

Abstract
BACKGROUND
Metabolic reprogramming is a hallmark of malignancy first recognized a century ago. In some cases, reprogrammed metabolic activities can be exploited to diagnose, monitor, and treat cancer. Stereotyped metabolic activities in cultured cancer cells–notably, aerobic glycolysis, glutamine catabolism, macromolecular synthesis, and redox homeostasis–support the requirements of exponential growth and proliferation. These pathways are under cell-autonomous control by oncogenic signaling and transcriptional networks. This has produced the widespread perception that a core set of fixed metabolic dependencies will prove to be excellent therapeutic targets across diverse cancer types. Several metabolic inhibitors designed to target these pathways have advanced into clinical trials.
ADVANCES
The past decade has brought numerous advances in our understanding of why tumors develop metabolic phenotypes that differ from adjacent, nonmalignant tissues and when these phenotypes represent actionable therapeutic vulnerabilities. Mechanistic insights into how the oncogenotype dictates metabolic patterns have exploded, aided by the ever-increasing use of advanced analytical techniques to characterize tumor metabolism in detail. This has led to the remarkable discovery of a few metabolic properties that can directly promote tumor initiation, including the accumulation of D-2-hydroxyglutarate in tumors with mutations in isocitrate dehydrogenase-1 and −2. Other advances have demonstrated the extraordinary amount of metabolic heterogeneity among human tumors and, in some cases, even within distinct regions of the same tumor. This heterogeneity results from a complex set of factors, including processes intrinsic and extrinsic to the cancer cell. Many of these studies have identified promising subtype-selective metabolic vulnerabilities in experimental models. However, they have cast doubt on the classical paradigm of convergent, oncogene-driven liabilities among histologically and genetically diverse tumors. Even more fundamentally, it has become increasingly clear that metabolic phenotypes and vulnerabilities evolve as tumors progress from premalignant lesions to locally invasive tumors to metastatic cancer. Microenvironmental and genetic factors appear to induce selective pressures that drive clonal evolution within tumors, and this can create or eliminate metabolic liabilities while facilitating cancer progression. During metastasis, for example, several studies demonstrate that cancer cells need to activate mechanisms to resist oxidative stress, or else these cells are culled by the oxidizing environment of the bloodstream. A major theme arising from recent research is that pathways that stimulate the growth of localized, treatment-naïve tumors are distinct from and in some cases irrelevant to the activities that drive mortality by supporting metastasis and therapy resistance.
OUTLOOK
The emerging view of cancer metabolism is that it is flexible and context-specific, with few fixed, broadly applicable liabilities. Understanding how reprogrammed metabolism supports tumor growth–and identifying which reprogrammed activities are most relevant to therapeutic liabilities–requires a more sophisticated view of how metabolic phenotypes evolve as cancer progresses. Advanced animal models that recapitulate the landmark events in human cancer progression will be instrumental in discovering the most important metabolic vulnerabilities. These animal studies will need to be complemented by increasing efforts to assess metabolism directly in human tumors through metabolomics, metabolic isotope tracers, and advanced techniques in metabolic imaging. Crucially, cooperative, multidisciplinary research is needed to translate findings from animal models into patients and from human cancer into mouse models for mechanistic studies and hypothesis testing. Ideally, work along these lines will generate efficient ways to detect predictive aspects of metabolic behavior in human tumors to aid in clinical trial design and to stratify patients to receive the most effective therapies. These efforts over the next decade should produce a more nuanced but ultimately more relevant and therapeutically actionable view of cancer metabolism.
Metabolic reprogramming is a hallmark of malignancy. As our understanding of the complexity of tumor biology increases, so does our appreciation of the complexity of tumor metabolism. Metabolic heterogeneity among human tumors poses a challenge to developing therapies that exploit metabolic vulnerabilities. Recent work also demonstrates that the metabolic properties and preferences of a tumor change during cancer progression. This produces distinct sets of vulnerabilities between primary tumors and metastatic cancer, even in the same patient or experimental model. We review emerging concepts about metabolic reprogramming in cancer, with particular attention on why metabolic properties evolve during cancer progression and how this information might be used to develop better therapeutic strategies.
Graphical Abstract
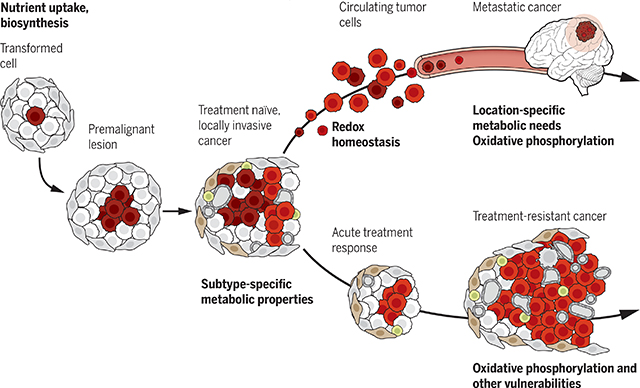
Metabolic evolution during cancer progression. Metabolic needs and vulnerabilities evolve throughout cancer progression. Early stages of tumor growth require nutrient uptake and biosynthesis, with additional subtype-selective metabolic needs emerging in locally invasive cancers. Tumors acquire dependence on new pathways during later stages of cancer progression, particularly metastasis and therapy resistance. These include potentially targetable liabilities such as dependence on mechanisms to resist oxidative stress and increased reliance on oxidative phosphorylation.
Cellular metabolism is a flexible network that allows tissues to meet demands for homeostasis and growth. In cancer, malignant cells acquire metabolic adaptations in response to a variety of cell-extrinsic and cell-intrinsic cues (Fig. 1). Some of these adaptations initiate the process of transformation, and some promote growth of malignant cells and render them susceptible to inhibitors of key pathways. Historically, most cancer metabolism research has focused on phenotypes observed in clinically detectable tumors or experimental models derived from them. The terms “cancer metabolism” and “metabolic reprogramming” are therefore usually taken to indicate a shared set of pathways observed in highly proliferative tumors or cancer cells. The Warburg effect, a preference for glycolysis and lactate secretion in the presence of oxygen, is an example of a metabolic property under cell-autonomous control by oncogenes in many proliferating cancer cells and tumors (1). Therapeutic opportunities might arise if cancer cells develop fixed addictions to the Warburg effect or other conserved pathways, while nonmalignant cells adapt to their inhibition.
Fig. 1. Intrinsic and extrinsic factors contribute to metabolic phenotypes in tumors.

Intrinsic factors include characteristics of the parental tissue as well as new properties arising in the malignant cells as a consequence of altered signaling and transcriptional networks. Extrinsic factors include metabolic stresses imposed by the microenvironment and metabolism of the patient.
Recent insights arising from advanced techniques in metabolic analysis and more attention to bona fide tumor metabolism in vivo suggest that a more nuanced view of metabolic reprogramming is needed. First, tumor cells require many of the same pathways and adaptations available to nonmalignant tissues, indicating that few metabolic activities are truly confined to tumors. Second, as the approaches used to assess metabolic phenotypes have become more informative, it has become clear that human tumors are metabolically heterogeneous. As a result, metabolic features and liabilities are subtype-selective rather than uniform across all cancers (2). This is consistent with the variable efficacy of therapies aimed at seemingly universal metabolic liabilities in cancer cells, including antifolates and other agents.
Third and most importantly for this Review, metabolic phenotypes and dependencies evolve as cancer progresses from preneoplastic lesions to localized, clinically apparent malignancies to metastatic cancer. Understanding cancer metabolism and identifying liabilities requires a sophisticated view of how metabolic phenotypes evolve over time. There is still value in characterizing which metabolic activities allow cells to maximize growth and proliferation and how such activities become chronically activated in cancer. However, the emerging view is that cancer metabolism is flexible and context-specific, and some of the most promising therapeutic targets are distinct from the pathways that support cell growth in culture or locally aggressive tumors. This Review focuses on recent conceptual advances in the metabolic basis of tumor progression, with particular attention to how evolving metabolic dependencies relate to therapeutic opportunities.
Premalignant lesions
Organ-specific metabolic phenotypes in nonmalignant tissues arise from the integrated effects of cell-intrinsic and cell-extrinsic factors. These include epigenetically regulated gene expression, cellular composition, tissue architecture, and in some cases commensal microbial populations, all of which vary from tissue to tissue (Fig. 1). Metabolic perturbations induce tissue-specific responses that support overall homeostasis. For example, fasting stress in mammals induces lipolysis in adipose tissue, ketogenesis in the liver, and ketone consumption in the brain and other organs. Understanding tissue-specific metabolic phenotypes is an important foundation for understanding cancer metabolism.
The initial stages of tumorigenesis occur within the metabolic constraints of the native tissue, which may otherwise be unperturbed. This explains two observations related to metabolic heterogeneity in cancer. First, although tumors are readily recognized through gene expression signatures, they retain transcriptomic features of the parental tissue so that tumors arising in the same organ are more similar to each other than tumors arising in different organs. A study that classified thousands of human tumors found that tissue of origin was a major factor defining DNA methylation and gene expression patterns (3). Gene expression signatures focused on the metabolic network also bear more similarity to the tissue of origin than to tumors from different organs (4). Second, although different oncogenes reprogram metabolism in different ways, the tissue in which the oncogene is expressed affects the execution of metabolic reprogramming. An excellent example involves expression of the human MYC oncogene in mice (5). Transgenic MYC induces metabolic changes in both liver tumors and lung tumors but activates glutamine catabolism in lung tumors and glutamine synthesis in liver tumors. Therefore, classifying tumors according to the oncogenic driver rather than the tissue of origin may obscure metabolic differences imposed by the original metabolic state of the tissue.
The tissue of origin and developmental context also determine whether potentially transforming mutations result in cancer initiation (6). Familial cancer syndromes provide an example of this phenomenon. In these diseases, patients inherit germline mutations in tumor suppressors and develop cancer when the other allele is lost through either mutation, deletion, or silencing. In the case of the classical tumor suppressor gene TP53, germline mutations result in a somewhat different tissue distribution of cancers than sporadic TP53-mutant cancers in patients who lack germline mutations (7). This suggests that the consequences of TP53 mutation depend on the developmental context.
Some metabolic enzymes are tumor suppressors, meaning that patients who inherit one mutant allele develop cancer when the other allele is lost in a susceptible cell. But only a small subset of cells are susceptible. Despite ubiquitous expression of the metabolic enzyme succinate dehydrogenase (SDH), germline mutations in subunits of this complex cause cancer only in a few sites, including neuroendocrine tissues (paraganglioma), the adrenal gland (pheochromocytoma), and the stomach and small intestine (gastrointestinal stromal tumors) (8–10). Similarly, germline mutations in the broadly expressed enzyme fumarate hydratase (FH) cause hereditary leiomyomatosis and renal cell carcinoma, a familial syndrome of uterine and cutaneous leiomyomas and kidney cancer, without susceptibility to more common tumors (11). Other cancers arise from inborn errors of metabolism, in which biallelic germline mutations in metabolic enzymes cause a widespread failure of metabolic homeostasis, but cancer is restricted to a subset of tissues specific for each enzyme (12).
The basis for such exquisite tissue specificity in these syndromes is unknown. One possibility is that the molecular hierarchy required for mutations in metabolic enzymes to result in malignancy is tissue specific. That is, some tissues may induce adaptive responses to constrain tumor initiation, or perhaps only a few cell types respond to the metabolic perturbation by becoming transformed. An alternative is that most cells simply cannot tolerate such perturbations and respond by undergoing senescence or cell death instead of transformation. These rare familial cancer syndromes are important because they provide opportunities to learn about tissue-specific mechanisms by which cells respond to genetically determined metabolic defects and why these responses include tumor initiation in the right context.
The concept of metabolic reprogramming as a cancer hallmark aligns with the idea that the mutations responsible for cancer initiation enable cells in nascent tumors to acquire metabolic properties that support cell survival, evasion of immune surveillance, and hyperplastic growth. This concept is well established for classical oncogenic drivers (such as MYC, KRAS, and others) that possess the ability to regulate metabolism in a cell-autonomous fashion. But it is unclear whether such mutations are necessary to establish metabolic properties that support tumor initiation. Some tumors lack recurrent mutations in canonical tumor suppressors and oncogenes but still harbor stereotyped metabolic properties that distinguish them from nonmalignant tissues. Clear cell papillary renal cell carcinoma (CCPAP), a generally low-grade kidney tumor, is one example (13). These tumors bear metabolomic signatures that suggest oxidative stress, suppressed oxidative metabolism, and depletion of mitochondrial DNA and RNA. Nevertheless, a detailed molecular analysis revealed a low genomic burden of point mutations and copy-number variants, no recurrent nonsynonymous mutations, and a DNA methylation pattern that resembles the nonmalignant kidney. Perhaps CCPAP tumors contain an initiating mutation yet to be discovered, or perhaps this mutation is lost before the tumor becomes clinically detectable. But another possibility is that the underlying metabolic perturbation phenocopies the effects of classical mutations. For example, chronic mitochondrial dysfunction in CCPAP may phenocopy loss of von Hippel-Lindau (VHL), the most commonly mutated tumor suppressor in clear cell renal cell carcinoma (ccRCC). Metabolite accumulation in the setting of dysfunctional mitochondria can inhibit VHL-dependent degradation of hypoxia-inducible factor-α (HIF-α) subunits, creating a pseudohypoxic state that mimics VHL loss (14).
Characterizing metabolic anomalies in premalignant lesions is challenging. First, these tumors usually escape clinical attention, so the literature on their metabolism is limited; there may also be bias against studies that fail to observe differences from parental tissue. Second, systemic metabolism influences cancer risk and probably the metabolism of premalignant lesions (Fig. 1). Many epidemiological studies report associations between obesity and cancer, and a large prospective analysis of more than 900,000 Americans found greater than a 50% increase in cancer mortality in patients with the highest body mass index (15, 16). The mechanisms linking cancer with obesity, diabetes, and other metabolic disorders are multifactorial. But because these diseases affect metabolism throughout the body, the particular pathways that promote cancer can be difficult to recognize in premalignant lesions. Despite these challenges, a few mechanistic relationships have been reported. Diabetes increases the frequency of KRAS variants in human pancreas, possibly through the effects of chronic hyperglycemia on nucleotide balance (17). Moreover, diets high in saturated fat potentiate the MYC transcriptional program in human and mouse prostate cancer, enhancing cell proliferation (18).
There are also several examples of specific metabolic changes in premalignant lesions relative to parental tissues. In mice, metabolic reprogramming in response to oncogenic KRas in pancreatic acinar cells promotes acinar-to-ductal metaplasia (ADM), a precursor to pancreatic ductal adenocarcinoma (PDAC) (19). Oncogenic KRas increases the levels of acetyl coenzyme A (CoA) and reactive oxygen species (ROS) before the development of ADM, and inhibiting these phenotypes blocks ADM (19, 20). Other studies in humans and mice report metabolic changes in premalignant colonic polyps, some of which are retained in adenocarcinomas (21, 22). Metabolic reprogramming precedes liver cancer in mice with MYC expression in hepatocytes (23). In early-stage, premalignant liver, MYC drives enhanced conversion of pyruvate to alanine, whereas later-stage malignant lesions convert pyruvate to lactate.
There is great interest in developing screening biomarkers for early-stage lesions, and altered metabolic states may be useful in this regard. In atypical adenomatous hyperplasias (AAHs) of the lung, premalignant lesions overexpress sodium-dependent glucose transporter 2 (SGLT2). SGLT2 does not transport the common cancer imaging tracer 18fluoro-2-deoxyglucose (FDG), and thus, these lesions are poor candidates for FDG-PET (positron emission tomography) imaging. But an alternative tracer, methyl-4-deoxy-4-[18F]-fluoro-α-d-glucopyranoside (Me4FDG), is taken up by SGLT and can identify AAH and low-grade adenocarcinomas of the lung (24).
A key question is whether early metabolic alterations produce targetable liabilities during tumor initiation. Leukemia initiation is sensitive to some aspects of early reprogramming. Mutations in isocitrate dehydrogenase isoform-1 (IDH1) and IDH2 generate the oncometabolite D-2-hydroxyglutarate (D-2HG), which inhibits epigenetic enzymes including histone demethylases and the Ten-eleven translocation (TET) family of 5-methylcytosine hydroxylases. These effects contribute to tumor initiation in acute myelogenous leukemia (AML) by resulting in persistent histone and DNA methylation and impaired cellular differentiation (25). Epigenetic effects also occur in gliomas with these same mutations (26). At least in AML, dependence on these mutant enzymes persists after the cancer becomes clinically apparent because mutant IDH inhibitors have therapeutic efficacy in humans (27). In mice, oncogenic neuroblastoma Ras viral oncogene homolog (NRas) cooperates with mutations in the epigenetic regulator enhancer of zeste homolog 2 (Ezh2) to produce aggressive leukemias. Ezh2 mutations result in enhanced expression of branched-chain amino acid transaminase-1 (Bcat1). This enzyme promotes cancer by allowing cells to generate a large leucine pool, activating the growth-stimulating mechanistic target of rapamycin (mTOR). Silencing Bcat1 reduces leukemia initiation, and overexpressing this enzyme promotes NRas-driven leukemia initiation even in cells that lack Ezh2 mutations (28). These studies demonstrate the functional and therapeutic relevance of metabolic reprogramming early in tumorigenesis.
Primary invasive cancers
Through excessive cell proliferation and acquisition of additional mutations, premalignant lesions progress to malignant tumors that arouse clinical attention. Much of what is known about cancer metabolism comes from primary, locally aggressive tumors confined to the parental tissue. The importance of cell survival and proliferation during progression from premalignant lesions to clinically detectable tumors may explain why several stereotyped metabolic properties repeatedly appear in cancer metabolism studies. We use the term “convergent properties” to describe these pathways because they appear to be the common result of myriad regulatory aberrations that culminate in tumorigenesis (2). Pathways involved in energy production (including the Warburg effect), macromolecular synthesis, and redox control are commonly reprogrammed by tumorigenic mutations in KRAS (29), TP53 (30), MYC (31), and many others (32). Perhaps these pathways appear convergent because clinically evident tumors–and the experimental models derived from them–would not arise without them.
Convergent metabolic pathways are supplied by abundant nutrients such as glucose and amino acids. A central concept in cancer metabolism is that malignant cells acquire an enhanced ability to feed themselves by activating nutrient uptake mechanisms (33). This cell-autonomous behavior is hardwired by mutations in the conserved mechanisms that govern cellular nutrient handling. Activating mutations in the phosphoinositide 3-kinase (PI3K) signaling pathway, which tethers growth factor signaling to activation of anabolic metabolism, are among the most common in human cancer (34). These mutations promote unrestrained growth by locking cells into a phenotype of nutrient uptake and anabolism with reduced dependence on extracellular growth factors for instruction. Mutations in the machinery that allows cells to sense nutrient availability and energetic status are also common. These include inactivating mutations in STK11, which encodes the serine-threonine kinase LKB1. LKB1 stimulates adenosine 5′-monophosphate (AMP)–activated protein kinase, which normally acts as a brake on anabolism under conditions of energetic stress. Growth factor signaling and nutrient availability converge on mTOR complex-1 (mTORC1), which integrates these signals to stimulate ribosome biogenesis, protein synthesis, and other pathways required for cell growth (35). Mutations that activate mTORC1 are common in primary invasive cancers and result in enhanced sensitivity to inhibitors of metabolic pathways under mTORC1 control (36).
The factors that regulate tumor metabolism, including the genomic landscape and microenvironment, evolve as the tumor progresses from premalignancy to overt cancer. Even the well-established effects of common oncogenic drivers may provide somewhat different benefits as the disease progresses. For example, oncogenic KRAS activates expression of the glucose transporter GLUT1 and enables high glycolytic rates when glucose is abundant (37). These mutations do not confer any particular advantage under glucose-replete conditions, but when glucose is scarce, mutant cells outcompete their wild-type counterparts and overtake the culture. Furthermore, simply culturing wild-type cells in low-glucose conditions selects for cells with enhanced GLUT1 expression, including clones that spontaneously acquire KRAS mutations (37). These findings indicate that the benefits provided by oncogenic mutations can be dictated by the metabolic milieu. For oncogenic drivers such as KRAS, mutations early in the process of tumorigenesis may provide a specific competitive advantage when nutrient delivery is compromised. In established tumors, ongoing expression of oncogenic KRas drives rapid growth by channeling glucose into pathways that promote macromolecule synthesis (29). It will be instructive to test whether dependence on metabolic effectors of KRas and other oncogenes evolves during progression to a malignant tumor.
Other metabolic properties diverge as tumors grow, resulting in remarkable metabolic heterogeneity in primary cancers (2). These divergent phenotypes reflect the effects of molecular heterogeneity in cancer cells and inconsistencies in the microenvironment (Fig. 1); the relative importance of these factors is an active area of investigation. A few studies have characterized the scope of heterogeneity defined by cell-intrinsic processes by profiling metabolism across panels of genetically diverse cancer cell lines. In one study, more than 80 lung cancer cell lines were cultured under identical conditions and subjected to 13C tracers and other assays to observe the scope of metabolic phenotypes defined purely by cell-intrinsic processes (38). Although all cells were derived from malignant lung tumors, their metabolic features were remarkably divergent. Correlating these features with orthogonal genomic, transcriptomic, and proteomic data revealed that some features [for example, patterns of nutrient utilization in the tricarboxylic acid (TCA) cycle] reflected single oncogenic drivers such as mutant KRAS or epidermal growth factor receptor (EGFR). Others resulted from the combined effects of multiple mutations in different genes acting in concert rather than single drivers, and still others could not easily be explained by the oncogenotype. Another study reached similar conclusions, identifying some genotype-phenotype correlations but failing to uncover predictable metabolic signatures for a number of common drivers (39). These studies imply that cancer cell metabolism is sensitive to combinatorial cell-intrinsic effects, such as the co-occurrence of multiple mutations and as-yet unknown processes.
Accumulation and selection of somatic mutations drives cancer progression and results in heterogeneous vulnerabilities. Truncal mutations are essential for cancer initiation, and subsequent mutations, including the order in which they arise, can influence tumor biology (6). KRAS is the most common oncogenic driver in human lung adenocarcinoma, but the behavior of KRAS-driven tumors is modified by mutations in other genes (Fig. 2). When coupled with oncogenic KRAS, mutations in STK11 result in aggressive malignant features, including metastasis and therapy resistance (40–42). This particular co-mutant state also influences metabolic liabilities. KRAS and STK11 mutations individually influence metabolism, but co-mutation of both genes causes new vulnerabilities to emerge, including enhanced dependence on pyrimidine metabolism and oxidative phosphorylation (OXPHOS) (43–45). The KRAS-STK11 metabolic phenotype is further modified by co-mutations in the tumor suppressor KEAP1 (kelch-like ECH associated protein 1), which encodes a negative regulator of the NRF2 (nuclear factor erythroid 2-related factor 2) antioxidant transcriptional program. The tendency for these three mutations to co-occur suggests that the metabolic state caused by mutations in KRAS and STK11 selects for the additional adaptation of enhanced antioxidant capacity satisfied by KEAP1 loss (Fig. 2) (46). In preclinical models, this antioxidant function requires glutamine catabolism, making tumors that contain all three mutations highly sensitive to glutaminase inhibitors (47).
Fig. 2. Accumulation of somatically acquired mutations changes tumor biology and causes metabolic liabilities to evolve.

KRAS mutations initiate tumorigenesis in the lung, driving nutrient uptake and anabolism. Mutation of STK11 increases key aspects of aggressive tumor biology, including metastatic efficiency and therapy resistance, but increases sensitivity to some metabolic inhibitors. KEAP1 mutations enhance resistance to oxidative stress by stimulating glutathione biosynthesis but induce dependence on glutaminase to supply precursors to produce glutathione. Cells are color-coded according to their genotype.
Interestingly, concomitant mutations in KRAS and STK11 also confer liabilities in PDAC, but the mechanisms are distinct from those in lung cancer. In mouse PDAC models, these mutations synergize to activate synthesis of serine and the methyl donor S-adenosyl methionine, a substrate for DNA methylation (48). This results in increased genome methylation in patterns that promote tumor growth. Inhibiting serine biosynthesis or DNA methyltransferases reduces tumor growth in these models.
The tumor microenvironment also evolves as clinically evident tumors arise from small, premalignant lesions. The microenvironment can impose a number of non-cell-autonomous pressures on cancer cells, including nutrient and oxygen deprivation, acidification of the extracellular space, and aberrant cell-matrix and cell-cell interactions (2, 49). Tumor progression requires that cancer cells tolerate these pressures and develop mechanisms to continue to proliferate. Oncogene-driven expression of nutrient transporters (24, 50), the ability to derive energy from diverse nutrient sources–including scavenged protein, recycled organelles, and necrotic debris (51–54)–and metabolic cooperativity among cancer cells or between cancer cells and stromal cells (55, 56) likely contribute to tumor cell fitness in stressful tumor microenvironments.
These cell-autonomous and microenvironmental processes not only affect metabolic differences among tumors but also give rise to regional metabolic heterogeneity within the same tumor. Progression of localized solid tumors in humans involves clonal expansion of cells with additional mutations and branched evolutionary growth. In ccRCC, sequencing multiple regions of primary tumors revealed that most somatically acquired mutations are not present in all areas (57). This produces regionally diverse combinations of mutations with cell-autonomous metabolic effects, including truncal VHL mutations followed by clonal mutations in mTOR, PTEN, and others. The effect of these combinations on metabolism is unknown. In human lung cancer, infusing patients with 13C-glucose during surgical resection of their tumors revealed marked regional metabolic heterogeneity despite the presence of truncal mutations in multiple regions (58, 59). In these tumors, local patterns of nutrient metabolism and metabolic gene expression correlated with local differences in perfusion, suggesting that the microenvironment and tumor cell genotype cooperate to regulate metabolism. We need better methods to assess regional metabolic phenotypes in human cancer because inconsistency of metabolic vulnerabilities across a tumor will limit the utility of metabolic therapies. Unless the vulnerability is established by a truncal mutation and is robust enough to withstand the mitigating effect of subsequent mutations and environmental factors, it may not represent a useful therapeutic target.
Metastatic cancer
Death from cancer is determined largely by metastasis rather than localized tumor growth at the primary site. Unlike metastatic cancer, localized tumors are often cured by means of surgery. Disseminated metastases cause neurological compromise, respiratory failure, thrombosis, and other potentially lethal complications. Metastasis requires that cells navigate a sequence of biological challenges, including escape from the primary tumor, survival in the circulation, colonization of distant organs, and growth into tumors at these remote sites. Many factors contribute to the metastatic capabilities of cancer cells (60–63). Metastasis also imposes metabolic requirements distinct from those that support cell growth, and inhibiting these activities reduces metastatic spread (Fig. 3).
Fig. 3. Tumor metabolism supports multiple steps of the metastatic cascade.

Bottlenecks occur at several steps of metastasis, and metabolic reprogramming supports successful navigation of some of these barriers. Extracellular acidification promotes intravasation of cells from the primary tumor. A major bottleneck occurs after cells enter the circulation, when survival requires mechanisms to produce NADPH and glutathione (GSH) to counteract oxidative stress. Successful seeding of distant organs and survival during dormancy may require harmony between the new microenvironment and the needs of cancer cells in micrometastatic lesions. Last, anabolic metabolism is reactivated during macrometastatic tumor growth.
The metastatic cascade begins with escape of potentially metastatic cells from the primary tumor. Intravasation into the blood or lymph involves degradation of extracellular matrix (ECM), migration away from the primary environment, and avoidance of immune surveillance. Metabolic factors are thought to contribute to these processes either by allowing cancer cells to acquire cell-autonomous properties associated with enhanced invasiveness or by altering the microenvironment. A convergent metabolic phenotype is the release of CO2, lactate, and other organic acids from metabolically active cancer cells; acidification of the extracellular space; and promotion of degradation of the ECM (Fig. 3). This involves decreasing the abundance of adherens junctions on cancer cells, allowing them to detach from adjacent cells and stimulating proteolytic enzymes that degrade the ECM (64).
Other metabolic adaptations promote the epithelial-mesenchymal transition (EMT), a multifactorial cell state involving loss of adhesion and enhanced migratory capabilities. Oncogene-dependent activation of uridine 5′-diphosphate (UDP)–glucose 6-dehydrogenase (UGDH) depletes UDP-glucose, resulting in enhanced expression of SNAIL–a transcription factor that promotes mesenchymal properties–and increased migration and metastasis in mice (65). Expression of asparagine synthetase (Asns), which converts aspartate into the conditionally essential amino acid asparagine, promotes breast cancer cell invasiveness and metastasis by supporting EMT because EMT-associated proteins have disproportionately high asparagine content. Silencing Asns or systemically depleting asparagine reduces metastasis to the lung in these models (66).
Metabolic stress in the microenvironment of the primary tumor can also influence metastasis. Hypoxic regions within tumors portend an increased risk of metastasis, and transcriptional programs downstream of HIF-1 and HIF-2 allow hypoxic breast cancer cells to intravasate and metastasize (67, 68). Rapid nutrient consumption by cancer cells is thought to deplete fuels such as glucose and glutamine for immune cells, resulting in a suboptimal environment for immune surveillance and possibly increasing the chance that invasive cancer cells will survive [reviewed in (69)].
Even small tumors shed cancer cells into the circulation, but only a small fraction of these cells (0.01%) are capable of forming metastatic lesions (70–73). This implies that major bottlenecks in metastasis occur after escape from the primary tumor. Antioxidant responses after loss of anchorage contribute to metastatic capabilities (Fig. 3). In cultured mammary epithelial cells, matrix detachment produces oxidative stress and results in cell death unless the stress can be mitigated by production of reduced nicotinamide adenine dinucleotide phosphate (NADPH) in the pentose phosphate pathway. This pathway provides reducing equivalents to regenerate ROS-detoxifying metabolites, such as glutathione (74). A redox shuttle enabling the transfer of NADPH from the cytosol into the mitochondria is required for maximal cancer cell growth after the loss of monolayer attachment in culture (75). In vivo, the oxidizing environment of the bloodstream makes antioxidant defense a major factor in metastatic efficiency. Suppressing oxidative stress with systemic antioxidants or cell-intrinsic activation of antioxidant pathways promotes metastasis in patient-derived melanoma xenografts (76) and in genetically modified mouse models of melanoma (77), breast cancer (78), and lung cancer (79–81). Not all tumor cells are susceptible to oxidative stress, however, and ROS may promote metastasis in some models (82–84). The exact role of ROS may depend on the stage and type of cancer (85).
Colonization of distant organs is another bottleneck in metastasis (Fig. 3) (70, 86–89). Colonization includes cell survival in dormant micrometastatic lesions and eventually reactivation of growth, leading to clinically apparent macrometastases (90, 91). The propensity of an organ to foster metastatic lesions is variable, with the liver, lung, brain, and bone being important metastatic sites in many cancers (92). Some primary cancers tend to metastasize to particular organs, a relationship called organotropism. Accessibility of the distant organ from the primary site by means of lymphatics and blood flow helps dictate which environments are most amenable to metastasis, but harmony between the metabolic needs of the cancer cell and the milieu of the organ also contributes. Abundant pyruvate in lung interstitial fluid in mice promotes breast cancer cell α-ketoglutarate synthesis, which stimulates collagen cross-linking by the α-ketoglutarate-dependent enzyme prolyl-4-hydroxylase (93). Excessive collagen cross-linking in the ECM improves the lung’s ability to support breast cancer metastasis. Metastatic ovarian cancer cells prefer fatty acids over other fuels, perhaps explaining why they frequently metastasize to the lipid-rich omental fat pad. In mice, preventing transfer of fatty acids from neighboring adipocytes to ovarian carcinoma cells reduces metastatic growth (94). Fatty acid oxidation also supports colonization of lymph nodes by some cancer cells (95). In these models, bioactive bile acids within the lymph node stimulate fatty acid oxidation through Yap (yes-associated protein)-mediated transcriptional reprogramming, and inhibiting either Yap or fatty acid oxidation reduces lymph node metastasis. Similarly, triple-negative breast cancers rely on fatty acid oxidation to maintain aberrant Src activity, which promotes metastasis (96).
Metabolic heterogeneity among cancer cells within a primary tumor can regulate both the overall metastatic efficiency and organotropism. A subpopulation of cells from human oral carcinomas expresses the lipid transporter CD36, which imports fatty acids for oxidation. CD36 expression is both necessary and sufficient for these cells to give rise to lymph node metastases at high efficiency (97). In a mouse mammary cancer model, cancer cells display heterogeneous metabolic properties that influence the site of metastasis, with metastases to the liver requiring the Hif-1 target pyruvate dehydrogenase kinase-1 (Pdk1), which promotes adaptation to hypoxia (98). In human melanoma, brain metastases are enriched for gene sets related to OXPHOS, and inhibiting OXPHOS in mouse models reduces metastases to the brain but not the lung (99). These findings indicate that specific metabolic adaptations promote organotropism in melanoma. Also in melanoma, expression of monocarboxylate transporter-1 (MCT1) defines a subpopulation of cells with high metastatic efficiency. In patient-derived xenografts and syngeneic models, MCT1-dependent lactate transport allows melanoma cells to maintain an intracellular pH and redox ratio that support the pentose phosphate pathway and mitigate oxidative stress (100).
The dormancy period between arrival of metastatic cells in a distant organ and appearance of macrometastasis varies widely. Glioblastoma metastases to the lung were only observed when the lungs of a deceased donor were transplanted into an immunosuppressed patient, suggesting an indefinite period of dormancy in immunocompetent hosts (101). How cancer cells survive during prolonged dormancy is incompletely characterized. When in vivo selection was used to identify latency-competent cancer cells, these tumors exhibited a quiescent-like state reminiscent of stem and progenitor cells (102). In a mouse pancreatic cancer model, ablating oncogenic Kras from established tumors revealed a fraction of cells that survive and eventually propagate. These dormant cells display enhanced OXPHOS relative to KRas-expressing cells, and inhibiting OXPHOS reduced tumor recurrence (103). Although this model did not assess metastasis, it does suggest a shift in metabolic dependencies during dormancy. Once dormant cells begin to proliferate, anabolic pathways involving biomass assimilation are presumably activated to support progression to macrometastasis (Fig. 3). It will be interesting to determine whether the growth-promoting metabolic network in macrometastases differs from the primary tumor.
Targeting metabolic liabilities
Tumors were classically thought to contain generalizable, hardwired metabolic vulnerabilities. But so far, this idea has not produced many metabolic therapies with broad and predictable efficacy in human cancer. The insights reviewed above indicate that cancer cell metabolism is flexible and heterogeneous, is responsive to cues that evolve during cancer progression, and thwarts efforts to target fixed liabilities. How then should we prioritize potential metabolic liabilities for further study and the development of new therapies?
A rare but crucial opportunity for intervention involves genetically defined metabolic alterations that contribute mechanistically to transformation. The most straightforward examples are mutations in metabolic enzymes that permanently alter cellular metabolism and promote the hallmarks of malignancy. Currently, IDH1 and IDH2 mutations in AML best illustrate this paradigm. Truncal mutations in either of these genes produce a cell-autonomous and persistent addiction to the resulting metabolic alteration (accumulation of D-2HG). These are appealing features for therapeutic targeting. Clinical efficacy of mutant IDH1 and −2 inhibitors has been demonstrated in AML patients, with clinical trials in solid tumors now under way. In addition to its epigenetic effects, D-2HG inhibits enzymes from the canonical metabolic network (104). Such enzymes include transaminases that produce glutamate for glutathione biosynthesis, rendering IDH-mutant gliomas susceptible to treatments that deplete glutamate and enhance oxidative stress (105). This may provide therapeutic opportunities in IDH1- and IDH2-mutant tumors beyond inhibiting mutant IDH.
FH and SDH mutations also create truncal metabolic aberrations. These mutations induce permanent restructuring of the TCA cycle and result in numerous metabolic liabilities in preclinical models (106–108). They also bring about nonintuitive liabilities that may provide opportunities for clinical intervention. In addition to their roles in the TCA cycle, high levels of succinate and fumarate in SDH- and FH-deficient cancers impair homologous recombination DNA repair (109). This repair pathway requires the α-ketoglutarate-dependent histone demethylases KDM4A and KDM4B. Like D-2HG, succinate and fumarate, at high concentrations, inhibit α-ketoglutarate-dependent enzymes, including KDM4A and KDM4B. Ineffective DNA repair in cancer cells that lack FH and SDH renders them sensitive to drugs that block DNA repair.
New clinical opportunities may also arise from studying the role of metabolic reprogramming in the therapy-resistant state. Acquired therapy resistance is a major factor leading to cancer-associated mortality, and metabolic alterations have the potential to contribute to therapy resistance. One example involves drug-tolerant persister cells that survive cytotoxic therapy through reversible, nonmutational mechanisms. Across a variety of cytotoxic treatments and cancer types, this persister state confers dependence on glutathione peroxidase 4 (GPX4), a lipid hydroperoxidase. Inhibiting GPX4 induces lipid peroxidation and death in persister cells and reduces the reemergence of tumors resistant to cytotoxic therapy (110).
In some cases, metabolic interactions between cancer cells and the microenvironment promote therapy resistance. Metabolic properties of non-small-cell lung cancers can reprogram stromal cells to induce resistance to EGFR inhibitors (111). Lactate export by the cancer cells induces neighboring fibroblasts to secrete hepatocyte growth factor, which activates its receptor tyrosine kinase c-MET on cancer cells. Consequently, the cancer cells sustain oncogenic signaling even in the presence of EGFR inhibitors (111). PDAC cells also co-opt the microenvironment to promote drug resistance. PDAC-induced macrophage polarization causes the macrophages to release deoxycytidine, which competitively inhibits the chemotherapeutic agent gemcitabine and lowers its therapeutic efficacy (112).
Some drugs induce systemic metabolic effects that complicate therapeutic responses. PI3K inhibitors suppress glucose uptake by the muscle and other tissues, resulting in elevated blood glucose and pancreatic insulin release after administration of the drug. The insulin surge reactivates PI3K signaling in the tumor, limiting the efficacy of PI3K inhibitors on tumor growth. Placing mice on a low-carbohydrate, ketogenic diet blunts the hyperglycemia and insulin release that accompany PI3K inhibition, and this improves the efficacy of PI3K inhibitors on tumor growth (113).
A recurrent theme in studies on cancer progression is the increased reliance of cancer cells on OXPHOS in advanced stages of disease. As discussed above, mouse PDAC cells subjected to prolonged withdrawal of oncogenic Kras in vivo require OXPHOS to emerge from dormancy (103). In chronic lymphocytic leukemia, acquired resistance to venetoclax–an inhibitor of the antiapoptotic protein B cell lymphoma-2 (BCL-2)–involves increased mitochondrial mass and enhanced OXPHOS (114). Inhibiting OXPHOS increases venetoclax sensitivity in culture and in vivo. OXPHOS also promotes resistance to the antimetabolite cytarabine in mouse models of AML (115). Cells persisting after cytarabine treatment and preexisting cells with innate cytarabine resistance both displayed enhanced OXPHOS. Cytarabine treatment in AML-bearing mice spares cells with high levels of OXPHOS, and cytarabine-resistant cells are susceptible to OXPHOS inhibitors (115). It is unclear why cells acquire enhanced dependence on OXPHOS during cancer progression and whether the underlying mechanism is the same in all contexts. Nevertheless, this form of metabolic reprogramming deserves further study because its appearance in diverse preclinical models implies a degree of generalizability and because an OXPHOS inhibitor with potent efficacy in mouse models is now in phase 1 clinical trials (116).
Is the therapeutic window for OXPHOS inhibitors wide enough, given that this pathway is so important in many other tissues? This is an open question. Recently described OXPHOS inhibitors display enhanced toxicity against cultured cancer cells compared with either nontransformed cells or other cancer cells with metabolic properties that allow them to compensate for OXPHOS impairment (116, 117). It is encouraging that these agents also suppress growth of susceptible tumors in mice, using doses that are well-tolerated over the short term. We currently do not know whether dosing can be sustained in large animals and humans so as to induce durable therapeutic responses without dose-limiting toxicities in the heart, muscle, brain, and other oxidative organs.
Similar issues arise with other reprogrammed pathways, most of which are not confined to tumors. Glutamine addiction is a common feature of cultured cancer cells, and early attempts to target this pathway in vivo used the glutamine analog 6-diazo-5-oxo-l-norleucine (DON). By inhibiting a myriad of enzymes that use glutamine as a substrate, DON kills cancer cells but is unacceptably toxic to the gastrointestinal tract and other tissues, limiting its clinical development. But a recent DON analog, JHU083, is a prodrug that is activated in the tumor microenvironment, improving the therapeutic window (118). In syngeneic mouse models, this drug markedly impairs glutamine consumption by cancer cells, suppressing cancer cell growth and increasing the availability of glutamine and other nutrients for T cells in the tumor microenvironment. This produces robust antitumor immunity that results in tumor regression.
A related challenge in advancing promising metabolic therapies into clinical practice is to identify the patients most likely to benefit from the drug. Metabolic heterogeneity among tumors makes this difficult, even if specific, potent, and well-tolerated inhibitors are available. Biomarkers better at predicting therapeutic responses are sorely needed, and advances in metabolic phenotyping of intact tumors suggest several paths forward (119). First, 2-HG accumulation in IDH1- and IDH2-mutant tumors is a rare example in which the metabolite directly reports the oncogenic driver and potential therapy (Fig. 4). Proton magnetic resonance spectroscopy can noninvasively track 2-HG abundance in IDH-mutant gliomas, predicting both disease progression and response to therapy through longitudinal 2-HG measurements (120). Tumors with detectable 2-HG at diagnosis could be stratified to receive inhibitors of mutant IDH in clinical trials.
Fig. 4. Prospects for using in vivo analysis to match tumor metabolism with metabolic therapies.

Several new approaches to assess metabolism in intact tumors, particularly with new imaging approaches, have been used in humans and experimental models to report subtype-selective metabolic properties, some of which correlate with therapeutic sensitivities. MRS, magnetic resonance spectroscopy; D-2HG, D-2-hydroxyglutarate; mIDH, mutant isocitrate dehydrogenase; MCT1, monocarboxylate transporter-1; 18F-BnTP, 4-[18F]fluorobenzy7l-triphenylphosphonium; Gln, glutamine; PET, positron emission tomography; GLS, glutaminase.
However, most metabolic therapies lack biomarkers, and conventional molecular analyses such as DNA and RNA sequencing usually prove to be inadequate surrogates for metabolism. But metabolic tracers can assess tumor metabolism in situ (Fig. 4). 13C tracers allow the fates of nutrients–for example, glucose and lactate–to be tracked, reporting metabolic pathways in intact tumors. Intravenous infusion of 13C-lactate into human non-small-cell lung cancer patients and mice bearing patient-derived melanomas identified tumors with a propensity for future metastasis, which required MCT1-dependent lactate transport in the mice (58, 100). Tracers labeled with 13C can also be imaged by using hyperpolarized magnetic resonance imaging (MRI), allowing for the noninvasive observation of activities such as the transfer of hyperpolarized 13C between pyruvate and lactate (121). In human prostate cancer, tumor grade correlates with MCT1 expression and with imaging of 13C-lactate after injection of hyperpolarized 13C-pyruvate (122). MCT1 inhibitors are in clinical development, and these studies suggest ways to identify patients who would benefit from them.
Last, new PET probes report aspects of tumor metabolism that are relevant to experimental therapeutics. Although 18FDG has been widely used in clinical oncology for 40 years, it has not been deployed to predict responses to specific metabolic therapies. A new probe, 4-[18F]fluorobenzyl-triphenylphosphonium (18F-BnTP), accumulates in tumors in proportion to their reliance on OXPHOS (123). In mouse models of lung cancer, 18F-BnTP imaging predicts sensitivity to OXPHOS inhibition, regardless of whether or not the tumors take up 18FDG. PET tracers detecting glutamine uptake have recently been assessed in human cancer, and this is relevant because of ongoing clinical trials for glutaminase inhibitors (124). In tumors that use glutaminase to convert glutamine to glutamate, glutaminase inhibition should deplete glutamate and increase the glutamine pool. In mouse models of breast cancer, both of these alterations were detected by using PET and other modalities after an acute period of glutaminase inhibition (125, 126).
Concluding remarks
Recent work in cancer metabolism has focused on assessing metabolic phenotypes in native microenvironments in humans and mice. This has produced a greater appreciation for metabolic heterogeneity among tumors, expanding the scope of metabolic dependencies beyond the classical pathways that dominate metabolism in culture. Evidence also indicates that metabolic phenotypes evolve as cancer progresses, with new dependencies emerging in the context of therapy resistance and metastasis. Future research should further explore these emerging vulnerabilities and devise ways to target them for therapy. This will require the use of experimental models that allow investigators to isolate and manipulate crucial cellular subsets such as dormant persister cells and cells at key points in the metastatic cascade. We also anticipate further advancements in methods to assess and quantify metabolic phenotypes in human cancers in vivo, including metabolomics, isotope tracing studies, and metabolic imaging. These efforts could ultimately allow clinical oncologists to tailor therapeutic strategies by matching the treatment with patient-specific tumor metabolism.
ACKNOWLEDGMENTS
We regret the inability to cite many excellent studies that have shaped our understanding of cancer metabolism. We thank members of the DeBerardinis laboratory for critically reading the manuscript and K. Regan for help with the figures.
Funding: R.J.D. is supported by the Howard Hughes Medical Institute, the National Cancer Institute, the Cancer Prevention and Research Institute of Texas, and the Moody Foundation. B.F. is a recipient of a postdoctoral fellowship from the Canadian Institutes of Health Research (MFE140911). A.S. is a Ruth L. Kirschstein Postdoctoral Fellow funded by the National Institute of Child Health and Human Development (F32HD096786).
Footnotes
Competing Interests: R.J.D. is an advisor for Agios Pharmaceuticals.
REFERENCES AND NOTES
- 1.Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). doi: 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim J, DeBerardinis RJ, Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 30, 434–446 (2019). doi: 10.1016/j.cmet.2019.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoadley KA, et al. , Yau C et al. , Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018). doi: 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu J et al. , Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat. Biotechnol. 31, 522–529 (2013). doi: 10.1038/nbt.2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuneva MO et al. , The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012). doi: 10.1016/j.cmet.2011.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine AJ, Jenkins NA, Copeland NG, The roles of initiating truncal mutations in human cancers: The order of mutations and tumor cell type matters. Cancer Cell 35, 10–15 (2019). doi: 10.1016/j.ccell.2018.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malkin D et al. , Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238 (1990). doi: 10.1126/science.1978757 [DOI] [PubMed] [Google Scholar]
- 8.Baysal BE et al. , Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000). doi: 10.1126/science.287.5454.848 [DOI] [PubMed] [Google Scholar]
- 9.Benn DE et al. , Novel succinate dehydrogenase subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene 22, 1358–1364 (2003). doi: 10.1038/sj.onc.1206300 [DOI] [PubMed] [Google Scholar]
- 10.Hao HX et al. , SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 (2009). doi: 10.1126/science.1175689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomlinson IP et al. , Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002). doi: 10.1038/ng849 [DOI] [PubMed] [Google Scholar]
- 12.Erez A, DeBerardinis RJ, Metabolic dysregulation in monogenic disorders and cancer - finding method in madness. Nat. Rev. Cancer 15, 440–448 (2015). doi: 10.1038/nrc3949 [DOI] [PubMed] [Google Scholar]
- 13.Xu J et al. , Abnormal oxidative metabolism in a quiet genomic background underlies clear cell papillary renal cell carcinoma. eLife 8, e38986 (2019). doi: 10.7554/eLife.38986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selak MA et al. , Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7, 77–85 (2005). doi: 10.1016/j.ccr.2004.11.022 [DOI] [PubMed] [Google Scholar]
- 15.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ, Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 348, 1625–1638 (2003). doi: 10.1056/NEJMoa021423 [DOI] [PubMed] [Google Scholar]
- 16.Lauby-Secretan B et al. , Body fatness and cancer-Viewpoint of the IARC Working Group. N. Engl. J. Med. 375, 794–798 (2016). doi: 10.1056/NEJMsr1606602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CM et al. , High glucose triggers nucleotide imbalance through O-GlcNacylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab. 29, 1334–1349.e10 (2019). doi: 10.1016/j.cmet.2019.02.005 [DOI] [PubMed] [Google Scholar]
- 18.Labbe DP et al. , High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 10, 4358 (2019). doi: 10.1038/s41467-019-12298-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liou GY et al. , Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336 (2016). doi: 10.1016/j.celrep.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrer A et al. , Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 9, 416–435 (2019). doi: 10.1158/2159-8290.CD-18-0567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backshall A et al. , Detection of metabolic alterations in non-tumor gastrointestinal tissue of the Apc(Min/+) mouse by (1) H MAS NMR spectroscopy. J. Proteome Res. 8, 1423–1430 (2009). doi: 10.1021/pr800793w [DOI] [PubMed] [Google Scholar]
- 22.Phillips CM et al. , Upregulation of cystathionine-β-synthase in colonic epithelia reprograms metabolism and promotes carcinogenesis. Cancer Res. 77, 5741–5754 (2017). doi: 10.1158/0008-5472.CAN-16-3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu S et al. , 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab. 14, 131–142 (2011). doi: 10.1016/j.cmet.2011.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scafoglio CR et al. , Sodium-glucose transporter 2 is a diagnostic and therapeutic target for early-stage lung adenocarcinoma. Sci. Transl. Med. 10, eaat5933 (2018). doi: 10.1126/scitranslmed.aat5933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figueroa ME et al. , Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010). doi: 10.1016/j.ccr.2010.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turcan S et al. , IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). doi: 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stein EM et al. , Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 130, 722–731 (2017). doi: 10.1182/blood-2017-04-779405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu Z et al. , Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov. 9, 1228–1247 (2019). doi: 10.1158/2159-8290.CD-19-0152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ying H et al. , Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670 (2012). doi: 10.1016/j.cell.2012.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bensaad K et al. , TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 (2006). doi: 10.1016/j.cell.2006.05.036 [DOI] [PubMed] [Google Scholar]
- 31.Shim H et al. , c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U.S.A. 94, 6658–6663 (1997). doi: 10.1073/pnas.94.13.6658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeBerardinis RJ, Chandel NS, Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 (2016). doi: 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlova NN, Thompson CB, The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016). doi: 10.1016/j.cmet.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goncalves MD, Hopkins BD, Cantley LC, Phosphatidylinositol 3-kinase, growth disorders, and cancer. N. Engl. J. Med. 379, 2052–2062 (2018). doi: 10.1056/NEJMra1704560 [DOI] [PubMed] [Google Scholar]
- 35.Saxton RA, Sabatini DM, mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017). doi: 10.1016/j.cell.2017.03.035 [DOI] [PubMed] [Google Scholar]
- 36.Valvezan AJ et al. , mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell 32, 624–638.e5 (2017). doi: 10.1016/j.ccell.2017.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yun J et al. , Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009). doi: 10.1126/science.1174229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen PH et al. , Metabolic diversity in human non-small cell lung cancer cells. Mol. Cell 76, 838–851.e5 (2019). doi: 10.1016/j.molcel.2019.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H et al. , The landscape of cancer cell line metabolism. Nat. Med. 25, 850–860 (2019). doi: 10.1038/s41591-019-0404-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calles A et al. , Immunohistochemical loss of LKB1 is a biomarker for more aggressive biology in KRAS-mutant lung adenocarcinoma. Clin. Cancer Res. 21, 2851–2860 (2015). doi: 10.1158/1078-0432.CCR-14-3112 [DOI] [PubMed] [Google Scholar]
- 41.Ji H et al. , LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810 (2007). doi: 10.1038/nature06030 [DOI] [PubMed] [Google Scholar]
- 42.Chen Z et al. , A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–617 (2012). doi: 10.1038/nature10937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim J et al. , CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172 (2017). doi: 10.1038/nature22359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shackelford DB et al. , LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 (2013). doi: 10.1016/j.ccr.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y et al. , Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 3, 870–879 (2013). doi: 10.1158/2159-8290.CD-13-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skoulidis F et al. , Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 5, 860–877 (2015). doi: 10.1158/2159-8290.CD-14-1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galan-Cobo A et al. , LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 79, 3251–3267 (2019). doi: 10.1158/0008-5472.CAN-18-3527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kottakis F et al. , LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539, 390–395 (2016). doi: 10.1038/nature20132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamphorst JJ et al. , Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015). doi: 10.1158/0008-5472.CAN-14-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao P et al. , c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009). doi: 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Commisso C et al. , Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). doi: 10.1038/nature12138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim SM et al. , PTEN deficiency and AMPK activation promote nutrient scavenging and anabolism in prostate cancer cells. Cancer Discov. 8, 866–883 (2018). doi: 10.1158/2159-8290.CD-17-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wyant GA et al. , NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758 (2018). doi: 10.1126/science.aar2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karsli-Uzunbas G et al. , Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014). doi: 10.1158/2159-8290.CD-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sonveaux P et al. , Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest 118, 3930–3942 (2008). doi: 10.1172/JCI36843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sousa CM et al. , Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483 (2016). doi: 10.1038/nature19084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerlinger M et al. , Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 (2012). doi: 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Faubert B et al. , Lactate metabolism in human lung tumors. Cell 171, 358–371.e9 (2017). doi: 10.1016/j.cell.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hensley CT et al. , Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). doi: 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smid M et al. , Subtypes of breast cancer show preferential site of relapse. Cancer Res. 68, 3108–3114 (2008). doi: 10.1158/0008-5472.CAN-07-5644 [DOI] [PubMed] [Google Scholar]
- 61.Wan L, Pantel K, Kang Y, Tumor metastasis: Moving new biological insights into the clinic. Nat. Med. 19, 1450–1464 (2013). doi: 10.1038/nm.3391 [DOI] [PubMed] [Google Scholar]
- 62.Disibio G, French SW, Metastatic patterns of cancers: Results from a large autopsy study. Arch. Pathol. Lab. Med. 132, 931–939 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Chen H, Shah AS, Girgis RE, Grossman SA, Transmission of glioblastoma multiforme after bilateral lung transplantation. J. Clin. Oncol. 26, 3284–3285 (2008). doi: 10.1200/JCO.2008.16.3543 [DOI] [PubMed] [Google Scholar]
- 64.Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK, Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 8, 1284–1291 (2002). [PubMed] [Google Scholar]
- 65.Wang X et al. , UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 571, 127–131 (2019). doi: 10.1038/s41586-019-1340-y [DOI] [PubMed] [Google Scholar]
- 66.Knott SRV et al. , Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381 (2018). doi: 10.1038/nature25465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Semenza GL, HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Invest. 123, 3664–3671 (2013). doi: 10.1172/JCI67230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H et al. , HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012). doi: 10.1038/onc.2011.365 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Kishton RJ, Sukumar M, Restifo NP, Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109 (2017). doi: 10.1016/j.cmet.2017.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luzzi KJ et al. , Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 153, 865–873 (1998). doi: 10.1016/S0002-9440(10)65628-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baccelli I et al. , Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 31, 539–544 (2013). doi: 10.1038/nbt.2576 [DOI] [PubMed] [Google Scholar]
- 72.Nagrath S et al. , Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007). doi: 10.1038/nature06385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stott SL et al. , Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2, 25ra23 (2010). doi: 10.1126/scitranslmed.3000403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schafer ZT et al. , Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009). doi: 10.1038/nature08268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jiang L et al. , Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016). doi: 10.1038/nature17393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Piskounova E et al. , Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). doi: 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Le Gal K et al. , Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 7, 308re8 (2015). doi: 10.1126/scitranslmed.aad3740 [DOI] [PubMed] [Google Scholar]
- 78.LeBleu VS et al. , PGC-1a mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003, 1–15 (2014). doi: 10.1038/ncb3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiel C et al. , BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 178, 330–345.e22 (2019). doi: 10.1016/j.cell.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 80.Sayin VI et al. , Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6, 221ra15 (2014). doi: 10.1126/scitranslmed.3007653 [DOI] [PubMed] [Google Scholar]
- 81.Lignitto L et al. , Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178, 316–329.e18 (2019). doi: 10.1016/j.cell.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishikawa K et al. , ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664 (2008). doi: 10.1126/science.1156906 [DOI] [PubMed] [Google Scholar]
- 83.Ferraro D et al. , Pro-metastatic signaling by c-Met through RAC-1 and reactive oxygen species (ROS). Oncogene 25, 3689–3698 (2006). doi: 10.1038/sj.onc.1209409 [DOI] [PubMed] [Google Scholar]
- 84.Porporato PE et al. , A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 (2014). doi: 10.1016/j.celrep.2014.06.043 [DOI] [PubMed] [Google Scholar]
- 85.Reczek CR, Chandel NS, The two faces of reactive oxygen species in cancer. Ann. Rev. Cancer Biol. 1, 79–98 (2017). doi: 10.1146/annurev-cancerbio-041916-065808 [DOI] [Google Scholar]
- 86.Wong CW et al. , Apoptosis: An early event in metastatic inefficiency. Cancer Res. 61, 333–338 (2001). [PubMed] [Google Scholar]
- 87.Cameron MD et al. , Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 60, 2541–2546 (2000). [PubMed] [Google Scholar]
- 88.Varghese HJ et al. , Activated ras regulates the proliferation/apoptosis balance and early survival of developing micrometastases. Cancer Res. 62, 887–891 (2002). [PubMed] [Google Scholar]
- 89.Naumov GN et al. , Persistence of solitary mammary carcinoma cells in a secondary site: A possible contributor to dormancy. Cancer Res. 62, 2162–2168 (2002). [PubMed] [Google Scholar]
- 90.Vanharanta S, Massague J, Origins of metastatic traits. Cancer Cell 24, 410–421 (2013). doi: 10.1016/j.ccr.2013.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Massague J, Obenauf AC, Metastatic colonization by circulating tumour cells. Nature 529, 298–306 (2016). doi: 10.1038/nature17038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Budczies J et al. , The landscape of metastatic progression patterns across major human cancers. Oncotarget 6, 570–583 (2015). doi: 10.18632/oncotarget.2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Elia I et al. , Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019). doi: 10.1038/s41586-019-0977-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nieman KM et al. , Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011). doi: 10.1038/nm.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee CK et al. , Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science 363, 644–649 (2019). doi: 10.1126/science.aav0173 [DOI] [PubMed] [Google Scholar]
- 96.Park JH et al. , Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 14, 2154–2165 (2016). doi: 10.1016/j.celrep.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pascual G et al. , Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017). doi: 10.1038/nature20791 [DOI] [PubMed] [Google Scholar]
- 98.Dupuy F et al. , PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589 (2015). doi: 10.1016/j.cmet.2015.08.007 [DOI] [PubMed] [Google Scholar]
- 99.Fischer GM et al. , Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov. 9, 628–645 (2019). doi: 10.1158/2159-8290.CD-18-1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tasdogan A et al. , Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 (2020). doi: 10.1038/s41586-019-1847-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Armanios MY et al. , Transmission of glioblastoma multiforme following bilateral lung transplantation from an affected donor: Case study and review of the literature. Neuro-oncol. 6, 259–263 (2004). doi: 10.1215/S1152851703000474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malladi S et al. , Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell 165, 45–60 (2016). doi: 10.1016/j.cell.2016.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Viale A et al. , Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). doi: 10.1038/nature13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lu C et al. , IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012). doi: 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McBrayer SK et al. , Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116.e25 (2018). doi: 10.1016/j.cell.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frezza C et al. , Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 477, 225–228 (2011). doi: 10.1038/nature10363 [DOI] [PubMed] [Google Scholar]
- 107.Lussey-Lepoutre C et al. , Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 6, 8784 (2015). doi: 10.1038/ncomms9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adam J et al. , A role for cytosolic fumarate hydratase in urea cycle metabolism and renal neoplasia. Cell Rep. 3, 1440–1448 (2013). doi: 10.1016/j.celrep.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sulkowski PL et al. , Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 50, 1086–1092 (2018). doi: 10.1038/s41588-018-0170-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hangauer MJ et al. , Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). doi: 10.1038/nature24297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Apicella M et al. , Increased lactate secretion by cancer cells sustains non-cell-autonomous adaptive resistance to MET and EGFR targeted therapies. Cell Metab. 28, 848–865.e6 (2018). doi: 10.1016/j.cmet.2018.08.006 [DOI] [PubMed] [Google Scholar]
- 112.Halbrook CJ et al. , Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 29, 1390–1399.e6 (2019). doi: 10.1016/j.cmet.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hopkins BD et al. , Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503 (2018). doi: 10.1038/s41586-018-0343-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guièze R et al. , Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid malignancies. Cancer Cell 36, 369–384.e13 (2019). doi: 10.1016/j.ccell.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Farge T et al. , Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 7, 716–735 (2017). doi: 10.1158/2159-8290.CD-16-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Molina JR et al. , An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 24, 1036–1046 (2018). doi: 10.1038/s41591-018-0052-4 [DOI] [PubMed] [Google Scholar]
- 117.Shi Y et al. , Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346 (2019). doi: 10.1038/s41586-019-0993-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leone RD et al. , Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021 (2019). doi: 10.1126/science.aav2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Faubert B, DeBerardinis RJ, Analyzing tumor metabolism in vivo. Ann. Rev. Cancer Biol. 1, 99–117 (2016). doi: 10.1146/annurev-cancerbio-050216-121954 [DOI] [Google Scholar]
- 120.Choi C et al. , Prospective longitudinal analysis of 2-hydroxyglutarate magnetic resonance spectroscopy identifies broad clinical utility for the management of patients with IDH-mutant glioma. J. Clin. Oncol. 34, 4030–4039 (2016). doi: 10.1200/JCO.2016.67.1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nelson SJ et al. , Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 5, 198ra108 (2013). doi: 10.1126/scitranslmed.3006070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Granlund KL et al. , Hyperpolarized MRI of human prostate cancer reveals increased lactate with tumor grade driven by monocarboxylate transporter 1. Cell Metab. 31, 105–114.e3 (2020). doi: 10.1016/j.cmet.2019.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Momcilovic M et al. , In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 575, 380–384 (2019). doi: 10.1038/s41586-019-1715-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Venneti S et al. , Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 7, 274ra17 (2015). doi: 10.1126/scitranslmed.aaa1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou R et al. , [18F](2S,4R)4-Fluoroglutamine PET Detects Glutamine Pool Size Changes in Triple-Negative Breast Cancer in Response to Glutaminase Inhibition. Cancer Res. 77, 1476–1484 (2017). doi: 10.1158/0008-5472.CAN-16-1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou R et al. , Glutamate-weighted chemical exchange saturation transfer magnetic resonance imaging detects glutaminase inhibition in a mouse model of triple-negative breast cancer. Cancer Res. 78, 5521–5526 (2018). doi: 10.1158/0008-5472.CAN-17-3988 [DOI] [PMC free article] [PubMed] [Google Scholar]