

Abstract
Introduction or background
Congenital insensitivity to pain (CIP) is caused by extremely rare Mendelian genetic disorders. CIP individuals demonstrate the unexpectedly severe consequences of painlessness. Although only a small number of causative conditions and genes are known, most have led to profound insights into human nociception. CIP gene discovery is catalyzing the manufacture of completely new classes of analgesics, and these are needed as alternatives to synthetic highly potent opioids.
Sources of data
Pubmed.gov peer-reviewed journal articles and reviews.
Areas of agreement
The importance of nerve growth factor-tropomyosin receptor kinase A (NGF-TRKA) signalling for nociceptor genesis and subsequent pain sensing.
New analgesics can be generated from knowledge of the NGF-TRKA nociceptor pathway.
Increased susceptibility to Staphylococcus aureus infection is a consequence of deficient NGF-TRKA signalling.
Mutations in the voltage-gated sodium channels SCN9A and SCN11A can cause congenital painlessness, and in contradistinction, other mutations can cause episodic neuropathic pain.
SCN9A/Nav1.7 is an analgesic target.
SCN11A/Nav1.9 is unlikely to be an analgesic target.
There are further Mendelian causes of painlessness to be discovered.
Areas of controversy
Which NGF-TRKA intracellular signalling pathways operate in nociceptor development and which in post-natal pain sensing?
Why have no clinically effective Nav1.7 antagonist been generated?
SCN9A-CIP causes analgesia, at least in part, through endogenous opioids.
Why do all CIP phenotypes involve a complete loss of all types of nociception?
Areas timely for developing research
PRDM12 as an analgesic target.
Discovery of the function and analgesic potential of new CIP genes.
Can NGF-TRKA be used in the treatment of S. aureus?
Keywords: congenital painlessness, pain genes, nociceptor development, nociceptor function
Background
Although not traditionally regarded as such, pain is a sense. Pain sensing is called nociception and, as with other senses, involves specialized neurons that are called nociceptors (‘nocere’ in Latin means ‘to harm’), see Figure 1. Nociceptors detect both actual and potential tissue damage of almost all types except ionising radiation. Potential tissue damage being defined as a stimulus that is of brief duration or intensity and does not cause significant cell damage, but if the stimulus continues cell damage and death will occur.
Fig. 1.
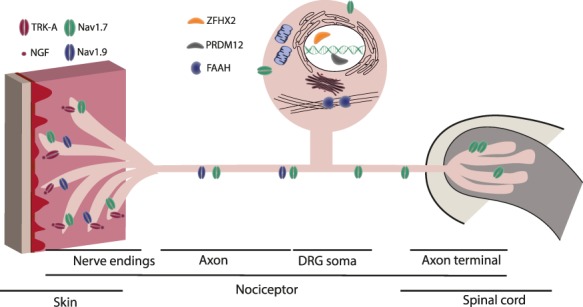
Schematic nociceptor showing subcellular location of proteins identified that cause Congenital Insensitivity to Pain. Voltage sodium channels, Nav1.7 and Nav1.9, are expressed at both the peripheral and central terminals as well as along axons of primary sensory neuron afferent.1 Nav1.7 is highly expressed in nociceptors where it is also found at the distal axon and in the presynaptic terminals in the dorsal horn of the spinal cord.2 NGF and it’s receptor TRK-A are expressed in the nociceptor nerve endings and play an important role in neuronal development and survival. Transcriptional factors, PRMD12 and ZFHX2, localize to the nucleus within the dorsal root ganglion.3 FAAH is found within the cytoplasm attached to the cytoskeleton.4 Abbreviations: DRG; dorsal root ganglion; Nav, voltage sodium channel; NGF, nerve growth factor; TRK-A, neutrophic tyrosine kinase receptor type 1; PRDM12, PR domain zinc finger protein 12; ZFHX2, Zinc finger homeobox protein 2; FAAH, Fatty-acid amide hydrolase 1.
Congenital insensitivity to pain (CIP) is an extremely rare human phenotype where no pain of any type is experienced during an affected individuals’ lifetime. For the senses of sight and hearing, more than a hundred Mendelian disorders are each known that cause a congenital loss of vision or sight. Understanding how these conditions cause blindness and deafness has considerably aided understanding of the normal mechanisms of human light and sound detection. Oddly, less than a dozen Mendelian disorders of CIP are known, but these conditions are also providing profound insights into the complexity and sophistication of human pain sensing.
CIP phenotypes
It is important to examine the known CIP phenotypes, see Table 1. All have features in addition to a lack of pain which aid in understanding the functions of each CIP gene. Together, they illustrate an important message—that complete inability to sense pain is not a panacea but causes a very considerable morbidity and mortality. Initially, there may be suggestions of abnormal pain sensing as vaccinations and accidents cause no concern. Coincident with teething in the first year of life, self-mutilation of lips, tongue, fingers and toes often occur. Pain behaviours, e.g. saying ‘ow’, are learnt during the first decade, but pain avoidance behaviours are not. This is particularly notable in males who typically do not learn what is dangerous for their body and can die from activities which pain would otherwise have prevented. Conversely, affected adult females tend to be risk-averse and behave more cautiously. Long bone fractures can heal, if allowed to do so, but it is the joint injuries that inevitably lead to Charcot’s joints, and hence progressive orthopaedic deformities. Of note, these are asymptomatic, often overlooked, but are present by 10 y, as limp and limb disuse is caused by pain not orthopaedic disruption in CIP. Corneal abrasions are an ever-present risk, and if unrecognized leading to degrees of blindness. Pain is essential in teaching us how to use our bodies optimally and avoid or respond to injuries, being permanently painless results in a significant morbidity and mortality.
Table 1.
CIP phenotypes
| Failure of nociceptor development: HSANs | Unresponsive nociceptors | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Disorder | HSAN4 | HSAN5 | HSAN8 | HSAN3 | HSAN2B | Not named | HSAN2D | HSAN7 | Marsili syndrome |
| OMIM number | 256 800 | 608 654 | 616 488 | 223 900 | 613 115 | Not assigned | 243 000 | 615 548 | 147 430 |
| Gene | NTRK1 | NGF | PRDM12 | IKBKAP | FAM134B | CLTCL1 | SCN9A | SCN11A | ZFHX2 |
| Cognition | Reduced | Can be reduced | Usually normal, occasionally reduced | Usually normal, occasionally reduced | Normal | Reduced | Normal | Normal | Normal |
| S. aureus infection risk | Increased | Increased | Increased | Increased | Increased | Increased | Normal | Normal | Normal |
| Additional phenotypic features | Anhidrosis* Absent temperature sensation | Anhidrosis* is present in most families. Reduced temperature sensation | Partial anhidrosis* Reduced temperature sensation Sometimes truncal pain sensing | Paroxysmal autonomic hypo and hyper- function dominates the clinical picture rather than the variable reduction in nociception | Hyperhidrosis, urinary incontinence, slow pupillary light reflexes Occasional mild motor neuropathy | Absent touch sense. Non- progressive severe learning disability | Anosmia (one family reported with progressive temperature and touch sensation) | Profound gastrointestinal hypomobility Neck and face itch Hypotonia and mild weakness Intolerance of temperature changes | Reduced ability to sense temperature and sweat occasional suffer from headache and visceral pains hyperthermia episodes |
| Prevalence of cases described† | Common | Very rare | Rare | Common | Very rare | One family | Common | Very rare | One family |
.
*Anhidrosis is an inability to sweat here due a failure of development of sweat glands.
†Common > 20 cases, rare 5–20 families, very rare < 5 families, one family reported.
CIP can be clinically divided into two groups: developmental disorders where nociceptors fail to develop or undergo early apoptosis through lack of trophic signals; and secondly, where nociceptors have developed and are in their correct anatomical position but are unable to respond to tissue-damage signals, see Table 1. The first group is known as Hereditary Sensory and Autonomic Neuropathies (HSANs) and involves nociceptive, sensory and autonomic features. In the HSANs peripheral sensory nerve, biopsy shows C and/or Aδ fibre loss. The commonest cause of developmental CIP is HSAN4.5 Cognition can be reduced in this group of conditions. Distinctive to HSANs is a significant susceptibility to Staphylococcus aureus infections manifest as grumbling skin infections, osteomyelitis and septic arthritis. This is a direct consequence of a lack of nerve growth factor/tropomyosin receptor kinase A (NGF/TRKA) signalling through an evolutionarily conserved pathway in macrophages where NGFβ is released in response to S. aureus. Activation of the pathway leads to enhanced macrophage phagocytosis and superoxide-dependent bacterial killing, and immune system activation through stimulated proinflammatory cytokine production.6
Individuals with second type of CIP are born with nociceptors that are unable to be activated by tissue-damage signals, see Tables 1 and 2. These phenotypes were originally contentious, as peripheral nerve biopsy showed all expected nerve fibre types present. It was not until 2006 with the discovery of the causative gene, SCN9A, that a clinical diagnosis of this type of CIP could be proven.7 Since then a further such Mendelian conditions have been described. Cognition is normal in this type of CIP, and there is no increased risk of S. aureus infections. The commonest condition within this group is CIP-SCN9A (also called channelopathy associated with CIP). In CIP-SCN9A the only additional feature is congenital anosmia (absence of the sense of smell), and many adults have gone undiagnosed, and/or unbelieved when they explain that they don’t feel pain.8
Table 2.
CIP genotypes
| Gene | NTRK1 | NGF | PRDM12 | IKBKAP | FAM134B | CLTCL1 | SCN9A | SCN11A | ZFHX2 |
|---|---|---|---|---|---|---|---|---|---|
| Inheritance pattern | AR | AR | AR | AR | AR | AR | AR | AD (de novo reported) | AD |
| Pre-natal expression pattern | First trimester sensory neuron precursors | Throughout pregnancy in PNS | First trimester sensory neuron precursors | First trimester neural crest precursors, spinal motor neurones | Sensory and autonomic ganglia | First trimester, in sensory neuron precursors | Increases in nociceptors and sympathetic enteric neurons throughout pregnancy | Nociceptors, enteric and sympathetic | Brain, spinal cord and dorsal root ganglia |
| Post-natal expression pattern | Nociceptors and monocytes | Widespread throughout CNS, nociceptors and white blood cells | Nociceptors, macrophages, dendritic cells | Throughout central and peripheral nervous system | Dorsal root ganglia, oesophagus, kidney, skeletal muscle | Low levels apart from skeletal muscle heart and testis with higher levels | Nociceptors sympathetic | Nociceptors, enteric and sympathetic | Nociceptors, throughout brain, lungs, testis/ovaries |
| Types of mutations reported | Non-sense, splicing and mis-sense (commonest) | Mis-sense and frame-shift | Mis-sense, non-sense and poly-alanine tract expansion | Nociceptor specific splicing | Truncating loss of function | Single mis-sense | Non-sense, mis-sense,6 splicing, frameshift, Exon and whole gene deletions | Recurrent mis-sense mutation (p.Leu765Pro) | Single mis-sense |
| Drug target potential* | Already in trials NCT02424942 | Already in trials NCT02697773 | Possibly | Already in trials NCT02876939 | Unlikely | Unlikely | Already in trials NCT01911975 | Unlikely | Possibly |
AD, autosomal dominant; AR, autosomal recessive; CNS, central nervous system.
*Example clinical trial from clinicaltrials.gov.
The genes that cause CIP
The genes causing each CIP phenotype are listed in Table 2, with the known mutation spectrum, inheritance pattern, developmental and post-natal tissue expression and the number of families and cases reported. Most are autosomal recessive disorders, with the exception of individuals with unique SCN11A mutations, and a single family with a dominant mutation in ZFHX2. Without doubt further CIP genes remain to be discovered.
Developmental CIP genes: causes of CIP because of the failure of nociceptors to develop.
Neurotrophic receptor tyrosine kinase 1 (NTRK1)
The first gene to be identified to cause a CIP was NTRK1 in 1996,5 see Table 2. Bi-allelic mutations in this gene were found by studying a group of families with the previously clinically defined condition HSAN type 4 (HSAN4).9NTRK1 stands for neurotrophic tyrosine kinase gene 1 and encodes for the protein originally called ‘Tropomyosin receptor kinase’ but now more descriptively named ‘High affinity nerve growth factor receptor’ (commonly known as TRKA). Many further cases and a large spectrum of mutations described including non-sense, splicing and mis-sense. Mis-sense mutations are the commonest; however, these may require functional analysis before their pathogenicity can be accepted.6 This, and basic cell biology studies, have confirmed the complexity of TRKA and its signalling pathways, and that whilst most mutations lead to a complete loss of function, contrary to expectations, some only affect one pathway.10,11
Nerve growth factor (NGF)
The tyrosine kinase receptor TRKA has a major ligand NGF (initially discovered and characterized by Rita Levi-Montalcini and Stanley Cohen for which they were awarded a Nobel prize in 1986). In 2004, a homozygous NGF mutation was reported to cause CIP in a large Swedish family.12 This delay between NGF being identified and the phenotype, its loss caused in humans, being described is probably because of the rarity of this CIP phenotype, and only a few further families have since been described each with different mis-sense or frame-shift NGF mutations. Functional studies of these mutations have shown either a destabilization of the cysteine knot structure of NGF (a structural motif characteristic of neurotrophins composed of a pair of disulphide cysteine bonds), or a failure of the intra-cellular proteolytic processing of NGF.10,13
PR domain zinc finger protein 12 (PRDM12)
In 2015 a new HSAN4-like phenotype was reported caused by bi-allelic mutations in PRDM12 (clinically known as HSAN6).14 PRDM12 functions as a transcriptional regulator. The clinical features that distinguish this new condition from HSAN4 are normal cognition (or occasionally mild cognitive delay) and a diminution on nerve biopsy of Aδ but not C fibres. This condition is the second commonest type of developmental CIP. Mutations are non-sense and more commonly mis-sense, and the few that have been functionally characterized abolish function. A rare mutational mechanism is an expansion of the C-terminal poly-Alanine tract from a normal range of 8–13 to >18 leading to PRDM12 aggregation. In 2019 two papers showed that PRDM12 was a key and non-redundant regulator of the generation of all types of nociceptors in utero. This is achieved through the activation of pro-neuronal transcription factors, including NEUROD1, BRN3A and ISL1, and the subsequent initiation and maintenance of the expression of TrkA.15,16
The following very rare autosomal recessive conditions cause congenital painlessness, none seem to yet inform us of unique pain pathways or guide us to new classes of analgesics. Familial dysautonomia (HSAN3) is an exceptionally odd condition where paroxysmal autonomic hyper- and hypo-function predominate the phenotype, and pain sensing is often significantly reduced but is not a predominant problem.17 A single Ashkenazi Jewish mutation occurs in >90% cases in the IKBKAP gene, c.2204 + 6 T > C, which causes a splicing defect confined only to autonomic and nociceptive neurons.18FAM134B (Hereditary Sensory and Autonomic Neuropathy type IIB) is a very rare causes of developmental CIP with prominent autonomic features.19 FAM134B facilitates endoplasmic reticulum degradation by autophagy leading to dysfunction and then death of sensory neurons.20CLTCL1 was described as causing CIP in a single family, with severe learning difficulties and a congenital absence of the sense of touch.21 CHC22 functions as a clathrin heavy chain in intracellular vesicle transport, resulting in a failure of extracellular neurotrophin release.22MPV17 mutations cause a CIP, but this is overshadowed by progressive liver failure, and progressive neurological decline presenting in the first year of life (mitochondrial DNA depletion syndrome 6).23 MPV17 is an inner mitochondrial membrane protein whose functions are essential to mitochondrial function.24
Non-functional nociceptors CIP genes: causes of CIP due to failure of nociceptors to respond to tissue-damage signals.
SCN9A is the gene encoding the protein voltage-gated sodium channel Nav1.7, and is highly expressed in all classes of nociceptor. Nav1.7 acts at the cell membrane of nociceptors and forms a channel capable of allowing only sodium ions to flow through a central pore from the exterior to the interior of the cell, with the pore being able to be open or closed dependent upon the voltage potential across the cell membrane, see Figure 2.7 Mutations in SCN9A cause conditions of both autosomal dominant excess pain and autosomal recessive painlessness.26 The dominant phenotypes were discovered first with a handful of families with either ‘Congenital Erthyromelalgia’ and ‘Familial paroxysmal extreme pain syndrome’, phenotypes of episodic severe neuropathic pain, which become more frequent and of greater severity with age, and both are caused by heterozygous mis-sense mutations in SCN9A.26 When these mutations are assessed by electrophysiology the channels are overactive. Either the channel opens prematurely (at lesser membrane depolarizations) or does not close properly (staying open during membrane repolarization). The activating effect of these mutations was not easily predictable by bio-informatics.27
Fig. 2.

Schematic diagram showing proteins that are involved in the differentiation process of nociceptor from neural crest cell to a mature neuron. PRDM12 and CLTCL1 are important in the differentiation from a neural crest cell to a precursor sensory neuron.3 NGF and TRK-A function in the differentiation of the nociceptor precursor to a mature nociceptor.15,16 Nav1.7 and Nav1.8 are upregulated in the fully developed nociceptor.25 Abbreviations: PRDM12, PR domain zinc finger protein 12; CLTCL1, clathrin heavy chain-like 1; NGF, nerve growth factor; TRK-A, tyrosine kinase receptor type 1; Nav, voltage sodium channel.
The recessive mutations in CIP individuals are assumed to cause non-functional changes to the Nav1.7 protein, see Table 2. Although this was proven to be so in the original reports, it has not always been subsequently. Non-sense, frame shift indels and canonical splice mutations can be assumed pathogenic without functional analysis.28 However, the functional consequences of mis-sense mutations cannot be reliably predicted to be non-functional, activating or harmless.28 Added to which SCN9A possesses a rare U12 intron, with different splice acceptor and donor sites compared to the canonical U2 splicing.29
So, activating mutations in SCN9A produce paroxysmal neuropathic pain, with no other obvious clinical features. And bi-allelic non-functional mutations cause congenital painlessness, with the only other consequence being anosmia. This makes SCN9A a critical and non-redundant essential element in pain sensing, although how this is achieved is still unclear.
SCN11A is the gene encoding the protein voltage-gated sodium channel Nav1.9; active at the cell membrane of nociceptors and acting as described above for Nav1.7. However, Nav1.9 has the role of setting the excitability of a nociceptor to incoming signals.30 It achieves this by altering the resting membrane potential to a more positive (making it easier to generate an action potential) or more negative (making it harder to generate). In this way, the protein was regarded as a modulator, and mouse knockout studies reported it had only a small effect on pain perception.30
A small number of autosomal dominant families with SCN11A mis-sense mutations that lead to increased activity of Nav1.9 were reported. The phenotype was of a variable pain syndrome including paroxysms of distal lower limb (mostly) extremity and abdominal pain, lasting days, and sometimes accompanied by sweating.31,32 Oddly, the painful episodes diminished with age in some families.31 This phenotype was surprising given the known functions of Nav1.9 and is similar to that caused by activating mutations in TRPA1 and SCN9A, with the exception that the pain decreases but not increases with age.33
Much more surprising was the report of a handful of cases with an extraordinary phenotype and the same de novo mutation in Nav1.9, p.(Leu811Pro).34 These individuals have CIP, but all presented with profound gastrointestinal hypomobility, usually necessitating intravenous feeding in the first year of life.35 During the first year the painlessness became apparent as did a profound itch affecting predominantly the neck and face, hypotonia and mild weakness, and an intolerance of temperature changes. The p.(Leu811Pro) mutation occurs in the last amino acid of the 2nd pore lining transmembrane domain of Nav1.9 (this transmembrane domain is called DII S6). Leucine-811 is conserved in all nine Nav1. channels. The mechanism of painlessness was unexpected, with the mutation causing excessive activity at resting voltages.34 This results in sustained depolarization of nociceptors, so nociceptors were unable to be triggered to fire action potentials. A second similar mutation was reported in a mother and two of her children with the Nav1.9 change p.(Leu1302Phe), in DIII S6 where leucine is again conserved in all Nav. channels; electrophysiology was not performed.36 A third similar mutation p.Leu396Pro was described in a boy with a very similar phenotype to p.(Leu811Pro). Again the mutation was de novo and electrophysiology showed a gain of function. The altered leucine being invariant in Nav1. channels and located at the distal end of DI S6 in Nav1.9.37 Taken together, these three SCN11A mutations that alter the last leucine of an S6 transmembrane domain all cause an unusual class of gain of function that inhibits a nociceptor from being able to generate action potentials.
Zinc finger homeobox 2 (ZFHX2)
A single remarkable three-generation family has been described with a heterozygous mis-sense mutation in ZFHX2 p.Arg1907Lys.38 This is putative transcription factor with the mutation within a homeodomain. The phenotype did not seem to involve a neuropathy, and the authors showed that a number of genes linked to pain had altered expression; however, the mechanism(s) by which this ZFHX2 caused a lack of pain sensing is currently unclear.
Fatty acid amide hydrolase (FAAH)
A woman with CIP was reported with deficient fatty-acid amide hydrolase 1 activity, one of two enzymes that metabolise N-acyl ethanolamines such as the endocannabinoid anandamide.39 Her phenotype was of a ‘pain-insensitive patient with a non-anxious disposition’ and was postulated to result from enhanced endocannabinoid signalling. There was no obvious genetic defect in FAAH2 on the X-chromosome.
So, how can these extremely rare genetic disorders help us understand the sophisticated and intricate mechanisms of pain, and provide new methods of diagnosing and treating pain?
Areas of agreement
The importance of NGF-TRKA signalling for nociceptor genesis
The recent finding that PRDM12 activated NTRK1 expression in nociceptor precursors further cemented the absolute importance of NGF-TRKA signalling for nociceptor genesis.15,16 Mutations found in patients have shown/confirmed the important parts/domains of proteins and help confirm which signalling pathways are necessary and which interacting partners are essential.11
New analgesics can be generated from knowledge of the NGF-TRKA nociceptor pathway
Somewhat surprisingly, the developmental pain genes NTRK1, NGF and PRDM12 (expressed and acting during early pregnancy) all have significant post-natal expression in nociceptors and white cells. NGF and TRKA have been shown to be involved in post-natal inflammatory pain sensing, and indeed antibodies blocking their interaction have emerged as a new class of analgesics.40 Initial trials have shown success in osteoarthritis and back pain.41 NGF has been shown to have a multitude of functions within cells other than nociceptors—mast cells, macrophages, keratinocytes and the central nervous system, but reassuringly no significant extra-nociceptor side effects have emerged.40,42 Interestingly, mutations found in painless patients have been used directly to generate potential new analgesics, such as in NGF where a patient mutations p.Arg121Try is unable to be cleaved from ProNGF to NGFBeta13 (and personal communication)”.
Increased susceptibility to S. aureus infection is a consequence of deficient NGF-TRKA signalling
Clinically, individuals who are either both without nociceptors (congenital HSANs and HSNs), or whose nociceptors degenerate (other HSNs, not covered by this review) will have frequent and chronic S. aureus infections. This feature was initially considered a consequence of the decreased cognition reported in HSAN4 and 5 but is consistent in all conditions where nociceptors or absent or degenerate; this includes PRDM12 and CLTCL1 CIPs.5,6,21,35
Mutations in the voltage-gated sodium channels SCN9A and SCN11A can cause congenital painlessness, and in contradistinction other mutations can cause episodic neuropathic pain
These nociceptor membrane proteins are essential for nociceptor function through their ability to respond to various tissue-damage signals and generate action potential, which are sent to the central nervous system. Nav1.7, Nav1.8 and Nav1.9 are prominently expressed in nociceptors; and whilst inactivating recessive mutations in SCN10A/Nav1.8 would be expected to include CIP in the resultant phenotype, none have been thus far reported (although activating mutations causing episodic pain have43).
SCN9A is an important analgesic target
The importance of SCN9A in pain sensing was initially almost overlooked, as knockout mice died during weaning, and a conditional knockout had only a partial pain phenotype.44 In retrospect, this was explained when the full human SCN9A-CIP phenotype was delineated; painlessness and anosmia, as anosmic mice fail to suckle and hence die.4 Since the 2006 discovery of SCN9A being a cause of CIP, it has been the subject of considerable activity by the Pharmaceutical industry, as not only do individuals with SCN9A-CIP feel no pain, they also feel no neuropathic pain, or visceral pain including painless childbirth. Furthermore, they have no cardiac, neurological or cognitive deficits, and despite SCN9A being expressed in the pancreatic Islets of Langerhans diabetes has not been reported.45 This suggested that a specific Nav1.7 antagonist would have only one side effect, a temporary loss of the sense of smell.
Mouse studies showed that SCN9A had to be knocked out both in dorsal root ganglia and in the sympathetic trunk to achieve the complete analgesia seen in humans.46 This supports anaesthetic practice where some types of neuropathic pain can only be treated with blockade of the sympathetic trunk and confirms the important role of the sympathetic nervous system in generating some forms of severe pain.
Because the importance of SCN9A in pain sensing, numerous molecules have been produced by pharmaceutical firms that antagonise Nav1.7, and some have been subject to early phase clinical trials.47,48
SCN11A is unlikely to be an analgesic target
This is because of the additional muscle, bowel, itch and temperature phenotypes of the individuals with SCN11A-CIP, and that these correlate with where the Nav1.9 protein is expressed.
There are further Mendelian causes of painlessness to be discovered
Most research teams have ‘unsolved cases’, and whilst some may be due to mutations that are not identified using Sanger and exome sequencing approaches in known genes, it is likely that others are undiscovered rare causes of Mendelian painlessness.
Areas of controversy
Which NGF-TRKA intracellular signalling pathways operate in nociceptor development and which in post-natal pain sensing?
NGF can exist as NGF or Pro-NGF, each can bind to not only TRKA, but also p75NTR and SORTILIN in nociceptors—and each has different signalling cascades that involve signalling endosomes. Active signalling endosomes are clathrin-coated organelles that are trafficked within the cell to axons, nucleus and dendrites, and are subject to further active processing and inhibition from lysosomal destruction. The complexity of this signalling repertoire and how it is used in development, pain signalling and S. aureus killing has yet to be unravelled.49–51
Why have no clinically effective Nav1.7 antagonist been generated?
A number of Pharmaceutical companies made Nav1.7 antagonists, including small molecules and antibodies. So far, at least six compounds have been abandoned following clinical trials, and three are still in the discovery phase of drug development. The reasons why these drugs have failed is unclear and three possible explanations have been suggested. Firstly, that 100% blockade of the Nav1.7 channel is needed to prevent pain and that a sufficient local concentration of the drug is not currently achievable. Secondly, that nociceptors use Nav1.7 at multiple sites on their membrane, including their spinal cord synapses to pass nociceptive signals to the central nervous system. However, all current Nav1.7 analgesics do not penetrate the blood brain barrier so these synapses are not ‘blocked’, and pain signals can still be generated. Thirdly, that lack of SCN9A causes a congenital absence of a part of the central pain pathways through lack of stimulation, so CIP-SCN9A individuals actually have a complex developmental form of CIP, of which non-functional peripheral nociceptors are just part.
SCN9A-CIP causes analgesia, at least in part, through endogenous opioids
Scn9a knockout mice are pain free. In these mice both μ- and δ-opioid receptors were necessary for painlessness, and endogenous encephalin production was increased.52,53 A corollary of these findings is that Nav1.7 antagonists may be opiate sparing, allowing much smaller doses of opioids to be used to achieve pain relief with a reduction in the risk of adverse side effects such as constipation and addiction.
Why do all CIP phenotypes involve a complete loss of all types of nociception?
No conditions have been described where one type of pain is absent, such as heat pain, or inflammatory pain—and mouse studies suggest that these phenotypes could exist.54,55 Whether this reflects the difficulty in ascertaining affected individuals or that nociceptors are always poly-modal (able to sense more than one type of tissue damage) is unclear. A further mystery is that different types of developmental CIP are associated with different selective nociceptor absences, e.g. Aδ in PRDM12, and predominantly C fibres in NTRK1 and NGF; and yet apparently identical painlessness results.
Areas timely for developing research
PRDM12 as an analgesic target
Although PRDM12 clearly acts in early human development, it is also expressed at significant levels in adult nociceptors. This suggests that it may have a post-natal role in pain, just as the developmental CIP genes NTRK1 and NGF do. It may ensure that a nociceptive phenotype continues to be imposed on neurons, if it maintains its prenatal role as a transcription factor. In this case, antagonists may have a role in chronic pain treatment by changing neuronal identity.
Discovery of the function and analgesic potential of new CIP genes
There are only nine voltage-gated sodium channel proteins—three of which cause Mendelian pain syndromes, but there are 27 calcium and 36 potassium voltage-gated channels—none of which are known to be involved in monogenic pain disorders. It would seem likely that there are undiscovered membrane ion channels that cause significant pain phenotypes that would be ‘druggable’. And also defining the function(s) of the rare causes of CIP may suggest further new analgesics, e.g. ZFHX2 to modulate nociceptor identity, MPV17 as a topical treatment for shingles and trigeminal neuralgia, and FAAH inhibition where pain and anxiety co-exist.
Can NGF-TRKA be used in the treatment of S. aureus?
S. aureus infection and asymptomatic carriage continue to be a significant medical problem, especially for hospitals, because of the emergence of multi-resistant strains. New treatments against S. aureus that bolstering the body’s own responses may be possible if molecular distinctions can be found between the functional pathways of NGF pain sensing and NGF intracellular bacterial killing.6
Conclusion
The individual, social, economic, and health care system consequences of pain are becoming more important as living standards rise, as more people have multi-pathologies and previously untreatable disorders, and as the population ages (about half of people aged over 65y have chronic pain56). Furthermore, the complexity of pain sensing and the differences between pain phenotypes is becoming clearer following considerable clinical and scientific research. However, the Pharmaceutical industry has produced further problems for pain sufferers with its advocacy of the potent synthetic opiates with increased dependency and addictive potential (note the infamous Jick NEJM letter57), and their aggressive marketing through non-specialists. This has led to an epidemic of synthetic opioid use (1 in 20 Americans are taking prescription opioids—four times the rate in the UK), and abuse. (In 2017 in USA there were 47 600 opioid-related deaths, compared to the 40 100 motor vehicle deaths and 40 000 gun deaths.58
The Pharmaceutical industry has also failed to produce new analgesics. Some of the reasons for this are the difficulties of classifying types of pain and responses to pain treatments, the myriad of molecules and pathways involved in pain, and that rodents are an imperfect model organism for studying adult-onset human neuropathic and chronic pain. The phenotype and genes that cause CIP, however, do give clear human targets for further research to both discover and understand pain pathways, and then to design targeted treatments. Despite Mendelian disorders of painlessness being extremely rare, they are enabling the creation of new types of analgesics for the benefit of all.
I.D. is a postdoctoral researcher (BSc, MSc, PhD) in the Cambridge Institute for Medical Research, University of Cambridge. She is experienced in electrophysiology and synaptic function, and interested in congenital insensitivity to pain and Mendelian neurodevelopmental disorders.
W.A.W. is currently a fifth year medical student at Keele Medical School intercalating in neuroscience planning to pursue a career in Neurology.
C.G.W. is a NHS Clinical Geneticist (MB ChB, FRCP, FMedSci) interested in childhood onset neurodevelopment disorders and pain genetics. He is also a Principal Investigator in the Cambridge Institute for Medical Research, https://www.cimr.cam.ac.uk.
Conflict of interest statement
The authors have no potential conflicts of interest.
Funding
ID is funded by the Cambridge NIHR Biomedical Research Centre.
References
- 1. Padilla F, Couble ML, Coste B, et al. Expression and localization of the Nav1.9 sodium channel in enteric neurons and in trigeminal sensory endings: implication for intestinal reflex function and orofacial pain. Mol Cell Neurosci 2007;35:138–52. [DOI] [PubMed] [Google Scholar]
- 2. Akin EJ, Higerd GP, Mis MA, et al. Building sensory axons: delivery and distribution of NaV1.7 channels and effects of inflammatory mediators. Sci Adv 2019;5:eaax4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nahorski MS, Chen YC, Woods CG. New Mendelian disorders of painlessness. Trends Neurosci 2015;38:712–24. [DOI] [PubMed] [Google Scholar]
- 4. Wei BQ, Mikkelsen TS, McKinney MK, et al. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem 2006;281:36569–78. [DOI] [PubMed] [Google Scholar]
- 5. Indo Y. Congenital insensitivity to pain with anhidrosis In: Adam MP, Ardinger HH, Pagon RA, et al. (eds.). GeneReviews® [Internet]. Seattle (WA): University of Washington, 1993,2020–08 [PubMed] [Google Scholar]
- 6. Hepburn L, Prajsnar T, Klapholz C, et al. A Spaetzle-like role for nerve growth factor in vertebrate immunity to Staphylococcus aureus. Science 2014;346:641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox J, Reimann F, Nicholas A, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006;444:894–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weiss J, Pyrski M, Jacobi E, et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 2011;472:186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Indo Y, Tsuruta M, Hayashida Y, et al. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet 1996;13:485–8. [DOI] [PubMed] [Google Scholar]
- 10. Franco M, Melero C, Sarasola E, et al. Mutations in TrkA causing congenital insensitivity to pain with anhidrosis (CIPA) induce misfolding, aggregation, and mutation-dependent neurodegeneration by dysfunction of the autophagic flux. J Biol Chem 2016;291:21363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shaikh S, Chen Y, Halsall S, et al. A comprehensive functional analysis of NTRK1 missense mutations causing hereditary sensory and autonomic neuropathy type IV (HSAN IV). Hum Mutat 2016;38:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Einarsdottir E, Carlssson A, Minde J. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet 2004;13:799–805. [DOI] [PubMed] [Google Scholar]
- 13. Shaikh S, Nahorski M, Woods C. A third HSAN5 mutation disrupts the nerve growth factor furin cleavage site. Mol Pain 2018;14:174480691880922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Y, Auer-Grumbach M, Matsukawa S, et al. Transcriptional regulator PRDM12 is essential for human pain perception. Nat Genet 2015;47:803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartesaghi L, Wang Y, Fontanet P, et al. PRDM12 is required for initiation of the nociceptive neuron lineage during neurogenesis. Cell Rep 2019;26:3484–3492.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desiderio S, Vermeiren S, Van Campenhout C, et al. Prdm12 directs nociceptive sensory neuron development by regulating the expression of the NGF receptor TrkA. Cell Rep 2019;26:3522–3536.e5. [DOI] [PubMed] [Google Scholar]
- 17. Axelrod F, Hilz M. Inherited autonomic neuropathies. Semin Neurol 2003;23:381–90. [DOI] [PubMed] [Google Scholar]
- 18. Anderson S, Qiu J, Rubin B. EGCG corrects aberrant splicing of IKAP mRNA in cells from patients with familial dysautonomia. Biochem Biophys Res Commun 2003;310:627–33. [DOI] [PubMed] [Google Scholar]
- 19. Kurth I, Pamminger T, Hennings J, et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet 2009;41:1179–81. [DOI] [PubMed] [Google Scholar]
- 20. Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015;522:354–8. [DOI] [PubMed] [Google Scholar]
- 21. Nahorski M, Al-Gazali L, Hertecant J, et al. A novel disorder reveals clathrin heavy chain-22 is essential for human pain and touch development. Brain 2015;138:2147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nahorski M, Borner G, Shaikh S, et al. Clathrin heavy chain 22 contributes to the control of neuropeptide degradation and secretion during neuronal development. Sci Rep 2018;8:2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spinazzola A, Santer R, Akman O, et al. Hepatocerebral form of mitochondrial DNA depletion syndrome. Arch Neurol 2008;65:1108–13. [DOI] [PubMed] [Google Scholar]
- 24. Alonzo J, Venkataraman C, Field M, et al. The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J Biol Chem 2018;293:20285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marmigère F, Ernfors P. Specification and connectivity of neuronal subtypes in the sensory lineage. Nat Rev Neurosci 2007;8:114–27. [DOI] [PubMed] [Google Scholar]
- 26. Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest 2007;117:3603–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waxman SG, Merkies ISJ, Gerrits MM, et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol 2014;13:1152–60. [DOI] [PubMed] [Google Scholar]
- 28. Schon K, Parker A, Woods CG. Congenital Insensitivity to Pain Overview In: Adam MP, Ardinger HH, Pagon RA, et al. (eds.). GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle, 1993,2020–18 [PubMed] [Google Scholar]
- 29. Shaikh SS, Nahorski MS, Rai H, et al. Before progressing from "exomes" to "genomes"… don't forget splicing variants. Eur J Hum Genet 2018;26:1559–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostman JA, Nassar MA, Wood JN, Baker MD. GTP up-regulated persistent Na+ current and enhanced nociceptor excitability require NaV1.9. J Physiol 2008;586:1077–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang XY, Wen J, Yang W, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet 2013;93:957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Han C, Yang Y, Te Morsche RH, et al. Familial gain-of-function Nav1.9 mutation in a painful channelopathy. J Neurol Neurosurg Psychiatry 2017;88:233–40. [DOI] [PubMed] [Google Scholar]
- 33. Kremeyer B, Lopera F, Cox JJ, et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 2010;66:671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leipold E, Liebmann L, Korenke G, et al. A de novo gain-of-function mutation in SCN11A causes loss of pain perception. Nat Genet 2013;45:1399–404. [DOI] [PubMed] [Google Scholar]
- 35. Woods C, Babiker M, Horrocks I, et al. The phenotype of congenital insensitivity to pain due to the NaV1.9 variant p.L811P. Eur J Hum Genet 2014;23:561–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phatarakijnirund V, Mumm S, McAlister WH, et al. Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9. Bone 2016;84:289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. King MK, Leipold E, Goehringer JM, et al. Pain insensitivity: distal S6-segment mutations in NaV1.9 emerge as critical hotspot. Neurogenetics 2017;18:179–81. [DOI] [PubMed] [Google Scholar]
- 38. Habib A, Matsuyama A, Okorokov A, et al. A novel human pain insensitivity disorder caused by a point mutation in ZFHX2. Brain 2017;141:365–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Habib A, Okorokov A, Hill M, et al. Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth 2019;123:e249–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Denk F, Bennett D, McMahon S. Nerve growth factor and pain mechanisms. Annu Rev Neurosci 2017;40:307–25. [DOI] [PubMed] [Google Scholar]
- 41. Schmelz M, Mantyh P, Malfait A, et al. Nerve growth factor antibody for the treatment of osteoarthritis pain and chronic low-back pain. Pain 2019;160:2210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miller RE, Malfait AM, Block JA. Current status of nerve growth factor antibodies for the treatment of osteoarthritis pain. Clin Exp Rheumatol 2017;107:85–7. [PMC free article] [PubMed] [Google Scholar]
- 43. Bennett DL, Clark AJ, Huang J, et al. Dib-hajj SD the role of voltage-gated sodium channels in pain Signaling. Physiol Rev 2019;99:1079–151. [DOI] [PubMed] [Google Scholar]
- 44. Nassar M, Stirling L, Forlani G, et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci 2004;101:12706–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Q, Chibalina M, Bengtsson M, et al. Na+current properties in islet α- and β-cells reflect cell-specific Scn3a andScn9a expression. J Physiol 2014;592:4677–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Minett M, Nassar M, Clark A, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun 2012;3:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emery E, Luiz A, Wood J. Nav1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets 2016;20:975–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rivara M, Zuliani V. Novel sodium channel antagonists in the treatment of neuropathic pain. Expert Opin Investig Drugs 2015;25:215–26. [DOI] [PubMed] [Google Scholar]
- 49. Marlin MC, Li G. Biogenesis and function of the NGF/TrkA signaling endosome. Int Rev Cell Mol Biol 2015;314:239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ye M, Lehigh K, Ginty D. Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. Elife. 2018;7 pii: e33012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Barford K, Keeler A, McMahon L, et al. Transcytosis of TrkA leads to diversification of dendritic signaling endosomes. Sci Rep 2018;8:4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Minett M, Pereira V, Sikandar S, et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun 2015;6:8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pereira V, Millet Q, Aramburu J, et al. Analgesia linked to Nav1.7 loss of function requires μ- and δ-opioid receptors. Wellcome Open Res 2018;3:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abrahamsen B, Zhao J, Asante CO, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 2008;321:702–5. [DOI] [PubMed] [Google Scholar]
- 55. Vandewauw I, De Clercq K, Mulier M, et al. A TRP channel trio mediates acute noxious heat sensing. Nature 2018;555:662–6. [DOI] [PubMed] [Google Scholar]
- 56. Häuser W, Schmutzer G, Hilbert A, et al. Prevalence of chronic disabling noncancer pain and associated demographic and medical variables: a cross-sectional survey in the general German population. Clin J Pain 2015;31:886–92. [DOI] [PubMed] [Google Scholar]
- 57. Porter J, Jick H. Addiction rare in patients treated with narcotics. N Engl J Med 1980;302:123. [DOI] [PubMed] [Google Scholar]
- 58.https://www.cdc.gov/drugoverdose/data/statedeaths.html