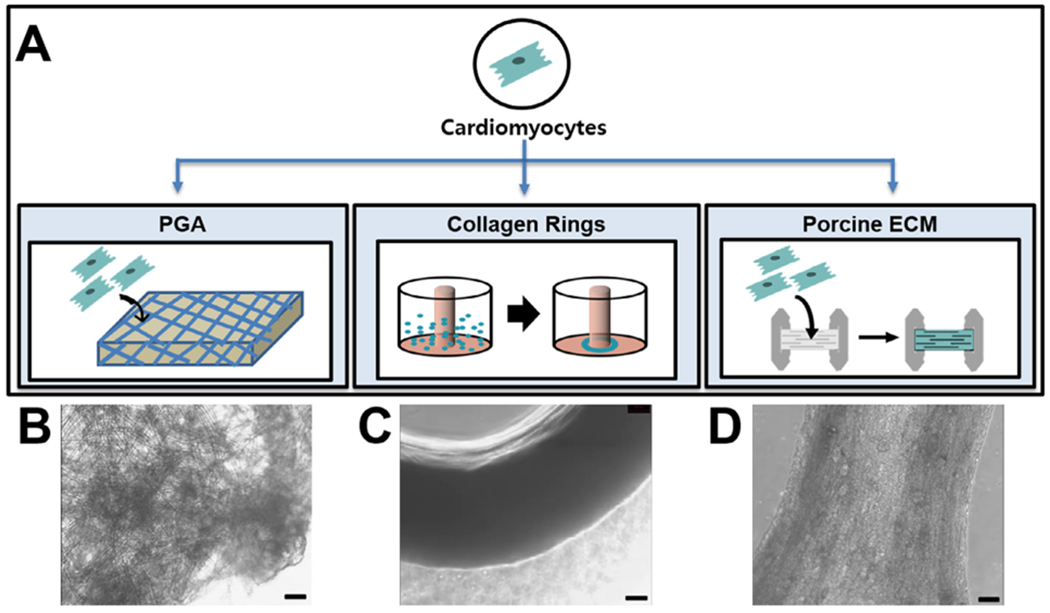
Design strategies for EHT system using hiPSC-CMs. (A) Schematic diagram depicting design strategy to produce EHTs. hiPSCs can be differentiated into beating hiPSC-CMs via temporal Wnt signaling modulation. hiPSC-CMs were then seeded into one of three tested scaffolds including degradable PGA, rat tail-derived collagen type I and decellularized porcine ECM with controllable fiber alignment (B, C and D) for the scaffold for engineering the cardiac tissue. For beating PGA, 0.7 million hiPSC-CMs were seeded onto 5 mm × 5 mm squares of PGA and cultured for two weeks. 1.2 million hiPSC-CMs were seeded into the wells in 2% agarose molds for the generation of beating cardiac rings. 1 million cells were seeded onto thin sections of decellularized porcine myocardial extracellular matrix. Scale bars = 100 μm.
The second strategy employed was a collagen gel suspension to generate EHTs. We have previously produced vascular smooth muscle cell rings for disease modeling applications using this method [22]. Interestingly, these vascular rings could be fused together to make long tube structures and theoretically cardiac-based rings could also be fused to form a functional TEPC. However, this strategy first needed to be validated for whether contractile EHT rings could be produced via an adaptation of this previously published method [22]. 1.2 × 106 hiPSC-CMs on differentiation Day 14 were mixed with a rat tail-derived collagen type I gel on a ring-shaped mold and cultured for two weeks (Fig. 2A, C). EHT rings showed relatively strong visual contractility in some batches, but it was rather inconsistent between batches (Supplementary Video S3, Supplementary Table S1). It has been noted in other EHT systems that a benefit in contractility can be achieved from the introduction of fibroblasts and endothelial cells, likely due to enhanced remodeling and paracrine signaling induced hypertrophy [29–31]. We hypothesized that a co-culture system with these cell types could improve the production of strong contractile rings.
EHT rings were prepared with either hiPSC-CMs and human umbilical vein endothelial cells (HUVECs) or commercially available HCFs in a collagen gel as previously described. However, distribution of hiPS-CMs in the ring was not improved with the introduction of HUVECs (Fig. S1). In contrast, the hiPSC-CMs and HCF co-culture rings showed a much more even distribution when compared to rings made with only hiPSC-CMs (Fig. S1). Despite the improvement of cell distribution seen with the addition of HCFs, neither of these co-culture systems were able to solve the previous issue of inconsistent contractility.
The third design strategy to produce robust EHTs adapted a natural cardiac ECM-based approach (Fig. 2A,D), previously established by our group [23]. Many researchers use decellularized ECM scaffolds to produce EHT of varied scales [23,32]. ECM-based biomaterials are noted to have naturally tuned biomechanical and biochemical properties in addition to an inherent topology to help guide the organization of the engineered tissue [23,33,34]. Proper alignment of myocardial fibers has been hypothesized to play a critical role in maximizing the contractile output of the cardiac tissue. Briefly, the left ventricles of pig hearts were isolated and processed into flat blocks and frozen on powdered dry ice before sectioning 150 μm sheets. These sheets were subsequently laser cut and attached to PTFE frames before decellularization using SDS detergent. Decellularized porcine ECM scaffolds were seeded with non-enriched hiPSC-CMs (1 × 106 cells) and statically cultured for 14 days before measuring the force output using a specialized apparatus (Fig. 3A).
Fig. 3.
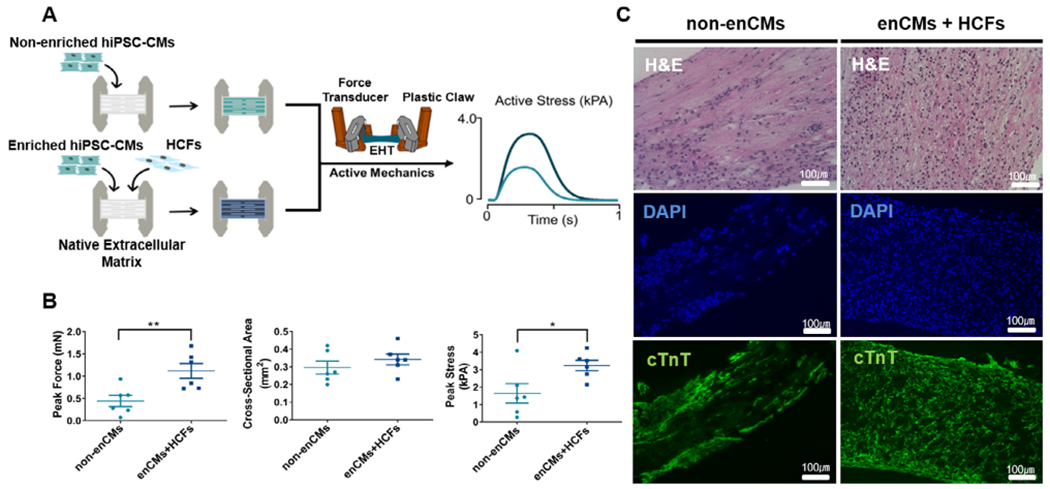
Histological comparison and contractile function of EHTs generated from two types of ceil compositions including non-enriched hiPSC-CMs and enriched hiPSC-CMs with HCFs. (A) Schematic diagram depicting design strategy for EHTs. (B) Peak force, cross-sectional area and peak stress of two EHT groups including non-enriched hiPSC-CMs (non-enCMs, 1 million) as well as enriched hiPSC-CMs (0.7 million) with HCF (0.3 million) (enCMs + HCFs). Peak force and peak stress generated in EHTs with HCFs were significantly increased (Student’s T-test; *: p < 0.05, **: p < 0.01) with no change in cross-sectional area (CSA) (Student’s T-test, p = 0.42). (C) EHTs made from either non-enCMs (1 million) alone or co-culture of enCMs (0.7 million) and HCFs (0.3 million) were stained with H&E and cTnT. Nuclei were stained by DAPI. Scale bars = 100 μm.
To move forward with development of an efficacious TEPC design strategy, a direct comparison of the contractility of each scaffold type was needed. In order to compare these EHTs, strain data was gathered from videos during contraction of hiPSC-CMs for each tested scaffold material (Supplementary Table S1). Upon quantification of contractility by measuring the global strain, a trend of consistent contractility with least variations was observed in the EHTs generated from the porcine ECM (strain value for PGA = 0.30±0.07; collagen ring = 1.72±1.39; porcine ECM = 2.56±0.44) as opposed to the PGA and collagen ring-based scaffolds (Supplementary Table S1). These data indicated that the porcine ECM scaffold material was the most suitable to use moving forward, when making a functional TEPC (Supplementary Table S1, Supplementary Video S4).
Similar to what was attempted in the collagen ring strategy, the effects of a co-culture system in our porcine ECM-based EHTs were hypothesized to improve contractility and remodeling. To investigate whether HCF may enhance the contractile function of EHTs, the force was measured in EHTs generated by non-enriched hiPSC-CMs alone and EHTs generated by enriched hiPSC-CMs co-cultured with HCFs (Fig. 3A). EHTs were generated by seeding enriched hiPSC-CMs (enCMs; 7 × 105 ) and HCFs (3 × 105 ) at a 7:3 ratio or non-enriched hiPSC-CMs alone (1 × 106 ) onto decellularized porcine ECM. EHTs generated by enCMs co-cultured with HCF showed stronger contractility, compared to group of non-enriched hiPSC-CMs alone (Fig. 3B). Moreover, H&E staining and immunohistochemical characterizations of these EHTs revealed even distribution and localization of hiPSC-CMs (Fig. 3C). Importantly, immunofluorescence staining of cTnT revealed significantly higher intensity of the myofilaments with qualitatively improved sarcomere directionality in EHTs generated by enCMs co-cultured with HCF as compared to non-enCMs (Fig. S2). These data suggested that EHTs generated by enCMs co-cultured with HCFs could be suitable candidate for beating conduit engineering in the subsequent experiments.
3.3. Generation of TEPC using scaled-up EHT and a robust biological adhesive
EHTs were generated with scaled-up scaffolds to wrap HUA and produce a TEPC (Fig. 4). Large (15 × 14.5 mm) scaffolds were seeded with 7 × 106 enriched hiPSC-CMs and 3 × 106 HCFs. Seeded EHTs were statically cultured in a PTFE frame for 4 days before being wrapped around a vessel (Fig. 4A–D).
Fig. 4.
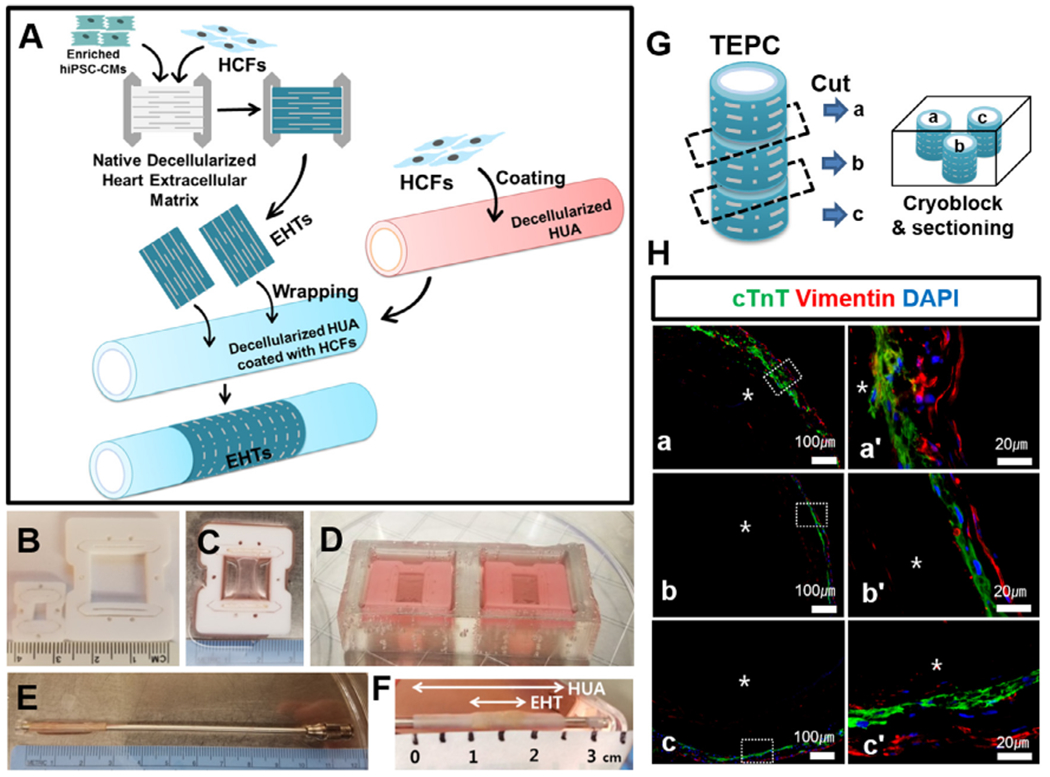
Scaled-up large scaffold and the generation of TEPC with deceiluiarized human HUA. (A) Schematic summary of wrapping EHT on the HUA using fibroblasts as a bio-natural glue. 1.5 million fibroblasts in 400 μL of media were coated on the HUA, after pre-coating with 0.1% gelatin for 30 min, in a petridish for 1 h EHTs were manually wrapped on the HUA to produce a TEPC. TEPC was cultured in T75 flask for five days. (B) Original frame (3 × 4 mm) and scaled-up frame (15 × 14.5 mm). (C and D) 15 × 14.5 mm of frame was employed for producing large scaffolds. A total 10 million cells (7 million enriched-hiPSC-CMs and 3 million of HCFs) were seeded onto a large scaffold in the seeding bath. EHTs were statically cultured in the Teflon frames for 4 days before wrapping a vessel. (E) Decellularized HUA located on the left end of the mandrel. (F) TEPC produced with EHTs and decellularized HUA. (G) TEPC was divided into three portions and embedded in OCT and sectioned using Leica cryostat, then mounted on a glass slide. (H) TEPC sections were stained with cTnT and vimentin. Images were acquired using a Nikon 80i microscope and captured using NIS-Elements AR software. The prime symbol “’“ denotes that the image was magnified from the box in a, b and c. Here TEPC lumen was indicated by an asterisk *. Scale bar=100 μm (a,b,c) and 20 μm (a’,b’,c’).
To maximize the contractile output of the TEPC, it was necessary to alter the compliance of the vascular scaffold before wrapping. Through this, the tissue would be able to more efficiently produce luminal pressure changes in vitro due to less of the work done by the EHT being used to contract the vascular scaffold component. To determine the most effective method for increasing compliance of the vascular scaffold, decellularized HUA were treated with either 0.3 mg/mL or 0.5 mg/mL of collagenase for 30 min at 37 °C. Small segments of treated HUA were taken for burst pressure measurements and mechanical testing. The stress-strain curves for 0.3 mg/mL and 0.5 mg/mL collagenase treated HUA showed an increased compliance and decreased burst pressure compared to the untreated control (Fig. S4). However, it was noted that the 0.5 mg/mL collagenase treated HUA was too weak and broke easily during mounting to the in vitro testing apparatus. Because the decellularized HUA treated by 0.3 mg/mL of collagenase was strong enough to mount into the in vitro apparatus, subsequent experiments were conducted using scaffolds treated under this condition. Additionally, decellularized HUA treated with 0.3 mg/mL collagenase demonstrated a suture retention strength of more than 100 g, indicating suitable mechanical strength for implantation (Fig. S5).
Next, a biological adhesive was necessary for stable adhesion of the engineered heart tissue to the umbilical artery. Several biocompatible glues were tested for adhesive efficacy and ease of handling, however most of them were not able to stably adhere the EHT to decellularized HUA (Supplementary Table S2, Fig. S6). Ultimately, we found that a coating of HCF on the exterior of the HUA was sufficient to attach the EHT (Fig. 4). Fibroblasts are known to secrete extracellular matrix proteins which are important mediators of cell attachment by the interaction between components of the ECM and specific cell surface proteins [35]. It is possible the EHT remained attached to the HUA through cell adhesion between cells in the EHT and ECM proteins secreted by the coated fibroblasts. 1.5 × 106 HCFs in 400 uL of media were coated on the HUA after pre-coating with 0.1% gelatin in a Petri dish for 1 h Cannulated HUA were submerged in 0.1% gelatin solution for 30 min in a sterile hood. In a 150 mm Petri dish, the concentrated 400 pL of HCF suspension was added dropwise along the HUA. The vessel was rotated every 15 min during a 1 h incubation period to allow fibroblasts to adhere. The coated HUA was then moved to a fresh dish, just prior to wrapping (Fig. 4E). An EHT was manually wrapped on the HUA to produce a TEPC (Fig. 4A) and was cultured in T75 flask for five days. On Day 5 post wrapping, wrapped EHTs were completely adhered to the HUA (Fig. 4F). TEPCs generated with EHT and HUA by coating with fibroblasts demonstrated good adhesion and beating efficacy at this timepoint (Supplementary Video S5). On Day 5, TEPCs were fixed and prepared for staining to probe cell distribution and viability. Three locations along the length of the TEPC were sectioned to provide a histological assessment of the tissue (Fig. 4G). The cTnT and vimentin staining revealed that the vimentin positive fibroblasts tended to localize along the abluminal edge of the TEPC, while the hiPSC-CM aligned themselves around the circumference of the vascular scaffold (Fig. 4H). Additionally, TUNEL analysis of the TEPC revealed ~98% of the cells remained viable through this initial development process (Fig. S7).
3.4. Measurement of lumen pressure changes of TEPCs
TEPCs were produced using optimized steps as determined by previous experiments. Decellularized HUA (~2.5 cm length) segments were treated with 0.3 mg/mL collagenase solution for 30 min at 37 °C before coating with HCF. Enlarged (15 × 14.5 mm) HCF co-cultured EHTs would be seeded and cultured as a tissue sheet for 4 days to allow the tissue to begin to spontaneously beat. These EHTs would then be manually wrapped around the collagenase treated HUA, coated with HCF, and statically cultured on a metal rod for 5 days. At the end of this production period, TEPCs would be mounted into an acute pacing apparatus to test force production (Fig. 5A).
Fig. 5.
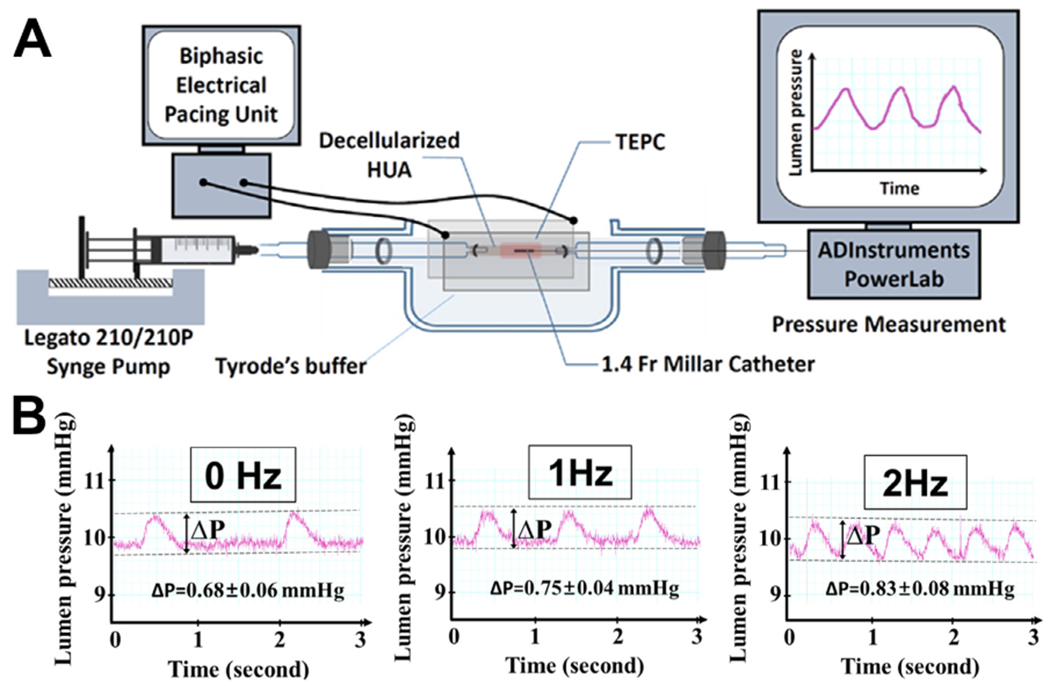
Measurement of pressure development of TEPCs. (A) A TEPC was sutured to the tube holding chamber. Both the lumen and surrounding environment of the TEPC were filled with Tyrode solution (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2 , 1.8 mM CaCl2. 0.2 mM Na2HPO4 , 12 mM NaHCO3. 5.5 mM D-glucose). The luminal starting pressure was controlled by injecting Tyrode solution via microinjector (Legato 210/210P Syringe Pump) and held at 10 mmHg. A Millar catheter was inserted from right side of the TEPC and pressure was measured by an external pressure amplifier. The measurement was performed in an incubator maintained at 5% CO2 and 37 °C. (B) Pressure generated by a TEPC (ΔP) under electrical pacing with 0, 1 and 2 Hz (100 mA current for 10 ms impulse).
To measure the lumen pressure evoked by the beating TEPC, the tissue was sutured into a glass dish that allowed it to be bathed in Tyrode’s solution which was also flowed through the lumen to apply a controllable pre-load. A TEPC was placed between two carbon electrodes to provide electrical field stimulation at 0, 1 or 2 Hz frequency. TEPC luminal pressure was maintained at 10 mmHg using a syringe microinjector during data collection. Without any electrical pacing, the TEPC was beating spontaneously once every two seconds with pressure changes around 0.68 mmHg on average. At 1 Hz frequency of electrical stimulation, TEPC contractions were captured and generated pressures at an average of 0.75 mmHg. Similarly, at 2 Hz frequency of electrical stimulation, the TEPC captured and generated an average pressure of 0.83 mmHg (Fig. 5B).
4. Discussion
We have established a working, standalone TEPC as a baseline for future explorations as a therapeutic Fontan conduit. The modular design and assembly methods for our construct allow each component to be fine-tuned with the goal of maximizing force output of the tissue. These modifications may include cellular composition of the EHTs, mechanical properties of the vascular scaffold and, potentially, EHT fiber alignment relative to the circumference of the vascular scaffold. This study is the first report demonstrating an in vitro system of lumen pressure evoked by a standalone TEPC construct generated from hiPSC-CMs. Through rigorous testing, our group found that a naturally derived ECM based EHT design strategy showed the most consistent and strong results in terms of contractile output. Additionally, the natural topography of these porcine ECM based EHTs encourages natural fiber alignment of the growing myocardial fibers [23]. This topography can be modified during EHT production to orient fibers at specific angles, relative to the circumference of the TEPC vascular scaffold. This would allow us to provide a range of fiber orientations could replicate native ventricle physiology, where these orientations are known to influence mechanical output and ejection fraction [36,37]. As detailed in this work, our EHTs are also optimizable in terms of cell composition and size. Enlarged dimension (15 × 14.5 mm) EHTs were produced in a 7:3 ratio of hiPSC-CMs and cardiac fibroblasts, respectively. A clear increase in contractile output of these EHTs was noted with the addition of fibroblasts as previously reported in other co-cultured EHT systems [29,38].
Previous attempts have been made to develop contractile conduits using cardiomyocyte cell sheet engineering [13,14,39]. One design strategy attempted to produce an independent and in vitro derived pulsatile conduit using a fibrin based scaffold wrapped with heart sheets derived from neonatal rat CMs [39]. This strategy yielded a spontaneously beating cardiac tube and demonstrated measurable inner pressure changes evoked by pulsatile conduits (average 0.11 mmHg, max 0.15 mmHg). Notably, our standalone TEPCs derived by wrapping human iPSC cardiomyocyte-based EHT around decellularized HUA generated markedly higher inner pressure (0.68 mmHg) than the neonatal rat CMs and fibrin tube based pulsatile conduit. Additionally, the electrical handling of the myocardium in this previously reported rat construct was only able to follow pacing stimulation of ~1 Hz frequency [39], indicating the need of enhancing electrical maturity of the tissue. Encouragingly, the herein reported hiPSC-based TEPC followed pacing frequencies up to 2 Hz (Fig. 5). Future electrical training of the human TEPC in the bioreactor is warranted and may further enhance the functional maturation of construct.
In a follow up study by the same group, neonatal rat cardiomyocyte sheets were wrapped around a resected thoracic aorta and transplanted to replace a segment of rat abdominal aorta [14]. This strategy allowed for in vivo training of the tissue under a high-pressure, aortic blood flow over a four week period where discernable pressure changes (average 5.9 mmHg) could be generated from spontaneous beating [14]. However, since Fontan conduits are implanted into the low-pressure mechanical environment of the vena cava, the pressure development of such construct may be markedly reduced in their direct to in vivo development system. Additionally, hiPSC derived cardiac sheets were wrapped around rat IVC in vivo to develop ectopic pressure. However, this approach only generated a slight pressure change of 0.05 mmHg and 0.27 mmHg 4 and 8 weeks post-implantation, respectively, based on graft natural pulsation [13]. It’s also worth noting that this straight in vivo wrapping approach does not address the clinical need, since Fontan operation requires an ectopic tubular construct to connect the IVC with the pulmonary artery. The current decellularized HUA scaffold coupled with hiPSC-CM EHTs provides a foundation for pulsatile conduit engineering for future clinical application.
It is important to note that the aim of our current study is not to investigate clinical applicability of the TEPC as a Fontan conduit. Rather, this work represents a proof of principle study for our proposed TEPC design strategy using hiPSC-CMs and lays the groundwork toward developing improved future therapy for SVD patients. There are limitations in our current study. The EHT design presented in this paper used a mixture of hiPSC-CMs mixed with commercially available cardiac fibroblasts. As the TEPC moves towards clinical translation, the cellular composition should progress to all hiPSC derived cell types. Future iterations of the TEPC will be composed entirely of cells derived from an hiPSC source. It is noted that endothelial cells contribute to maturation of hiPSC-CMs [40,41]. With this in mind, addition of vascular endothelial cells into our EHTs may increase contractile output of the tissue. In terms of the vascular scaffold used to produce the TEPC, there are two opposing characteristics that must be balanced for optimal performance. These would be the strength of the tube to facilitate patent blood flow, and the malleability of it to allow for efficient contraction by the myocyte component. It is possible that the mechanics of the HUA could be further tuned with new protease treatments, or a synthetic scaffold material with more finely controllable mechanics could be explored.
Moving forward, cellular maturation of the TEPC via the application of biomechanical stretch and electrical stimuli to train TEPCs in vitro will be paramount to further enhance their contractile force and thus increase the capacity to improve pulmonary circulation in vivo. Straight to in vivo strategies for contractile conduit production displayed increases in hypertrophy markers, of both neonatal rat cardiomyocyte and hiPSC based tissues, that was attributed to pulsation of the myocardial tube due to blood flow [13,18]. Because these tissues are ultimately intended for use in the Fontan circulation, it is unlikely that the lowered systemic venous pressures noted in SVD patients will be sufficient to train pulsatile conduits. Therefore, a robust in vitro training regimen will be necessary for proper TEPC production. Additionally, multiple cardiomyocyte layers and the introduction of endothelial cells will be necessary for producing larger luminal forces, as shown in previous reports [13,14,31,39]. Because the TEPC construct is intended to be trained in vitro, prior to implantation, a microvascular network is likely necessary to support the tissue during this training phase and shorten the time to anastomose to the host vasculature. Providing an in vitro training regimen will allow for a second level of optimization to improve the efficiency of force generation of the tissue. Further, work on developing valvular TEPCs to ensure unidirectional blood flow in addition to in vivo assessments of longevity and effect of the constructs on local hemodynamics warrants future consideration. For the TEPC to be viable as a therapeutic, it must be capable of increasing local flow rates as a vena cava interposition graft. This will require implantation and maintained patency of the TEPC construct, which is determined by the vascular scaffold used during production. The decellularized HUA scaffold reported in this study displays relevant parameters to allow safe implantation of the construct and has been used in previous applications as an interposition graft [24,42]. Additionally, potential therapeutic benefits will be directly linked to contractility of the TEPC which, in turn, is reliant on mature cardiomyocytes residing in the tissue. This necessity underscores the importance of the use of bioreactor training methods in vitro prior to in vivo assessments of therapeutic benefit. The versatile design strategy of this standalone TEPC has been developed with these goals in mind, and is readily transferable into bioreactor and in vivo systems. In summary, current data suggests a strong potential for future applications of the TEPC. Many avenues of exploration have opened, and our group is optimistic about the future of this product. We believe the development of this TEPC will set the stage for developing a curative, reconstructive surgical option (as opposed to a palliative procedure) for patients born with single ventricle anomalies.
5. Conclusion
Herein, we have reported a modular design of a standalone, functioning TEPC. These tissues are developed with compositionally optimized EHTs and collagenase treated, decellularized HUA. We have demonstrated that decellularized porcine ECM scaffolds seeded with hiPSC-CMs exhibited strong contractility that was highly reproducible and scalable. Additionally, the incorporation of HCF significantly increased the tissues’ contractility from decellularized porcine ventricle heart tissue-derived EHTs that contained hiPSC-CMs. Compositionally optimized EHTs were scaled-up to wrap around decellularized HUA and produce a TEPC. After statically culturing the TEPC on a metal rod in a T75 flask for five days, the tissue was capable of generating observable pressure increases in vitro spontaneously and while following electrical stimulation. Contraction frequency of the tissue closely followed electrical stimuli at both 1 Hz and 2 Hz frequency while developing an average pressure increase of 0.75 mmHg and 0.83 mm Hg, respectively. We believe this provides a solid foundation to work from in the goal of producing a therapeutic TEPC to treat patients with single ventricle disorders. Future iterations of this construct will include mechanical and electrical training regimens to further enhance force production of the tissue.
Supplementary Material
Statement of significance.
Single Ventricle Cardiac defects (SVD) are a form of congenital disorder with a morbid prognosis without surgical intervention. These patients are treated through the Fontan procedure which requires vascular conduits to complete. Fontan conduits have been traditionally made from stable or biodegradable materials with no pumping activity. Here, we propose a tissue engineered pulsatile conduit (TEPC) for use in Fontan circulation to alleviate excess strain in SVD patients. In contrast to previous strategies for making a pulsatile Fontan conduit, we employ a modular design strategy that allows for the optimization of each component individually to make a standalone tissue. This work sets the foundation for an in vitro, trainable human induced pluripotent stem cell based TEPC.
Acknowledgments
We appreciate Dr. George Tellides and Dr. Sang-Joon Ahn for the discussion of optimal vascular scaffold. We also thank the Qyang group members for their valuable feedback for this line of research. We also acknowledge financial support from DOD 11959515, 1R01HL116705, 1R01HL131940 (all to YQ), P.D. Soros Fellowship for New Americans and a NIH/NIGMS Medical Scientist Training Program Grant (T32GM007205) (both to LRS), NIH 1F31HL143928-01 (to CWA), The American Heart Association (AHA) Postdoctoral Fellowship (19POST34381048) (to MHK), AHA Postdoctoral Fellowship (19P0ST34450100) (to JL), and 1R01HL136590 (to SGC).
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.actbio.2019.10.019.
References
- [1].Hoffman JIE, Kaplan S, The incidence of congenital heart disease, J. Am. Coll. Cardiol 39 (2002) 1890–1900, doi: 10.1016/S0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- [2].Leary PWO, Prevalence, clinical presentation and natural history of patients with single ventricle, 16 (2002) 31–38. [Google Scholar]
- [3].Samánek M, Children with congenital heart disease: probability of natural survival, Pediatr. Cardiol 13 (1992) 152–158, doi: 10.1007/BF00793947. [DOI] [PubMed] [Google Scholar]
- [4].Ridderbos FJS, Wolff D, Timmer A, Van Melle JP, Ebels T, Dickinson MG, Timens W, Berger RMF, Adverse pulmonary vascular remodeling in the Fontan circulation, J. Hear. Lung Transpl 34 (2015) 404–413, doi: 10.1016/j.healun.2015.01.005. [DOI] [PubMed] [Google Scholar]
- [5].Book WM, Gerardin J, Saraf A, Clinical phenotypes of Fontan failure: implications for management, Congenit Heart Dis 11 (4) (2016) 296–308, doi: 10.1111/chd.12368. [DOI] [PubMed] [Google Scholar]
- [6].de Leval MR, Deanfield JE, Four decades of Fontan palliation, Nat. Rev. Cardiol 7 (2010) 520–527, doi: 10.1038/nrcardio.2010.99. [DOI] [PubMed] [Google Scholar]
- [7].Tara S, Kurobe H, Rocco KA, Maxfield MW, Best CA, Yi T, Naito Y, Breuer CK, Shinoka T, Well-organized neointima of large-pore poly(L-lactic acid) vascular graft coated with poly(L-lactic-co-ε-caprolactone) prevents calcific deposition compared to small-pore electrospun poly(L-lactic acid) graft in a mouse aortic implantation model, Atherosclerosis 237 (2014) 684–691, doi: 10.1016/j.atherosclerosis.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roh JD, Sawh-Martinez R, Brennan MP, Jay SM, Devine L, Rao DA, Yi T, Mirensky TL, Nalbandian A, Udelsman B, Hibino N, Shinoka T, Saltzman WM, Snyder E, Kyriakides TR, Pober JS, Breuer CK, Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling, Proc. Natl. Acad. Sci 107 (2010) 4669–4674, doi: 10.1073/pnas.0911465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hibino N, McGillicuddy E, Matsumura G, Ichihara Y, Naito Y, Breuer C, Shinoka T, Late-term results of tissue-engineered vascular grafts in humans, J. Thorac. Cardiovasc. Surg. 139 (2010) 431–436 e2, doi: 10.1016/j.jtcvs.2009.09.057. [DOI] [PubMed] [Google Scholar]
- [10].Shin’oka T, Matsumura G, Hibino N, Naito Y, Watanabe M, Konuma T, Sakamoto T, Nagatsu M, Kurosawa H, Midterm clinical result of tissue-engineered vascular autografts seeded with autologous bone marrow cells, J. Thorac. Cardiovasc. Surg. 129 (2005), doi: 10.1016/j.jtcvs.2004.12.047. [DOI] [PubMed] [Google Scholar]
- [11].Huang L, Schilling C, Dalziel KM, Xie S, Celermajer DS, McNeil JJ, Win-law D, Hons M, Hornung TS, Radford DJ, Grigg LE, Bullock A, Wheaton GR, Justo RN, Blake J, Bishop R, Du Plessis K, Udekem Y, Du Plessis K, D’Udekem Y, Hospital inpatient costs for single ventricle patients surviving the Fontan procedure, Am. J. Cardiol. 120 (2017) 467–472, doi: 10.1016/j.amjcard.2017.04.049. [DOI] [PubMed] [Google Scholar]
- [12].Gewillig M, Goldberg DJ, Failure of the fontan circulation, Heart Fail. Clin 10 (2014) 105–116, doi: 10.1016/j.hfc.2013.09.010. [DOI] [PubMed] [Google Scholar]
- [13].Seta H, Matsuura K, Sekine H, Yamazaki K, Shimizu T, Tubular cardiac tissues derived from human induced pluripotent stem cells generate pulse pressure in vivo, Sci. Rep 7 (2017) 45499, doi: 10.1038/srep45499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sekine H, Shimizu T, Yang J, Kobayashi E, Okano T, Pulsatile myocardial tubes fabricated with cell sheet engineering, Circulation (2006) 114, doi: 10.1161/CIRCULATIONAHA.105.000273. [DOI] [PubMed] [Google Scholar]
- [15].Ruan J-L, Tulloch NL, Razumova MV, Saiget M, Muskheli V, Pabon L, Reinecke H, Regnier M, Murry CE, Mechanical stress conditioning and electrical stimulation promote contractility and force maturation of induced pluripotent stem cell-derived human cardiac tissueclinical perspective, Circulation 134 (2016) 1557–1567, doi: 10.1161/CIRCULATIONAHA.114.014998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mihic A, Li J, Miyagi Y, Gagliardi M, Li SH, Zu J, Weisel RD, Keller G, Li RK, The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes, Biomaterials 35 (2014) 2798–2808, doi: 10.1016/j.biomaterials.2013.12.052. [DOI] [PubMed] [Google Scholar]
- [17].Stoppel WL, Kaplan DL, Black LD, Electrical and mechanical stimulation of cardiac cells and tissue constructs, Adv. Drug Deliv. Rev 96 (2016) 135–155, doi: 10.1016/j.addr.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Biermann D, Eder A, Arndt F, Seoudy H, Reichenspurner H, Mir T, Riso A, Kozlik-Feldmann R, Peldschus K, Kaul MG, Schuler T, Krasemann S, Hansen A, Eschenhagen T, Sachweh JS, Towards a tissue-engineered contractile Fontan-conduit: the fate of cardiac myocytes in the subpulmonary circulation, PLoS ONE 11 (2016) 1–10, doi: 10.1371/journal.pone.0166963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lim JWE, Bodnar A, Proteome analysis of conditioned medium from mouse embryonic fibroblast feeder layers which support the growth of human embryonic stem cells, Proteomics 2 (2002) 1187–1203 doi:. [DOI] [PubMed] [Google Scholar]
- [20].Gui L, Dash BC, Luo J, Qin L, Zhao L, Yamamoto K, Hashimoto T, Wu H, Dardik A, Tellides G, Niklason LE, Qyang Y, Implantable tissue-engineered blood vessels from human induced pluripotent stem cells, Biomaterials 102 (2016) 120–129, doi: 10.1016/j.biomaterials.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Luo J, Qin L, Kural MH, Schwan J, Li X, Bartulos O, Cong X-Q, Ren Y, Gui L, Li G, Ellis MW, Li P, Kotton DN, Dardik A, Pober JS, Tellides G, Rolle M, Campbell S, Hawley RJ, Sachs DH, Niklason LE, Qyang Y, Vascular smooth muscle cells derived from inbred swine induced pluripotent stem cells for vascular tissue engineering, Biomaterials 147 (2017) 116–132, doi: 10.1016/j.biomaterials.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dash BC, Levi K, Schwan J, Luo J, Bartulos O, Wu H, Qiu C, Yi T, Ren Y, Campbell S, Rolle MW, Qyang Y, Tissue-Engineered vascular rings from human iPSC-Derived smooth muscle cells, Stem Cell Rep 7 (2016) 19–28, doi: 10.1016/j.stemcr.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schwan J, Kwaczala AT, Ryan TJ, Bartulos O, Ren Y, Sewanan LR, Morris AH, Jacoby DL, Qyang Y, Campbell SG, Anisotropic engineered heart tissue made from laser-cut decellularized myocardium, Sci. Rep 6 (2016) 32068, doi: 10.1038/srep32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Gui L, Muto A, Chan SA, Breuer CK, Niklason LE, Development of decellularized human umbilical arteries as small-diameter vascular grafts, Tissue Eng. Part A 15 (2009) 2665–2676, doi: 10.1089/ten.tea.2008.0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP, Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating WNT/β-catenin signaling under fully defined conditions, Nat. Protoc 8 (2013) 162–175, doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K, Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes, Cell Stem Cell 12 (2013) 127–137, doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- [27].Huang AH, Niklason LE, Engineering biological-based vascular grafts using a pulsatile bioreactor, J. Vis. Exp 1 (2011) 5–10, doi: 10.3791/2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Watanabe M, Shin’oka T, Tohyama S, Hibino N, Konuma T, Matsumura G, Kosaka Y, Ishida T, Imai Y, Yamakawa M, Ikada Y, Morita S, Tissue-engineered vascular autograft: inferior vena cava replacement in a dog model, Tissue Eng 7 (2001) 429–439, doi: 10.1089/10763270152436481. [DOI] [PubMed] [Google Scholar]
- [29].Tiburcy M, Hudson JE, Balfanz P, Schlick SF, Meyer T, Chang Liao M-L, Levent E, Raad F, Zeidler S, Wingender E, Riegler J, Wang M, Gold JD, Kehat I, Wettwer E, Ravens U, Dierickx P, van Laake L, Goumans M-J, Khadjeh S, Toischer K, Hasenfuss G, Couture LA, Unger A, Linke WA, Araki T, Neel B, Keller G, Gepstein L, Wu JC, Zimmermann W-H, Defined engineered human myocardium with advanced maturation for applications in heart failure modelling and repair, Circulation 116 (2017) 024145 CIRCULATIONAHA, doi: 10.1161/CIRCULATIONAHA.116.024145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Roberts MA, Tran D, Coulombe KLK, Razumova M, Regnier M, Murry CE, Zheng Y, Stromal cells in dense collagen promote cardiomyocyte and microvascular patterning in engineered human heart tissue, Tissue Eng. Part A 22 (2016) 633–644, doi: 10.1089/ten.tea.2015.0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iyer RK, Odedra D, Chiu LLY, Vunjak-Novakovic G, Radisic M, Vascular endothelial growth factor secretion by nonmyocytes modulates connexin-43 levels in cardiac organoids, Tissue Eng. Part A 18 (2012) 1771–1783, doi: 10.1089/ten.tea.2011.0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ott HC, Matthiesen TS, Goh S-K, Black LD, Kren SM, Netoff TI, Taylor D.a, Perfusion-decellularized matrix: using nature’s platform to engineer a bioartificial heart, Nat. Med 14 (2008) 213–221, doi: 10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- [33].Sreejit P, Verma RS, Natural ecm as biomaterial for scaffold based cardiac regeneration using adult bone marrow derived stem cells, Stem Cell Rev. Rep 9 (2013) 158–171, doi: 10.1007/s12015-013-9427-6. [DOI] [PubMed] [Google Scholar]
- [34].Williams C, Sullivan K, Black LD, Partially digested adult cardiac extracellular matrix promotes cardiomyocyte proliferation I, Adv. Healthc. Mater 4 (2015) 1545–1554, doi: 10.1002/adhm.201500035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dalton SL, Marcantonio EE, Assoian RK, Cell attachment controls fibronectin and alpha 5 beta 1 integrin levels in fibroblasts. Implications for anchorage-dependent and -independent growth, J. Biol. Chem 267 (1992) 8186–8191, doi: 10.1038/nature16966. [DOI] [PubMed] [Google Scholar]
- [36].Palit A, Bhudia SK, Arvanitis TN, Turley GA, Williams MA, Computational modelling of left-ventricular diastolic mechanics: effect of fibre orientation and right-ventricle topology, J. Biomech 48 (2015) 604–612, doi: 10.1016/j.jbiomech.2014.12.054. [DOI] [PubMed] [Google Scholar]
- [37].Sommer G, Schriefl AJ, Andra M, Sacherer M, Viertler C, Wolinski H, Holzapfel GA, Biomechanical properties and microstructure of human ventricular myocardium, Acta Biomater 24 (2015) 172–192, doi: 10.1016/j.actbio.2015.06.031. [DOI] [PubMed] [Google Scholar]
- [38].Li Y, Asfour H, Bursac N, Age-dependent functional crosstalk between cardiac fibroblasts and cardiomyocytes in a 3D engineered cardiac tissue, Acta Biomater 55 (2017) 120–130, doi: 10.1016/j.actbio.2017.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kubo H, Shimizu T, Yamato M, Fujimoto T, Okano T, Creation of myocardial tubes using cardiomyocyte sheets and an in vitro cell sheet-wrapping device, Biomaterials 28 (2007) 3508–3516, doi: 10.1016/j.biomaterials.2007.04.016. [DOI] [PubMed] [Google Scholar]
- [40].Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H, Murry CE, Growth of engineered human myocardium with mechanical loading and vascular coculture, Circ. Res 109 (2011) 47–59, doi: 10.1161/CIRCRESAHA.110.237206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Talman V, Kivela R, Cardiomyocyte-Endothelial cell interactions in cardiac remodeling and regeneration, Front. Cardiovasc. Med 5 (2018) 101, doi: 10.3389/fcvm.2018.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gui L, a Chan S, Breuer CK, Niklason LE, Novel utilization of serum in tissue decellularization, Tissue Eng. Part C. Methods 16 (2010) 173–184, doi: 10.1089/ten.tec.2009.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.