

Abstract
Competitive antagonists for ionotropic glutamate receptors (iGluRs) are highly valuable tool compounds for studying health and disease states in the central nervous system. However, only few subtype selective tool compounds are available and the discovery of antagonists with novel iGluR subtype selectivity profiles remains a profound challenge. In this paper, we report an elaborate structure-activity-relationship (SAR) study of the parental scaffold 2,3-trans-3-carboxy-3-phenylproline by the synthesis of 40 new analogs. Three synthetic strategies were employed with two new strategies of which one being a highly efficient and fully enantioselective strategy based on C(sp3)-H activation methodology. The SAR study led to the conclusion that selectivity for the NMDA receptors was a general trend when adding substituents in the 5’ position. Selective NMDA receptor antagonists were obtained with high potency (IC50 values as low as 200 nM) and 3–34 fold preference for GluN1/GluN2A over GluN1/GluN2B-D NMDA receptors.
Keywords: Ionotropic glutamate receptors, NMDA receptor antagonist, Proline analogs, C(sp3)-H activation, Electrophysiology
Graphical Abstract

Introduction
(S)-Glutamate (Glu) belongs to the pool of common amino acids, but is the major excitatory neurotransmitter in the mammalian central nervous system (CNS).1 Upon axon firing, Glu is released into the synaptic cleft where it binds to pre- and postsynaptic Glu receptors. These are divided into two main classes: the metabotropic Glu receptors (mGluRs) that produce a slower signal transduction through second messenger systems, and ionotropic Glu receptors (iGluRs) that are responsible for fast signal transmission. Based on ligand selectivity studies, iGluRs have been divided into the three groups, named the α-amino-3-hydroxy-5-methyl-4-isoxazolepropinic acid (AMPA) receptors (subunits GluA1–4), the kainic acid (KA) receptors (subunits GluK1–5) and N-methyl-D-aspartate (NMDA) receptors (subunits GluN1, GluN2A-D, and GluN3A,B).2 Imbalance of Glu signaling has been correlated to a number of neurological and/or psychiatric diseases like anxiety,3 depression,4,5 migraine,6,7 pain,8 and schizophrenia9–11. Furthermore, it has been shown that elevated Glu signaling is neurotoxic and ultimately leads to neuronal death.12–14 Thus, iGluRs are also thought to be involved in the disease mechanism of neurodegenerative diseases such as Alzheimer’s disease,15,16 Huntington’s disease,17 amyotrophic lateral sclerosis (ALS),18 cerebral stroke,19 and epilepsy.8,20
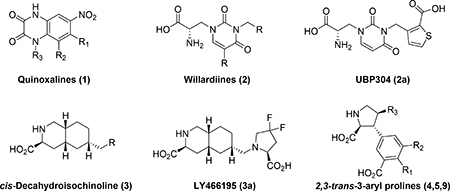
The iGluRs have been of long interest in academic and industrial research with the aim to develop fully subtype selective antagonists, as such pharmacological tools are attractive for studying and understanding of the role and function of these receptors in the healthy and diseased brain. Different approaches have been taken from design and synthesis of non-competitive channel blockers,21,22 negative allosteric modulators23 to orthosteric antagonists. With focus here on the latter class, amino acid bioisosters such as quinoxalinedione (1) and its congeners24–27 have been investigated intensely, but most importantly structurally modified amino acid analogs. Representative selective iGluR antagonist scaffolds are willardiines (2),28–30 cis-decahydroisoquinolines (3),7,31–34 2,4-trans-prolines35 and the more recently identified class by us 2,3-trans-3-aryl prolines (4).36 However, despite extensive synthetic efforts, and a vast number of x-ray crystal structures and modeling, only subtype selective antagonist have been developed for GluK1 (e.g. UBP30428,37 and LY4661957). Thus, there is still a profound need for the development of subtype-selective antagonists for the GluA1–4 and GluK2–5 subunits.
In previous studies, we have shown that introduction of a substituent in the 4’ position of 4a significantly influences its iGluR binding affinity profile.38 While a hydroxyl group (4b) results in a high nanomolar affinity for GluK3 (Ki= 0.87 μM) with 10–5 fold preference over GluK2/GluK1, a methyl group (4c) eliminates all affinity for AMPA and KA receptors leaving mid-range micromolar affinity for native NMDA receptors (Ki= 17 μM). The halogen series (F, Cl and Br (4d-f))39 unveiled a clear shift towards selectivity for native NMDA receptors with the 4’-chloro and 4’-bromo substituents being optimal for binding (Ki= 0.63 and 0.62 μM, respectively). Subsequently, 4e was shown to be a full antagonist with preference for GluN1/GluN2A and furthermore shown to enhance the anti-cancer effect of sorafenib in vitro.39 For the 5’ position, neither a hydroxyl group (5a) nor large lipophilic groups (not shown) improved the binding affinity significantly or modified the selectivity profile positively.38 Directly on the proline skeleton the 2,4-cis-4-position was substituted with an n-propyl group (9a) to which resulted in high nanomolar affinity for GluK1 (Ki= 0.62 μM) but only with a 4-fold preference over GluK3.38
Herein, we report further SAR investigations of 4a in our search for subtype selective iGluR antagonists. Furthermore, a new and efficient route for the stereoselective synthesis of 4a and its analogs is described utilizing a C(sp3)-H activation-arylation strategy as the key step.
Results and discussion
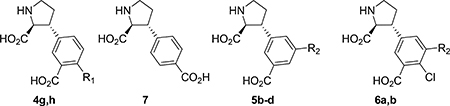
From the x-ray crystal structure of 5a in GluA2-LBD (Figure 1A), is it seen that the 4’-position directs substituents into close contact with receptor residue Leu671 (GluA2 numbering). Our previous SAR study (4a-f, Table 1) also shows that different substituents in that position greatly influence the binding affinity profile. We therefore sought to expand the SAR study further by synthesis of the 4’-amino analog 4g and the 4’-carboxylic acid analog 4h. Along this line, we also decided to reposition the vital 3’-carboxylic acid group to the 4’-position (compound 7, Figure 2).
Figure 1.


A) X-ray crystal structure of 5a (green) which holds a 5’-OH substituent, in the LBD of GluA2 (PDB: 4YMA).38 Receptor surface is displayed in grey. B) X-ray crystal structure of 5a (green) in GluA2 (PDB: 4YMA). Protein surface is colored in grey. The 5’-position directs towards cave-shaped space with a depth of 10–12Å.
Table 1.
Generalized iGluR antagonist compound classes 1–4 and binding affinities of selected antagonists UBP304 (2a), LY466195 (3a), and 4a-e at native AMPA, KA, and NMDA receptors (rat synaptosomes) and cloned homomeric receptors GluK1–3. All values in μMa
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Native iGluRs | Homomeric GluK receptorsb | ||||||||
| R1 | R2 | R3 | AMPAa (IC50) | KAa (IC50) | NMDAb (Ki) | GluK1 (Ki) | GluK2 (Ki) | GluK3 (Ki) | |
| 2a28,37 | -- | -- | -- | 0.012 | >100 | 111 | |||
| 3a7 | -- | -- | -- | 0.05 | -- | 8.9 | |||
| 4a36 | H | H | H | 51 | 22 | 6 | 4.3 | >100 | 8.1 |
| 4b38 | OH | H | H | 2 | 1.4 | 1 | 4.8 | 10–100 | 0.87 |
| 4c39 | Me | H | H | >100 | >100 | 17 | >100 | >100 | >100 |
| 4d39 | F | H | H | > 100 | 59 | 4.6 | 12 | >100 | 11 |
| 4e39 | Cl | H | H | >100 | >100 | 0.63 | 154 | >100 | 131 |
| 4f39 | Br | H | H | >100 | >100 | 0.62 | > 100 | > 100 | > 100 |
| 5a38 | H | OH | H | 6.3 | 6.7 | 4.1 | 3.7 | 32 | 2.1 |
| 9a38 | H | H | n-Pr | 48 | 22 | 25 | 0.62 | 81 | 2.2 |
Radioligands: AMPA, [3H]AMPA; KA, [3H]KA; NMDA, [3H]CGP-39653; GluK1, [3H]NF608; GluK2 and GluK3, [3H]KA.
Figure 2.

Overview of herein designed and synthesized analogs of iGluR antagonist 4a: series 4–6,8,9 and compound 7.
The 5’-position of 4a (series 5) directs substituents into a 10–12 Å deep hydrophilic cone-shaped cavity (Figure 1B). We decided to explore further the SAR of substituents with hydrogen bonding properties by the synthesis of analogs 5c (COOH) and 5d (1,2,3-triazole) (Figure 2). In addition, the hydrogen bonding capable 5’-aryl groups pyridines 5e-g and pyrimidines 5h,i were included in the SAR study (Figure 2). In addition to the aforementioned bisaryl analogs, we decided to design conformational relieved analogs by incorporation of a tethering nitrogen atom (compound 5j), which to be further functionalized to sulfonamides 5k,l and benzamide 5m (Figure 2).
Finally, analog 6b was designed which comprise substituents in both the 4’ and 5’-positions, based on the NMDA receptor-selective analog 4f (4’-Cl). The 5’-bromo analog 6a was included in the SAR study as it was easily accessible from a synthetic intermediate (discussed later).
At this stage, we recognized that the 5’-carboxylate group could easily be coupled with an amino acid or short peptide to explore the described cave in the receptor, in a diversity oriented way. For simplicity, we decided to firstly synthesize analogs with only one amino acid, series 8a-s. Depending on the results, additional analogs should be prepared.
The last series of compounds (9b-e) focus on the modification of the C4-side chain (R3) of 9a. Although the 2,4-cis stereochemistry is opposite to the observed 2,4-trans stereochemistry of naturally occurring ligands (e.g. kainate,40 domoate41,42), the binding mode of 9a in LBD of GluK1 (PDB: 4YMB) indicated a good fit of the propyl chain in the binding pocket (Figure 3). Therefore, the 2,4-cis stereochemistry remained in our design of new C4-analogs. As widely accepted in the field, the LBD of iGluRs adopt an open conformation when antagonists are bound.43 As indicated in Figure 3, on the right-hand site of the receptor a cavity is formed that can be reached by extending the propyl chain. Additionally, the crystal structure demonstrated that the propyl chain is in close proximity to the side chain of Glu441 (GluK1 numbering). In the agonist (closed) conformation of the LBD, Glu441 stabilizes interdomain contact.44–46 However, due to the open conformation of the LBD stabilized by binding of 9a this interaction is lost.38 We envisioned that alkyl amines 9b,c would be able to interact with Glu441 through electrostatic interactions as well as hydrogen bonding interactions. Additionally, 9c will be able to reach into the channel. Furthermore, alcohol (9d) and carboxylic acid 9e were designed to replace the role of Glu441 and engage with the extensive hydrogen bonding network in this region and thereby further stabilize the open, inactive conformation of the LBD.
Figure 3.

Compound 9a in the GluA1-LBD (PDB: 4YMB).
Chemistry
4’-Carboxylic acid analog 7 was synthesized stereoselectively in accordance with our previously described route (Scheme 1).38 1,4-Addition of the corresponding arylic cuprate to enantiopure enone 10 gave adduct 11 as one diastereomer in 84%. Reduction of the lactam by borane in THF (to give 11a not shown) followed by removal of the TBS ethers afforded diol 12 in 52% yield over two steps. Oxidation with RuCl3, gave the corresponding diacid (12a not shown) and after removal of the BOC group by TFA, the target compound 7 was obtained in 86% over two steps.
Scheme 1. Synthesis of analog 7 from enantiopure enone 10a.

aReagent and conditions: (a) nBuLi, ((3-bromobenzyl)oxy)(tert-butyl)dimethylsilane, CuCN, thiophene, Et2O, −78 °C to −42 °C, (84%); (b) BH3·Et2O, THF, 0 °C to reflux, 18h, (59%); (c) 1N TBAF, THF, rt, 18h, (88%); (d) NaIO4, RuCl3·H2O, H2O/MeCN/EtOAc, 0 °C, 1.5h, (86%); (e) TFA, DCM, rt, overnight, 99%.
Analogues 5e-i were synthesized in a similar fashion as 7, with the only change being that the copper catalyzed 1,4-addition was replaced by a rhodium(I) catalyzed 1,4-addition.39 The appropriate boronic ester 17 was prepared from 13 in four steps (Scheme 2).
Scheme 2. Synthesis of boronic acid 17a.

aReagents and conditions: (a) MeOH, H2SO4, reflux; (b) pinacol, Et2O; (c) LiAlH4, THF, 0–20 °C, (86% over three steps); TBDMSCl, imidazole, DMAP, DMF (quantitative).
With boronic ester 17 in hand, rhodium(I) catalyzed 1,4-addition to enantiopure enone 10 gave adduct 18 in a full diastereoselective fashion (Scheme 3). Subsequent reduction of the lactam functionality of 18 was attempted using several reported conditions,36 with dimethylsulfide-borane complex being superior (intermediate 18a, not shown). The two silyl ethers were cleanly removed with TBAF to provide diol 19 in 42% over 2 steps. Initially, we pursued direct oxidation of diol 12 to warrant late stage differentiation followed by only a deprotection step, but unfortunately all attempts thereto failed. Consequently, aryl group insertion was performed on 19 by standard Suzuki cross coupling chemistry in 63–87%. Ruthenium-catalyzed oxidation of diols 20a-e to their corresponding diacid was accompanied by side reactions mostly undesired N-oxidation. After purification, removal of the Boc group with TFA in DCM or BBr3 in DCM by pyridine analogues 5e-g in quantitative yields, while purification of 5h and 5i proved more difficult, thus resulting in low yields.
Scheme 3. Synthesis of 5e-ia.

aReagents and conditions: (a) Boronic ester 17, Rh(I), CsCO3, (86%); (b) DMS·BH3, THF, reflux, (62%); (c) TBAF, THF, (68%); (d) Pd-catalyst, Ar-B(OR)2, solvent, (Xantphos), heating, (31–87%); (e) RuCl3, NaIO4, H2O, MeCN, EtOAc; (f) TFA, DCM, then 1M HCl, (6–98 %); (g) BBr3, DCM, (4%).
For the synthesis of analogs 5j-m a racemic diastereoselective approach was taken due to efficiency (Scheme 4). Firstly, (S)-O-methyl-proline was converted to the corresponding enone 21 as described by Huy.47 Bromination with NBS gave bromine 22 in 80%, which was coupled with boronic acid 23 to give 24 in high yield. Reduction of the alkene over Pd/C yielded racemic 2,3-cis di-amine which was protected in situ with Boc anhydride to give aniline (±)-cis-25 in 50% over two steps. Derivatization to sulfonamides (±)-cis-26b,c and benzamide (±)-cis-26d proceeded smoothly and epimerization of the alpha center was performed with 1N NaOMe in (trans:cis 10:1 to >20:1). To obtain (±)-cis-27a, (±)-cis-25 was treated with LDA and quenched with NH4Cl. Deprotection of (±)-trans-27a-c was achieved in 4N HCl to afford the target analogs (±)-5j-l. Benzamide (±)-trans-27d was deprotected to give 5m in a two-step procedure, first ester hydrolysis with LiOH followed by, N-Boc was removed with TFA in DCM (20% over three steps).
Scheme 4. Synthesis of 5j-ma.

aReagents and conditions: (a) i) Et3N, NCS; ii) Pyr, CbzCl, (55%); (b) NBS, DABCO, (80%); (c) 23, PdCl2dppf, Cs2CO3, THF/H2O, 80 °C, (85%); (d) H2 (g), Pd/C, MeOH, rt, (quantitative); (e) Boc2O, DCM, rt, (50% over two steps); (f) RCl, pyridine, rt, overnight, (85–92%); (g) i) (for 26a) LDA, THF, −78 °C to 0 °C, 10 min, (50%, d.r. 1.3:1) or (ii) (for 26b-d) 25wt% NaOMe in MeOH, MeOH, reflux, 24h, (quantitative); (h) 4N HCl, reflux, 24h, (16–74%); (i) i) LiOH, MeOH/H2O, ii) TFA, DCM, (20%); (j) MeOH, SOCl2, (73%).
For the remaining target molecules 4g-i, 5b-d and 6a,b the two above described synthetic routes were not easily applicable due to functional group incompatibilities. Furthermore, the cuprate approach (Scheme 1 and 3) is troubled by a high number of protection and deprotection steps, while the second approach (Scheme 4) lacks enantioselective control. Therefore, a CH-activation approach was pursued to access 5a and the remaining analogs designed (Scheme 5).
Scheme 5.

Retrosynthetic analysis for CH-activation strategy
To secure a global and mild deprotection the amino and carboxylic acid functionalities of 6e were planned protected as their tert-butyl carbamate and tert-butyl ester, respectively (Scheme 5). Intermediate 28 should derive from 29 by a sequence of amide hydrolysis, alpha carbon epimerization and esterification. A similar analog to 29 was recently reported obtained from 30 by a stereoselective Pd-catalyzed C-H activation-arylation protocol of amide 30 with the corresponding aryl iodide 31.48,49 Amide 30 originates from commercially available N-Boc-D-proline.
Starting from commercially available N-Boc-D-proline, amide formation was performed with 8-aminoquinoline (8-AQ) in the presence of EDCI and HOBt in 85% (Scheme 6). The key C-H activation-arylation step was carried out according to reported conditions to afford 29 in 45% yield.48,49 The 8-AQ directing group was then removed under basic conditions with 10 equiv. NaOH in EtOH at 100 °C.49 Fortunately, due to the high stability of the amide the alpha center fully epimerized under these forcing conditions and gave 32 as a single enantiomer. The subsequent simultaneous protection of both carboxylic acids as tert-butyl esters turned out rather challenging. Various conditions were attempted and resulted in tert-butyl 2,2,2-trichloroacedimidate (TBTA) as superior reagent to afford 28a in 70%. To summarize, the new strategy delivers key intermediate 28 in only four steps (21% yield) and full diastereoselectivity.
Scheme 6. Synthesis of intermediate 28aa.

aReagents and conditions: (a) 8-Aminoquinoline, EDCI, HOBt, DCM, rt, 24h, (85%); (b) Methyl-3-bromo-5-iodobenzoate, 10 mol% Pd(OAc)2, 30 mol% PivOH, Ag2CO3, toluene, reflux, 3d, (40–48%); (c) NaOH, EtOH, reflux, 24h, (95%); (d) TBTA, DCM, rt, 24h, (74%).
To confirm that the epimerization had indeed taken place, 4a was resynthesized (Scheme 7). Firstly, 30 was reacted with methyl-3-iodobenzoate to give 33. Subsequent epimerization and amide hydrolysis with NaOH in EtOH was followed by Boc deprotection with TFA in DCM to afford 4a in 74% over 2 steps. Its 1H-NMR spectrum proved identical to an authentic sample of 4a obtained by 1,4-cuprate strategy, thus proving the 2,3-trans relationship. The 2,3-cis-4a diastereomer was obtained by amide hydrolysis under acidic conditions with 40% aqueous H2SO4 and the chemical shift of the alpha proton of 2,3-cis-4a was shown to be significantly different (4.15 ppm for 4a vs 4.45 ppm for 2,3-cis-4a; measured in D2O).
Scheme 7. Synthesis of 4a and 2,3-cis-4aa.

aReagents and conditions: (a) NaOH, EtOH, reflux, 24h, (95%); (b) TFA, DCM, rt, (74%); (c) 40% H2SO4, H2O, (27%).
For analogs 4g and 4h the C-H activation-arylation was performed with aryl iodide 36 (Scheme 8). C-H-activation-arylation of 30 with 36 gave 34a in 61%. After epimerization and amide hydrolysis 35a was further converted to 35b with NaN3 in the presence of Cs2CO3, CuI and EtOH. Furthermore, 35a was protected as dimethyl ester and subjected to Pd-catalyzed oxidative carbonylation50 conditions to install 4’-carboxylate. However, this approach only yielded in dehalogenation of the 4’-position. The obtained intermediates 35a-b were deprotected with TFA in DCM to afford 4g in 39% and 4h in 14% over two steps, respectively. In an alternative approach to diacid analog 4i the diacid was installed by preparation of the 38. As 38 is not commercially available, it was synthesized from 37 in three steps by esterification, reduction and subsequent Sandmeyer51 reaction to afford 38 in 30% overall yield. C-H activation-arylation with 38 afforded 34c in 37% which was further treated with NaOH in EtOH, followed by deprotection with TFA in DCM. The final compound was purified by preparative HPLC and transformed into its tri sodium salt with 1N NaOH to afford 4i in 72%. To ensure that cyclization to a cyclic anhydride has not occurred during purification and isolation of the compound, the final product was treated with benzyl amine. Analysis by LCMS and HPLC confirmed that no amide formation had occurred.
Scheme 8. Synthesis of 4f-ha.

aReagents and conditions: (a) 36 or 38, 10 mol% Pd(OAc)2, 30 mol% PivOH, Ag2CO3, toluene, reflux, 3d, (61% and 37%); (b) NaOH, EtOH, reflux, 24h, (86% and 86 %); (c) NaN3, Cs2CO3, CuI, EtOH, reflux, 3d, (d) TFA, DCM (14–72%); (e) SOCl2, MeOH, reflux, overnight, (70%); (f) Pd/C, H2, MeOH, (95%); (g) I2, tert-BuNO2, MeCN, toluene, 0 °C, then rt, 18h (46%).
Analogue 5b (Scheme 9) was obtained by direct deprotection of 28a, while 5’-carboxylic acid 5c was obtained by first converting 28a to its corresponding 5’-carboxylate 28b by a Pd-catalyzed oxidative carbonylation50 in 90%. Deprotection of 28b was carried out with TFA in DCM and cleanly afforded 5c.
Scheme 9. Synthesis of 5b,ca.

aReagents and conditions: (a) HCOOH, Ac2O, Et3N, Pd(OAc)2, Xantphos, 80 °C, (90%); (b) TFA, DCM, (77% and 75%, respectively).
Sonogashira coupling52 of 23a (Scheme 10) with trimethylsilylacetylene (TMSA) alkyne and subsequent deprotection with TBAF afforded alkyne 35a in 66% yield over two steps. CuAAC53 with TMSN3 afforded triazole 36a in 65%. Finally, deprotection with TFA in DCM afforded 5d in 92%.
Scheme 10. Synthesis of 5da.

aReagents and conditions: (a) TMSA, PdCl2(PPh3)2, CuI, Et3N, 60 °C, 12h, (77%); (b) 1N TBAF, THF, rt, 30min (82%); (c) TMSN3, CuI, DMF-MeOH, (65%); (d) TFA, DCM, (92%)
The synthesis of 6a,b (Scheme 11) commenced with iodination of commercially available 43 followed by esterification to give tetrasubstituted aryl iodide 44 (69% over two steps). Coupling of highly functionalized aryl iodide 44 with 30 under CH-activation conditions, gave 45 in 31% and underlines the power of this synthetic strategy. By following the before established sequence of basic hydrolysis, epimerization and esterification with TBTA intermediate 46a was obtained in 46% over two steps. Subsequent Pd-catalyzed oxidative carbonylation50 gave carboxylate 46b. Global deprotection of 46a,b with TFA yielded free amino acids 6a,b in 96% and 93% yield, respectively.
Scheme 11. Synthesis of 6a,ba.

aReagents and conditions: (a) NIS, TfOH, (99%); (b) SOCl2, MeOH, reflux, ON, (70%); (c) 44, 10 mol% Pd(OAc)2, 30 mol% PivOH, Ag2CO3, toluene, reflux, 3d, (31%); (d) NaOH, EtOH, reflux, 24h, (88%); (e) TBTA, DCM, (52%); (f) HCOOH, Ac2O, Et3N, Pd(OAc)2, Xantphos, 80 °C, (63%); (g) TFA, DCM, (96% and 93%, respectively).
The synthesis of the 19 amino acid conjugates 8a-s commenced from 5’-carboxylic acid 23a and suitably protected amino acid under coupling reaction conditions, followed by suitable deprotection (Scheme 12). For details see supporting information.
Scheme 12. Synthesis of amino acid conjugates 8a-s from intermediate 23aa.

aReagents and conditions: (a) EDCI, HOBt, AA-OtBu, DMF, rt, 24h, (65–99%); (b) TFA, DCM, rt, overnight (33–97%); (c) (i) 1N LiOH, MeOH, rt, overnight, (ii) TFA, DCM, rt, overnight, (46%).
For the synthesis of analogs 9b-e comprising a C4’-substituent, the C-H activation-arylation strategy was applied, but starting from methyl-Boc-D-pyroglutamate (Scheme 13). Deprotonation with LHMDS and alkylation with allylbromide gave a 2:1 mixture of the trans:cis diastereomers, which were separated on flash column chromatography to afford 47 in 36% as a single diastereomer. Lactam reduction was performed with super-hydride, followed by treatment with BF3 and Et3SiH to give 48 in 72% over two steps. Ester hydrolysis and amide coupling with 8-AQ in the presence of EDCI and HOBt proceeded in 72% over two steps. The following C-H-activation-arylation step with methyl-3-iodobenzoate provided 49 in 36%. Epimerization and amide hydrolysis with NaOH in EtOH, followed by esterification with TBTA gave protected alkene, which was converted to aldehyde 50 by treatment with OsO4 and NaIO4 (19% over three steps). To summarize, aldehyde 50 was obtained in nine steps in 12% overall yield from commercially available methyl-Boc-D-pyroglutamate.
Scheme 13. Synthesis of intermediate aldehyde 50a.

aReagents and conditions: (a) LHMDS, then allylbromide, THF, DMPU, −78 °C to −40 °C, 4h, (36%); (b) (i) LiEt3SiH, then H2O2; (ii) BF3·Et2O, Et3SiH, (72%); (c) LiOH, H2O, THF, (quantitative); (d) EDCI, HOBt, 8-Aminoquinoline, DCM, rt, overnight, (72%); (e) Methyl-3-iodobenziate, Pd(OAc)2, PivOH, toluene, reflux, 3d, (35%); (f) (i) NaOH, EtOH, 100 °C, overnight, (ii) TBTA, DCM, rt, overnight, (35% over two steps); (g) OsO4, NaIO4, THF, H2O, rt, 4h, (53%).
Reductive amination of key aldehyde 50 with MeNH2·HCl or BnNH2·HCl in the presence of NaBH3CN gave secondary amines 51a,b in 48% and 70%, respectively (Scheme 14). Furthermore, aldehyde 50 was reduced to alcohol 51c with NaBH4 in MeOH in 83%, and acid 51d was obtained by Pinnick oxidation of aldehyde 50 in 94%. Global deprotection of 51a-d was achieved with TFA in DCM to provide target compounds 9b-e in 53–99%, respectively.
Scheme 14. Synthesis of 9b-e from aldehyde 50.a.

aReagents and conditions: (a) MeNH2·HCl, NaBH3CN, MeOH, rt, overnight, (48%); (b) BnNH2·HCl, NaBH3CN, MeOH, (70%); (c) NaBH4, MeOH, rt, 30min, (83%); (d) NaO2Cl, THF:H2O:tert-BuOH 1:1:1, 2-methyl-butene, phosphate buffer, (94%); (e) TFA, DCM, rt, overnight, (53–99%).
To summarize, a total of 40 new analogues of 4a were synthesized by three different strategies. On the comparison of these, the C-H activation-arylation approach is by far the most efficient and also allows for full enantiocontrol of the product.
Pharmacological Characterization
All analogues synthesized were first characterized in radioligand binding assays at native iGluRs and cloned rat homomeric subtypes, GluK1–3 (Table 2). Furthermore, analogs 5e-i were characterized at cloned homomeric GluA1 and GluA2, and analogs which showed good to high affinity for the NMDA receptors (Ki= <1 μM) were furthermore investigated in functional assays at the four GluN1/GluN2A-D NMDA receptor subtypes.
Table 2.
Chemical structures and binding affinity data for analogues 4g,h,5b-d,6a,b and 7 at native iGluRs (rat synaptosomes) and homomeric recombinant rat iGluRs. All values in μM.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Native iGluRs (synaptosomes)b | Cloned homomeric iGluRsc | ||||||
| AMPA IC50 | KA IC50 | NMDA Ki | GluK1 Ki | GluK2 Ki | GluK3 Ki | ||
| 4g | R1= NH2 | >100 | >100 | >100 | (70 ± 16)d | (77 ± 5)d | (64 ± 4)d |
| 4h | R1= CO2H | >100 | >100 | 1.3 [5.88 ± 0.06] | >100 | >100 | >100 |
| 7 | -- | >100 | >100 | 8.5 | >100 | >100 | >100 |
| 5b | R2= Br | 26 [4.60 ± 0.04] | 10 [4.99 ± 0.04] | 2.4 [5.63 ±0.05] | 5.88 ± 0.60 | >100 | 2.42 ± 0.11 |
| 5c | R2= CO2H | 36 [4.45 ± 0.04] | 63 [4.21 ± 0.05] | 0.48 [6.32 ± 0.03] | 12.5 ± 1.7 | >100 | 15.7 ± 3.1 |
| 5d | R2= triazole | 21 [4.69 ±0.04] | 20 [4.71±0.06] | 0.83 [6.08±0.05] | >50 | >100 | 10–50 |
| 6a | R2= Br | >100 | >100 | 1–10 | ≥100 | ≥100 | ≥100 |
| 6b | R2= CO2H | >100 | >100 | 0.16 [6.82 ± 0.08] | ≥100 | ≥100 | >50 |
-,--: Values not determined, Radioligands used: AMPA, [3H]AMPA; KA, [3H]KA; [3H]CGP-39653; GluK1, [3H]NF608; GluK2 and GluK3, [3H]KA
Data are mean values of three to six individual experiments performed in triplicate. For AMPA and KA: pIC50 values with SEM in brackets. For NMDA: pKi values with SEM in brackets.
Mean values ± SEM of at least three experiments conducted in triplicate at 12–16 drug concentrations.
% specific binding mean values ± SD of three experiments at 10 μM ligand concentration.
Surprisingly, 4’-amino analog 4g displayed significantly reduced affinity for all iGluRs compared to the 4’-hydroxy analog 4b (Table 1). In contrast, introduction of a carboxylic acid group in the 4’-position (compound 4h) resulted in a fully selective, low micromolar affinity NMDA receptor ligand. In comparison with compound 7, it is interesting to observe that both carboxylate group are required for the observed shift to full NMDA receptor selectivity.
With a bromine in the 5’-position (compound 5b) no significant change in the pharmacological profile was observed, the 5’carboxylic acid analogs 5c showed a 9-fold increase in affinity for the NMDA receptors (Ki= 0.48 μM). Although mid-micromolar affinity was observed for both AMPA and KA receptors, 5c displays a 75-fold selectivity for NMDA receptors. Likewise, 5’-Triazole 5d showed a 7-fold higher affinity for native NMDA receptors (Ki= 0.83 μM) compared to 4a (Ki= 6.0 μM) but only a 24-fold preference for NMDA over KA (IC50= 20 μM) and AMPA (IC50= 21 μM) receptors.
The group of six membered 5’-heteroaryl-analogues 5e-i, all had no significant change in their profile amongst native iGluRs homomeric GluK1–3. From docking studies of the group, it became apparent that the heteroaromoatic group is in close proximity to residues that are not conserved in GluA1 and GluA2 (GluA1: Ala666, Glu665; GluA2: Ser673, Asp672). For this reason, we decided to determine the binding affinity of 5e-i at GluA1 and GluA2. A general trend of 3–4-fold higher affinity for GluA1 over GluA2 was observed, which regrettably could not motivate subsequent design and SAR of additional analogs.
The installation of amine (±)-5j as well as sulfonamide (±)-5k,l and amide linker (±)-5m did not lead to a significant change in binding affinity profile. Mid micromolar affinities persisted for (±)-5j-l at native AMPA and NMDA receptors, while affinity for native and cloned KA receptors varied over the micromolar range (IC50= 16 to >100 μM and Ki= 4 to >100 μM)).
Compounds 6a and 6b, which are hybrid structures of 4e and 5b,c maintained their selectivity for the NMDA receptors, and notably 6b showed an additive effect of the 4’-chloro substituent rendering it the most selective highest affinity NMDA receptor ligand in this series (Ki= 160 nM).
The 19 analogs 8a-s which comprise an amino acid coupled in the 5’-position showed a general preference for binding to native NMDA receptors (Ki = 0.5–10 μM) over native AMPA (IC50= 5–50 μM) and native KA (IC50= 10–100 μM) receptors (Table 3). In more detail, the SAR study shows that introduction of an alkyl substituent, analogs 8a-e, lead to a size-dependent decrease in binding affinity for native NMDA receptors (8a, Ki= 0.60 μM to 8e, Ki= 5–10 μM). Perhaps unexpected, analog 8k, which comprises Glu maintained high nanomolar affinity for native NMDA receptors, and in fact displayed higher selectivity over AMPA and KA receptors compared to 8a. On the other hand, installation of hydroxyl groups in analogs 8h and 8i showed no significant effect. Histidine analog 8q was found to be the best GluK3-preferring ligand of this series (Ki= 6.23 ± 2.69), but also lysine analog 8o which displayed low micromolar affinity for both GluK1 and GluK3. Unfortunately, amide analogs 8l and 8n were both unstable in the assay buffer, and thus a clear pharmacological profile could not be obtained.
Table 3.
Chemical structures and binding affinities of analogues 5e-m at native iGluRs (rat synaptosomes) and homomeric recombinant iGluRs. All values in μM.a
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Native iGluRs (synaptosomes)b | Cloned homomeric iGluRsc | ||||||||
| AMPA IC50 | KA IC50 | NMDA Ki | GluA1 Ki | GluA2 Ki | GluK1 Ki | GluK2 Ki | GluK3 Ki | ||
| 5e |  |
40 [4.40±0.05] | 20 [4.71±0.05] | 3.8 [5.42±0.01] | 19 | 55 | 20 ± 1.8 | >100 | 6.4 ± 0.41 |
| 5f | 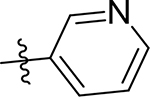 |
29 [4.57±0.13] | 23 [4.65±0.03] | 3.1 [5.51±0.02] | 15 | 40 | 25 ± 1.2 | >100 | 5.9 ± 0.62 |
| 5g | 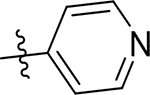 |
27 [4.581±0.07] | 25 [4.61±0.05] | 1.7 [5.76±0.01] | 12 | 33 | 22 ± 2.3 | >100 | 6.5 ± 0.92 |
| 5h | 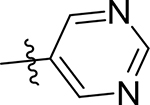 |
59 [4.30±0.13] | >100 | 10 [4.98±0.01] | 22.7 | 82.9 | 100 | 100 | 13 ± 0.17 |
| 5i | 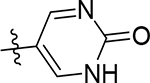 |
13 [4.92±0.13] | >100 | 4.1 [5.39±0.03] | 10 | 56 | 76 ± 1.8 | >100 | 16 ± 0.98 |
| (±)-5j | -NH2 | 52 [4.29±0.07] | 86 [4.07±0.04] | 14 [4.87±0.05] | -- | -- | 32.7 ± 7 | >100 | 19.3 ± 0.9 |
| (±)-5k | 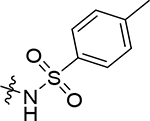 |
23 [4.65±0.09] | >100 | 15 [4.82±0.03] | -- | -- | 19 ± 2.8 | 19c | 10.9 ± 0.5 |
| (±)-5l | 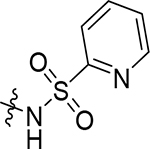 |
27 [4.57±0.04] | >100 | 36 [4.46±0.08] | -- | -- | 27.6 ± 0.6 | ∼100 | 14.0 ± 0.9 |
| (±)-5m | 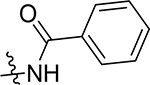 |
24 [4.63±0.04] | 16 [4.81±0.03] | 6.9 [5.16±0.04] | -- | -- | 18.5 ± 1.8 | ∼100 | 3.95 ± 0.2 |
-,--: Values not determined, Radio ligands used: AMPA, [3H]AMPA; KA, [3H]KA; [3H]CGP-39653; GluA1, [3H]AMPA; GluA2, [3H]AMPA]; GluK1, [3H]NF608; GluK2 and GluK3, [3H]KA
Data are mean values of three to six individual experiments performed in triplicate. For AMPA and KA: pIC50 values with SEM in brackets. For NMDA: pKi values with SEM in brackets.
Mean values ± SEM of at least three experiments conducted in triplicate at 12–16 drug concentrations.
n=1.
In comparison to the 2,4-cis-4-propyl analog 9a, N-methyl amine analog 9b showed a general decrease in affinity for all the iGluRs. But interestingly, on the increase of size the N-benzyl analog 9c showed affinity for GluK1 and GluK3 in low micromolar range (Ki= 2.98 and 2.46 μM, respectively). This could indicate that the phenyl ring can occupy the cavity in the open conformation of the LBD, but also the increased lipophilicity of the ligand serves to lower its desolvation energy. The 4-ethyl hydroxy analog 9d lead to a reduction of affinity for GluK1 compared to 9a, and finally carboxylic acid 9e led to complete depletion of affinity for any of the iGluRs (IC50 >100 μM, Ki >100μM). This might be explained by charge-charge repulsion between the negatively charged Glu465 residue, that is in close proximity to the acid side chain of 9e. To summarize for series 9, the positive effect of favorable polar interactions of the ligand with the receptor did not was not crystallize in an increase of affinity. Furthermore, the data does not indicate energetically favorable interactions of the ligand with the water network in the receptor to stabilize the receptor ligand complex.
Pharmacological characterization at the four GluN1/GluN2A-D NMDA receptors
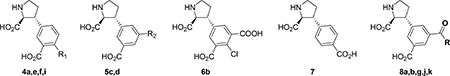
The SAR study had identified a total of eight new analogs 4h, 5c, 5d, 8a,b,g,j,k which display ~1 micromolar binding affinity for native NMDA receptors. Previously published 4’-bromo analog 4g falls within this range and was therefore also included here. Furthermore, compound 7 was also of interest as this analog represents a new lead for NMDA receptor ligands. To investigate the functional properties across the GluN1/GluN2A-D NMDA receptor subtypes, they were subsequently characterized in a two-electrode voltage-clamp recording assay (Xenopus oocytes) (Table 6). All analogs were full antagonists at the four NMDA receptor subtypes with Ki values in the high nanomolar to low micromolar range. In general, all analogs displayed a preference for GluN1/2A over the remaining three NMDA receptor subtypes. The degree of selectivity for GluN1/2A was determined to be 3–6 fold over GluN1/2B and GluN1/2C, while enhanced preference/selectivity over GluN1/2D was observed (4–33 fold). The best selectivity profiles were demonstrated for analogs 4h and 5c, which displayed a 32 to 34-fold selectivity for GluN1/2A over GluN1/2D, but only 6 to 10-fold selectivity over GluN1/2B and GluN1/2C. Regrettably, analog 7 did not display a novel selectivity profile, but analog 6b is worth paying attention to as it is the most potent analog of the series (Ki= 200 nM at GluN1/GluN2A). From the data, it is also noticeable that the amino acid analogs 8a,b,g,j,k followed the same trend as when increasing bulk size of the lead structure 4a by addition of traditional (functional) groups in the 5’-position.
Table 6.
Chemical structures and functional characterization at GluN1/GluN2A-D NMDA receptors. All values in μM.
 | ||||||
|---|---|---|---|---|---|---|
| R | Native NMDA (Ki) | GluN1/2A (IC50) | GluN1/2B (IC50) | GluN1/2C (IC50) | GluN1/2D (IC50) | |
| 4a | R1= H | 6.0 | 11.8 ± 0.23 | 34.5 ± 0.97 | 91.2 ± 2.60 | > 100 |
| 4e39 | R1= Cl | 0.63 | 4.7 | 10 | 24 | 41 |
| 4g | R1= Br | 0.6239 | 1.83 ±0.11 | 6.15 ± 0.17 | 21.4 ± 1.09 | 25.5 ± 1.46 |
| 4h | R1= COOH | 1.3 | 0.75 ± 0.03 | 4.24 ± 0.13 | 5.17 ± 0.12 | 24.3 ± 1.46 |
| 5c | R2= COOH | 0.48 | 0.63 ± 0.01 | 4.37 ± 0.17 | 6.27 ± 0.13 | 21.2 ± 0.37 |
| 5d | R2= triazole | 0.83 | 1.36 ± 0.06 | 3.51 ± 0.18 | 7.28 ± 0.17 | 18.5 ± 1.54 |
| 6b | -- | 0.16 | 0.20 ± 0.02 | 0.75 ± 0.04 | 1.33 ± 0.03 | 5.15 ± 0.23 |
| 7 | -- | 8.5 | 45 ± 1.5 | 165 ± 14.1 | 193 ± 6.1 | 159 ± 8.7 |
| 8a | Gly | 0.60 | 1.32 ± 0.05 | 5.76 ± 0.22 | 9.94 ± 0.52 | 44.30 ± 7.90 |
| 8b | L-Ala | 0.87 | 1.64 ± 0.07 | 7.17 ± 0.17 | 11.55 ± 0.62 | 33.84 ± 4.47 |
| 8g | L-Tyr | 0.96 | 1.87 ± 0.08 | 1.73 ± 0.05 | 9.70 ± 0.48 | 25.20 ± 1.46 |
| 8j | L-Pro | 1.1 | 1.00 ± 0.06 | 2.89 ± 0.10 | 4.50 ± 0.22 | 34.79 ± 12.25 |
| 8k | L-Glu | 0.83 | 0.61 ± 0.06 | 3.89 ± 0.22 | 2.61 ± 0.07 | 23.50 ± 1.00 |
Inhibition of recombinant NMDA receptor subtypes measured using two-electrode voltage-clamp recordings. IC50 values for inhibition of current responses activated by co-application of 100 μM glycine and 1 μM Glu to Xenopus oocytes expressing recombinant rat GluN1/GluN2A-D NMDA receptors were determined from 5–6 oocytes and Ki values were estimated using the Cheng-Prusoff relationship54 and previously determined Glu EC50 values55. Ki values are mean ± SEM
Conclusion
In conclusion, we have reported the SAR study of 40 new 3-aryl prolines as ligands for the iGluRs. Their synthesis was achieved by via three different synthetic routes: The first one being previously reported strategy based on a cuprate or ruthenium(I)-catalyzed 1.4 Michael addition which delivered analogs 5e-i and 7 as pure enantiomers. The second strategy was disclosed herein and is a racemic approach to afford analogs (±)-5j-m in an expedient way starting from readily available bromo enone 22. The third and newly developed strategy takes advantage of C(sp3)-H activation and delivered enantiopure analogs 4g-i,5b-d,6a,6b and 8a-s in a concise and fully stereoselective fashion starting from (R)-proline.
With the work presented herein, the SAR of this class of iGluR antagonists now shows that potent and selective NMDA receptor subtype antagonists are obtained by introduction of substituents in the 5’position, and that an additional chloro or bromo substituent in the 4’-position enhances potency. In terms of NMDA receptor subtype selectivity profile, the GluN1/GluN2A subtype is generally favored, and most interestingly, the 5’-amino acid analogs (series 8) demonstrate that large groups can be accommodated in this position, fitting into the cave-like space in the receptor, and maintaining nanomolar binding affinity. Based on data disclosed herein, it is reasonable to suggest that the observed selectivity for and potency at the NMDA receptors can be further improved and/or directed towards one of the other NMDA receptor subtypes, by continuing the SAR studies of the 5’-position.
Experimental Section
Chemistry
All reactions involving dry solvents or sensitive agents were performed under an argon atmosphere and glassware was flame dried under vacuum prior to use. Commercially available chemicals were used without further purification. DCM, THF and DMF were dried using a SG WATER solvent purification system (commercialized by Pure Process Technology). MeOH was dried by standing over 4 Å molecular sieves for a minimum of 48h. Reactions were monitored by analytical thin-layer chromatography (TLC, Merck silica gel 60 F254 aluminum sheets) or by HPLC. Flash chromatography was carried out using Merck silica gel 60A (40–63 μm). For dry column vacuum chromatography (DCVC), Merck silica gel 60 (15–40 μm) and a standard setup was used. 1H NMR spectra were recorded at 400 or 600 MHz and 13C NMR spectra at 100 or 150 MHz on a Bruker Avance III or Bruker Avance III HD, respectively. Chemical shifts (δ) are reported in ppm relative to TMS. For 13C NMR in D2O was added 1% CD3OD as internal reference. HPLC was performed using a Dionex UltiMate 3000 pump and photodiode array detector (210 and 254 nm, respectively) installed with an XTerra MS C 18 3.5 μ m, 4.6 mm × 150 mm column, using a 5 → 95% MeCN gradient in H2O containing 0.1% TFA. For HPLC control, data collection and data handling, Chromeleon software v. 6.80 was used. Preparative HPLC was carried out on an Agilent Prep HPLC systems with Agilent 1100 series pump, Agilent 1200 series diode array, multiple wavelength detector (G1365B), and Agilent PrepHT High Performance Preparative Cartridge Column (Zorbax, 300 SB-C18 Prep HT, 21.2 × 250 mm, 7 μm). LC-MS spectra were recorded using either an Agilent 1200 series solvent delivery system equipped with an autoinjector coupled to an Agilent 6400 series triple quadrupole mass spectrometer equipped with an electrospray ionization source or Waters Aquity UPLC-MS with dual wavelength detection with electrospray ionization. Gradients of 5% aqueous MeCN + 0.1% HCO2H (solvent A), and 95% aqueous MeCN + 0.05% HCO2H (solvent B) were employed. IR spectra were recorded on a Perkin-Elmer 801 spectrophotometer. Optical rotation was measured using a Perkin-Elmer 241 Spectrometer, with Na lamp (D line, 589 nm). Compounds were dried under high vacuum or freeze dried using a Holm & Halby, Heto LyoPro 6000 freeze drier. The purity of compounds submitted for pharmacological characterization was determined by HPLC to be >95%.
Synthesis 4’-carboxy analog 7
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(4-(((tert-butyldimethylsilyl)oxy)-methyl)phenyl)-5-oxopyrrolidine-1-carboxylate (11)
Solution A: Thiophen (487 mg, 5.79 mmol, 1.90 equiv) was dissolved in dry Et2O (8 mL) in a dry vial under N2. The mixture was cooled to 0 ˚C and dropwise added 2.03 M n-BuLi (2.85 mL, 5.79 mmol, 1.90 equiv) over the course of 15 min. The mixture was left to stir at 0 °C for 15 min. before removed from the ice bath and stirred at r.t. for 1 h (a white, colloid precipitate was formed). (3-Bromobenzyl)oxy)(tert-butyl)dimethylsilane (1.15 g, 3.82 mmol, 1.25 equiv) was dissolved in dry Et2O (30 mL) in a dry flask under N2. The mixture was cooled to −78 ˚C and dropwise added 1.55 M t-BuLi (4.95 mL, 7.67 mmol, 2.51 equiv) over the course of 20 min. The mixture was left to stir at −78 ˚C for 60 min. (The solution became yellow/brownish coloured and unclear) before a suspension of CuCN (342 mg, 3.82 mmol, 1.25 equiv) in dry Et2O (5 mL) was added dropwise over the course of 5 min. The mixture was left to stir at −78 ˚C for 10 min, then 5 min at r.t. and 10 min at −42 ˚C before recooled to −78 ˚C over the course of 10 min. The mixture was dropwise added to Solution A (7.5 mL, 3.8 mmol, 1.3 equiv) over the course of 15 min and left to stir at −78 ˚C for 10 min, at r.t. for 5 min, at −42 ˚C for 10 min before recooled to −78 ˚C over the course of 10 min. A solution of enone 10 (1.00 g, 3.05 mmol, 1.00 equiv) dissolved in Et2O (4 mL) was dropwise added over the course of 20 min. (The mixture instantly turned bright yellow). The mixture was left to stir at −78 ˚C for 15 min., at r.t. for 5 min. and at −42 ˚C for 60 min before quenched the reaction was quenched with sat. NH4Cl (6 mL) and stirred at −42 ˚C for additional 10 min. The mixture was transferred to a separation funnel containing sat. NaHCO3 (75 mL) and EtOAc (40 mL). The aqueous layer was extracted with EtOAc (2 × 50 mL) and the combined organic phases was washed with brine (75 mL), dried over MgSO4, filtered and concentrated in vacuo. The mixture was purified using DCVC (dia. = 7, 75 mL fractions, 0 – 4% EtOAc in heptanes) to yield the title compound as colorless oil that solidified upon standing (1.41 g, 84%). 1H NMR (300 MHz, CDCl3) δ 0.08 (3H, s), 0.10 (3H, s), 0.12 (6H, s), 0.92 (9H, s), 0.96 (9H, s), 1.54 (9H, s), 2.53 (1H, dd, J = 18, 3 Hz), 3.15 (1H, dd, J = 18, 10 Hz), 3.45 (1H, dm, J = 9 Hz), 3.80 (1H, dd, J = 11, 2 Hz), 4.00 (1H, dd, J = 11, 4 Hz), 4.06 (1H, m), 4.72 (2H, s), 7.15 (2H, d, J = 8 Hz), 7.28 (2H, d, J = 8 Hz) 13C NMR (75 MHz, CDCl3) δ −5.2, −5.0, 18.4, 18.7, 26.1, 26.2, 28.3, 38.7, 40.2, 63.8, 64.7, 67.0, 83.1, 126.3, 126.8, 140.3, 142.9, 149.8, 174.2 LCMS m/z [M + H]+ calc: 450.3, found: 450.3; [a]22D: −27.55 (c = 0.65 g/100 mL; EtOAc).
(2S,3R)-tert-Butyl 2-(((tert-butyldimethylsilyl)oxy)methyl)-3-(4-(((tert-butyldimethylsilyl)oxy)-methyl)phenyl)pyrrolidine-1-carboxylate (11a)
Lactam 11 (1.43 g, 2.35 mmol, 1.00 equiv) was dissolved in dry THF (15 mL) and 1M BH3.THF complex (30.0 mL, 30.0 mmol, 11.7 equiv) was added over the course of 5 min. The mixture was refluxed under N2 for 18 h. The mixture was cooled to 0 ˚C and dropwise added H2O (6 mL) over the course of 2 h, NaOH (2M, 30 mL) dropwise over the course of 40 min and H2O2 (30 %, 10 mL) over the course of 15 min (organic/aqueous ratio important). After 5 min the mixture was remove from the icebath and left to stir at r.t. for 1.5 h. The mixture was poured into sat. NaHCO3 (100 mL) and EtOAc (50 mL). After separation the aqueous phase was reextracted with EtOAc (2 × 75 mL) and the combined organic phases was washed with brine (100 mL), dried over MgSO4, filtered, concentrated in vacuo and purified using DCVC (dia= 4 cm, 30 mL fractions, 0 – 4% EtOAc in PhMe) to yield the title compound (809 mg, 59%) as a colorless oil. 1H NMR (300 MHz, CDCl3) δ 0.06 (6H, s), 0.13 (6H, s), 0.92 (9H, s), 0.97 (9H, s), 1.23 – 1.38 (1H, m), 1.50 (9H, s), 1.83 – 1.98 (1H, m), 2.18 – 2.34 (1H, m), 3.25 – 3.45 (1H, m), 3.47 – 3.80 (4H, m), 3.85 (0.53H, m), 4.04 (0.41H, m), 4.73 (2H, s), 7.10 – 7.23 (2H, m), 7.23 – 7.30 (2H, m). 13C NMR (75 MHz, CDCl3 δ −5.1, −5.0, 18.4, 18.7, 26.1, 26.2, 28.8, 31.8, 33.0, 45.4, 46.3, 46.5, 47.2, 61.4, 62.9, 64.9, 65.6, 65.8, 79.1, 79.5, 126.4, 127.1, 127.3, 139.6, 142.1, 142.7, 154.2, 154.3 LCMS m/z [M + H]+ calc: 436.3, found: 436.3 [a]22D: +7.27 (c = 0.87 g/100 mL; EtOAc).
(2S,3R)-tert-Butyl 2-(hydroxymethyl)-3-(4-(hydroxymethyl)phenyl)pyrrolidine-1-carboxylate (12)
The protected intermediate 11a (722 mg, 1.34 mmol, 1.00 equiv) was dissolved in dry THF (10 mL) and added 1M TBAF (5 mL). The mixture was left to stir under nitrogen atmosphere for 18 h. The mixture was transferred to a separation funnel containing EtOAc (50 mL) and sat. NaHCO3 (50 mL). The aqueous phase was reextracted with EtOAc (2 × 50 mL). The pooled organic phases were washed with brine (75 mL), dried over MgSO4, filtered and concentrated in vacuo. The mixture was purified by DCVC (dia = 3 cm, 30 mL fractions, 0 – 100% EtOAc in heptanes) to yield 12 (364 mg, 88%) as a clear, colorless oil. 1H NMR (300 MHz, CDCl3) δ 1.48 (9H, s), 1.91 (1H, m), 2.12 (1H, m), 2.91 (1H, m), 3.20 – 3.45 (2H, m), 3.51 – 3.80 (3H, m), 3.88 (1H, m), 4.57 (2H, s), 5.20 (1H, m), 7.15 (2H, d, J = 8 Hz), 7.25 (2H, d, J = 8 Hz) 13C NMR (75 MHz, CDCl3) δ 28.6, 32.9, 47.1, 47.4, 64.5, 65.5, 66.8, 80.6, 127.3, 127.5, 139.8, 140.0, 156.6 LCMS m/z [M + H]+ calc: 252.1, found: 252.1 (- t-Bu); [a]22D: +3.98 (c = 0.43 g/100 mL; EtOAc).
(2S,3R)-3-(3-(Benzyloxy)-5-carboxyphenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (12a)
Suspension B: NaIO4 (1.92 g, 8.98 mmol, 9.87 equiv) and RuCl3.H2O (8 mg, 0.037 mmol, 0.04 equiv) were suspended in H2O (6 mL) and stirred at r.t. for 1 min. prior to use.
Alcohol 12 (280 mg, 0.91 mmol, 1.00 equiv) was dissolved in CH3CN (5 mL) and EtOAc (5 mL), cooled to 0 ˚C and dropwise added suspension B over the course of 15 min. The flask containing suspension A was washed with H2O (2 mL), which was added to the mixture over the course of 5 min. The mixture was left to stir at 0 ˚C for 1.5 h. The mixture was filtered into a separation funnel through a plug of celite, which was afterwards washed with water (20 mL) and EtOAc (2 × 20 mL). The pH was adjusted to 1–2 using a couple of drops of conc. HCl and sat. NaCl (5 mL) added for separation of the two phases. After separation, the aqueous phase was extracted with EtOAc (2 × 20 mL). The pooled organic phases were washed with brine (50 mL), dried over MgSO4, filtered, concentrated in vacuo and purified using DCVC (dia = 3 cm, 25 mL fractions, 0 – 50% EtOAc in heptane containing 2% AcOH) to yield the title compound (263 mg, 86%) as a white foam. 1H NMR (300 MHz, DMSO) δ 1.35 (6H, s), 1.42 (3H, s), 2.00 (1H, m), 2.21 (1H, m), 3.33 – 3.50 (2H, m), 3.50 – 3.62 (1H, m), 4.06 (0.66H, d, J = 8 Hz), 4.09 (0.33H, d, J = 7 Hz), 7.41 (0.66H, d, J = 8 Hz), 7.43 (1.33H, d, J = 8 Hz), 7.88 (2H, d, J = 8 Hz) 13C NMR (75 MHz, DMSO) δ 28.0, 28.2, 32.1, 32.7, 46.0, 46.1, 48.2, 49.3, 65.1, 65.4, 79.1, 127.3, 127.5, 129.4, 129.5, 145.8, 146.1, 152.7, 153.2, 166.9, 172.8, 173.3 LCMS m/z [M + H]+ calc: 236.1, found: 236.1 (- Boc); [a]22D: +81.90 (c = 0.33 g/100 mL; EtOAc).
(2S,3R)-3-(4-Carboxyphenyl)pyrrolidine-2-carboxylic acid trifluoro acetic acid (7)
The Boc protected diacid 12a (215 mg, 0.64 mmol, 1.00 equiv) was dissolved in DCM (5 mL) and TFA (4 mL) was added. The mixture was stirred for 18h at rt under argon atmosphere. The mixture was concentrated in vacuo to yield a clear, colorless foam. The mixture was dissolved in H2O (4 mL) and a white, amorph solid precipitated. The solid was dried to yield 112 mg. 1H NMR (300 MHz, NaOD in D2O) δ 1.48 (1H, m), 1.80 (1H, m), 2.59 (1H, m), 2.67 – 2.80 (2H, m), 3.00 (1H, d, J = 9 Hz), 6.94 (2H, d, J = 8 Hz), 7.39 (2H, d, J = 8 Hz) 13C NMR (75 MHz, NaOD in D2O) δ 36.0, 46.9, 51.7, 70.7, 128.1, 130.0, 135.0, 147.3, 176.1, 181.7. [a]22D: +21.97 (c = 0.47 g/100 mL; 2M NaOH).
Synthesis of Analogues 5e-i
3-Bromo-5-(methoxycarbonyl)phenyl)boronic acid (14)
Commercially available 3-borono-5-bromobenzoic acid 13 (500 mg, 2.04 mmol, 1 equiv.) was dissolved in anhydrous MeOH (20 mL, 0.1 M) to which H2SO4 (50 μL, 18 M) was added slowly. Resulting mixture was refluxed 20h and after cooling to room temperature concentrated to dryness in vacuo. The crude product was used without further purification in the next step. 1H NMR (400 MHz, DMSO) δ 8.37 (dd, J = 1.5, 1.0 Hz, 1H), 8.18 (dd, J = 2.1, 1.0 Hz, 1H), 8.08 (dd, J = 2.1, 1.6 Hz, 1H), 3.87 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.2, 141.0, 133.6, 132.9, 131.2, 121.6, 52.4. (C-B(OH)2 absent).
Methyl 3-bromo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (15)
Crude boronic acid 14 (9.69 g, 37.4 mmol, 1 equiv.) was suspended in Et2O (80 mL, 0.46 M) and pinacol (5.31 g, 44.9 mmol, 1.2 equiv.) was added. The reaction mixture was stirred at rt until it became a clear solution (1.5h). The mixture was partitioned between Et2O and brine. The aqueous layer was extracted with Et2O and combined organic phase dried over MgSO4, filtered and concentrated in vacuo. All attempts to separate excess pinacol were unsuccessful and thus the crude mixture was used in the next step. 1H NMR (400 MHz, DMSO) δ 8.19 – 8.17 (m, 1H), 8.17 (t, J = 1.8 Hz, 1H), 7.97 (dd, J = 2.1, 1.0 Hz, 1H), 3.88 (s, 3H), 1.31 (s, 12H).
3-Bromo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (16)
Crude methyl 3-bromo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (15) (11.42 g, 37.4 mmol, 1 equiv.) was dissolved in anhydrous THF (200 mL, 0.18 M) and cooled to 0 °C. After 10 minutes was added LiAlH4 (2.84 g, 74.8 mmol, 2 equiv.) in small portions. The reaction mixture was allowed to slowly warm up to rt and stirred for 14h. The reaction was quenched by slow addition of 1M HCl and the aqueous later was extracted three times with Et2O. The organic layers were combined, washed with 1M HCl, dried over MgSO4, filtered and concentrated in vacuo to yield the title compound as a white solid (8.93 g, 86% over three steps). 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, J = 2.0, 1.0 Hz, 1H), 7.70 – 7.67 (m, 1H), 7.64 (dd, J = 1.7, 0.8 Hz, 1H), 4.68 (s, 2H), 1.34 (s, 12H). 13C NMR (101 MHz, DMSO) δ 145.2, 134.7, 131.8, 131.1, 121.5, 84.1, 61.9, 24.6. C-B is absent.
((3-Bromo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl)oxy)(tert-utyl)dimethylsilane (17)
Alcohol (16) (26.2 g, 84 mmol, 1 equiv.) was dissolved in anhydrous DMF (250 mL, 0.34 M). To the clear solution was added tert-butyldimethylsilyl chloride (TBDMSCl, 25.3 g, 168 mmol, 2 equiv.), imidazole (17.1 g, 251 mmol, 3 equiv.) and N,N-dimethylpyridine-4-amine (DMAP, 1.02 g, 8.3 mmol, 0.1 equiv.). The reaction mixture was stirred at rt for 21h. Then, the reaction was quenched with 1M HCl and the aqueous layer was extracted with EtOAc. The combined organic layers were washed 4 times with 1M HCl, dried over MgSO4, filtered and concentrated in vacuo to afford the title compound as yellowish syrup used without further purification due to instability on silica column chromatography (35.8 g, quantitative yield). 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 1.2 Hz, 1H), 7.63 – 7.61 (m, 1H), 7.59 – 7.58 (m, 1H), 4.71 (s, 2H), 1.34 (s, 12H), 0.94 (s, 9H), 0.10 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 143.3, 136.10, 132.1, 130.8, 122.7, 84.3, 64.3, 26.1, 25.8, 25.0, −5.11. C-B is absent.
(2S,3R)-tert-Butyl 3-(3-bromo-5-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)-2-(((tert-butyldimethylsilyl)oxy)methyl)-5-oxopyrrolidine-1-carboxylate (18)
In an optimized procedure, enone (10) (10.0 g, 30.5 mmol, 1 equiv.), boronic ester (17) (20.9 g, 48.9 mmol, 1.6 equiv.), [Rh(cod)Cl]2 (753 mg, 1.53 mmol, 0.05 equiv.) and Cs2CO3 (15.9 g, 48.9 mmol, 1.6 equiv.) were charged to a dry round-bottom flask with argon atmosphere. Contents were evacuated and purged with argon four times followed by addition of anhydrous, Ar-degassed THF (292 mL, 0.1 M) and subsequently Ar-degassed H2O (881 μL, 48.9 mmol, 1.6 equiv.). The reaction mixture was stirred at rt for 24h at which point it was partitioned between EtOAc and H2O after cooling to rt. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The obtained residue was purified by flash column chromatography (heptane:EtOAc, 20:1) to afford the title compound as colorless syrup (16.60 g, 86%). 1H NMR (600 MHz, CDCl3) δ 7.39 – 7.36 (m, 1H), 7.20 (t, J = 1.7 Hz, 1H), 7.03 (t, J = 1.8 Hz, 1H), 4.67 (s, 2H), 4.04 (dt, J = 3.8, 1.9 Hz, 1H), 3.99 (dd, J = 10.5, 3.8 Hz, 1H), 3.78 (dd, J = 10.5, 2.1 Hz, 1H), 3.41 (dt, J = 9.6, 2.0 Hz, 1H), 3.12 (dd, J = 18.0, 9.7 Hz, 1H), 2.49 (dd, J = 18.0, 2.6 Hz, 1H), 1.52 (s, 9H), 0.93 (s, 9H), 0.90 (s, 9H), 0.09 (s, 6H), 0.07 (s, 3H), 0.06 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 173.6, 145.0, 146.4, 144.8, 128.4, 128.1, 123.2, 122.6, 83.4, 66.6, 64.3, 63.8, 39.9, 38.7, 28.3, 26.1, 26.0, 18.6, 18.4, −5.11, −5.12, −5.34, −5.35.
(2S,3R)-tert-Butyl 3-(3-bromo-5-(((tert-butyldimethylsilyl)oxy)methyl)phenyl)-2-(((tert-butyldimethylsilyl)oxy)methyl)pyrrolidine-1-carboxylate (18a)
To a solution of pyrrolidinone (18) (236 mg, 375 μmol, 1 equiv.) in anhydrous THF (3 mL, 0.12 M) was added borane dimethylsulfide complex (107 μL, 10.5 M, equiv.) at rt followed by refluxing for 2h. The solution was diluted with Et2O and quenched with saturated NH4Cl solution. The organic layer was washed with H2O, brine, dried over MgSO4 and filtered through a plug of celite. The residue was purified using DCVC (0–17% EtOAc in heptane) to afford the title compound as colorless syrup (143 mg, 62%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 7.32 (s, 1H), 7.22 (s, 1H), 7.07 (s, 1H), 4.67 (s, 2H), 4.03 – 3.59 (m, 4H), 3.47 (dd, J = 12.1, 7.2 Hz, 1H), 3.40 – 3.27 (m, 1H), 2.25 (td, J = 12.6, 7.2 Hz, 1H), 1.94 – 1.81 (m, 1H), 1.48 (s, 9H), 0.94 (s, 9H), 0.88 (s, 9H), 0.10 (s, 6H), 0.04 (s, 3H), 0.04 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 154.3, (146.3 + 145.9), (144.1 + 144.1), (129.0 + 128.9), 127.4, (123.9 + 123.7), 122.7, (79.8 + 79.4), (65.5 + 65.4), 64.4, (63.1 + 61.7), (47.1 + 46.6), (46.5 + 45.5), (32.8 + 31.8), 28.7, 26.1, 26.0, (18.5 + 18.3), −5.11, −5.25.
(2S,3R)-tert-Butyl 3-(3-bromo-5-(hydroxymethyl)phenyl)-2-(hydroxymethyl)pyrrolidine-1-carboxylate (19)
To a solution of 18a (13.45 g, 26.8 mmol) in THF (250 mL, 0.1 M) was added TBAF (131 mL, 1 M, 5 equiv.) and the solution was stirred at rt for 7h. The reaction mixture was diluted with EtOAc, washed with saturated NaHCO3 and brine and subsequently dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash chromatography (heptane-EtOAc 1:2) to afford the title compound as a colorless oil (7.04 g, 68%). 1H NMR (600 MHz, CDCl3) δ 7.35 (s, 1H), 7.26 (s, 1H), 7.13 (s, 1H), 4.58 (s, 2H), 3.91 – 3.83 (m, 1H), 3.74 – 3.68 (m, 1H), 3.64 (d, J = 8.6 Hz, 1H), 3.57 (dd, J = 11.5, 6.4 Hz, 1H), 3.32 (dd, J = 16.1, 9.6 Hz, 1H), 2.93 – 2.82 (m, 1H), 2.17 – 2.08 (m, 1H), 1.96 – 1.85 (m, 1H), 1.47 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 156.7, 144.0, 143.4, 129.6, 128.6, 124.6, 122.9, 80.8, 66.8, 65.5, 64.1, 47.4, 47.1, 32.8, 28.6.
Suzuki cross coupling - General procedure A
Bromine 19 (294 mg, 761 μmoL, 1 equiv.) and boronic acid (3.0 mmol, 4 equiv.) were charged to a two-necked flask and contents were purged with N2. Pd(PPh3)4, (44 mg, 38 μmol), degassed PhMe (3.8 mL), degassed EtOH (1.7 mL) and Na2CO3 (1.7 mL, 2 M) were added under a stream of N2. The reaction mixture was stirred at 90 °C for 5.5h and then cooled to rt. The contents were transferred to brine and extracted four times with EtOAc. The organic layers were concentrated in vacuo and purified by flash column chromatography (EtOAc:MeOH, 19:1) to give desired product. *Common for these compounds was a strong tendency to form EtOAc containing solids regardless of drying duration.
Suzuki cross coupling - General procedure B
Bromine 19 (100 mg, 259 μmol, 1 equiv.), boronic acid (311 μmol, 1.2 equiv.), and Pd(PPh3)4 (15.0 mg, 13.0 μmol (0.05 equiv.) was charged to an argon-purged vial to which was added dioxane (1.7 mL). The contents were degassed with argon for 2 minutes before degassed Na2CO3 (647 μL, 1.28 mmol, 2.1 equiv.) was added and the vial was sealed with a septum-fitted cap. The reaction mixture was stirred at 95 °C for 4h and then cooled to rt. The contents were diluted with EtOAc, dried over MgSO4, filtered and concentrated in vacuo. The crude was purified by flash column chromatography (EtOAc:MeOH, 24:1) to afford the desired compound. Common for these compounds was a strong tendency to form EtOAc containing solids.
(2S,3R)-tert-Butyl-2-(hydroxymethyl)-3-(3-(hydroxymethyl)-5-(pyridin-2-yl)phenyl)pyrrolidine-1-carboxylate (20a)
Bromide 19 (257 mg, 665 μmol, 1 equiv.), MIDA-boronate, 6-methyl-2-(pyridin-2-yl)-1,3,6,2-dioxazaborocane-4,8-dione, (311 mg, 1.33 mmol, 2 equiv.), Pd(OAc)2 (7.5 mg, 33 μmol, 0.05 equiv.) and Xantphos (39 mg, 66.5 μmol, 0.1 equiv.) were charged to a round bottom flask. The contents were evacuated and backfilled with argon four times followed by addition of degassed dioxane (8.3 mL, 0.08 M). Degassed K3PO4 solution (1.66 mL, 3 M, 7.5 equiv.) was added and the resulting suspension was stirred at 50 °C for 20h. After allowing the reaction mixture to cool to rt, it was diluted with EtOAc and the organic layer was washed with brine. The aqueous layer was extracted with EtOAc three times. Subsequently, the organic layers were combined, dried over MgSO4, filtered and concentrated in vacuo. The resulting crude was purified by flash column chromatography (EtOAc:MeOH, 19:1) to afford the title compound as white foam (78 mg, 31%*, inseparable from PPh3 by-product, 13% compared to product). 1H NMR (400 MHz, CDCl3) δ 8.65 (ddd, J = 4.8, 1.6, 1.0 Hz, 1H), 7.79 (s, 1H), 7.77 (s, 1H), 7.75 – 7.61 (m, 2H), 7.29 (s, 1H), 7.25 – 7.20 (m, 1H), 4.71 (s, 2H), 4.01 (s, 1H), 3.86 – 3.69 (m, 2H), 3.63 (dd, J = 11.6, 6.5 Hz, 1H), 3.35 (ddd, J = 10.9, 9.9, 6.5 Hz, 1H), 3.06 – 2.93 (m, 1H), 2.22 – 2.11 (m, 1H), 2.02 – 1.94 (m, 1H), 1.49 (s, 9H).
(2S,3R)-tert-Butyl-2-(hydroxymethyl)-3-(3-(hydroxymethyl)-5-(pyridin-3-yl)phenyl)pyrrolidine-1-carboxylate (20b)
General procedure A. White solid (184 mg, 63%). 1H NMR (600 MHz, CDCl3) δ 8.83 (s, 1H), 8.61 (d, J = 4.0 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.47 (s, 1H), 7.38 (dd, J = 7.7, 4.8 Hz, 1H), 7.36 (s, 1H), 7.32 (s, 1H), 4.78 (s, 2H), 4.01 (t, J = 8.0 Hz, 1H), 3.78 (m, 2H), 3.66 (dd, J = 11.0, 6.7 Hz, 1H), 3.39 (td, J = 10.6, 6.5 Hz, 1H), 2.99 (dd, J = 15.8, 7.2 Hz, 1H), 2.24 – 2.17 (m, 1H), 1.85 – 1.80 (m, 1H), 1.51 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 148.8, 148.4, 142.6, 138.9, 138.9, 136.5, 134.8, 126.0, 125.9, 124.8, 123.9, 123.8, 80.9, 67.4, 66.1, 65.2, 48.1, 47.3, 32.0, 28.6.
(2S,3R)-tert-Butyl 2-(hydroxymethyl)-3-(3-(hydroxymethyl)-5-(pyridin-4-yl)phenyl)pyrrolidine-1-carboxylate (20c)
General procedure A. White solid (249 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 8.49 (dd, J = 4.7, 1.4 Hz, 2H), 7.46 (s, 1H), 7.41 (dd, J = 4.6, 1.6 Hz, 2H), 7.32 (s, 1H), 7.29 (s, 1H), 4.69 (s, 2H), 3.99 – 3.90 (m, 1H), 3.75 – 3.66 (m, 2H), 3.61 (dd, J = 11.5, 5.9 Hz, 1H), 3.38 – 3.28 (m, 1H), 3.09 – 2.97 (m, 1H), 2.22 – 2.11 (m, 1H), 1.98 – 1.87 (m, 1H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 150.5, 149.8, 148.4, 143.3, 138.6, 126.7, 125.1, 124.2, 121.8, 121.5, 80.6, 66.9, 65.2, 64.3, 47.5, 47.1, 32.8, 28.5.
(2S,3R)-tert-Butyl-2-(hydroxymethyl)-3-(3-(hydroxymethyl)-5-(pyrimidin-5-yl)phenyl)pyrrolidine-1-carboxylate (20d)
General procedure B. White foam (81 mg, 81%). 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H), 8.86 (s, 2H), 7.43 (s, 1H), 7.34 (s, 1H), 7.30 (s, 1H), 5.05 (broad s, 1H), 4.74 (s, 2H), 3.94 (m, 1H), 3.81 – 3.66 (m, 2H), 3.62 (dd, J = 11.4, 6.2 Hz, 1H), 3.36 (ddd, J = 10.9, 9.7, 6.6 Hz, 1H), 3.32 – 3.26 (broad s, 1H), 3.02 (m, 1H), 2.18 (m, 1H), 2.00 – 1.94 (m, 1H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.6, 156.7, 155.0, 143.5, 142.8, 135.0, 134.3, 126.6, 125.5, 124.3, 80.8, 67.1, 65.5, 64.5, 47.6, 47.2, 33.0, 28.6.
(2S,3R)-tert-Butyl 2-(hydroxymethyl)-3-(3-(hydroxymethyl)-5-(2-methoxypyrimidin-5-yl)phenyl)-pyrrolidine-1-carboxylate (20e)
General procedure B. White foam (90 mg 84%). 1H NMR (600 MHz, CDCl3) δ 8.70 (s, 2H), 7.41 (s, 1H), 7.31 (s, 1H), 7.28 (s, 1H), 4.95 (broad s, 1H), 4.78 (d, J = 5.4 Hz, 2H), 4.07 (s, 3H), 4.02 – 3.97 (m, 1H), 3.88 – 3.73 (m, 2H), 3.66 (dd, J = 10.5, 7.7 Hz, 1H), 3.39 (td, J = 10.6, 6.4 Hz, 1H), 2.99 (dd, J = 15.6, 8.1 Hz, 1H), 2.24 – 2.18 (m, 1H), 2.07 – 1.98 (m, 1H), 1.79 (t, J = 5.8 Hz, 1H), 1.51 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 165.4, 157.6, 142.9, 135.6, 132.3, 132.2, 132.1, 128.7, 128.6, 128.1, 80.9, 67.4, 66.1, 65.2, 55.3, 48.1, 47.3, 33.2, 28.6.
Oxidation of alcohols - General procedure C
NaIO4 (1.13 g, 5.28 mmol, 8.2 equiv.) and RuCl3 xH2O (8.0 mg, 3.9 μmol, 0.006 equiv.) in H2O (8.0 mL, 0.08 M) was added to a stirred solution of the corresponding diol (645 μmol, 1 equiv.) in MeCN/EtOAc (4.5 mL/4.5 mL (0.07 M) at 0 °C. The mixture was stirred at 0 °C for 2h and then diluted with EtOAc. MgSO4 was added at 0 °C and the suspension was filtered through a plug of silica. The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography (EtOAc-MeOH 19:1 + 1% AcOH) to afford the desired compound unless otherwise stated.
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-2-yl)phenyl)pyrrolidine-2-carboxylic acid
General procedure C. White solid (54%). Rotamer 1: 1H NMR (600 MHz, MeOD) δ 8.64 (d, J = 4.7 Hz, 1H), 8.50 (d, J = 1.4 Hz, 1H), 8.15 (s, 1H), 8.04 (s, 1H), 7.93 – 7.89 (m, 2H), 7.39 (ddd, J = 10.6, 7.0, 3.9 Hz, 1H), 4.31 (d, J = 6.9 Hz, 1H), 3.73 (ddt, J = 12.6, 8.6, 4.3 Hz, 1H), 3.69 – 3.63 (m, 1H), 3.63 – 3.57 (m, 1H), 2.44 – 2.35 (m, 1H), 2.21 – 2.13 (m, 1H), 1.45 (s, 9H). Rotamer 2: 1H NMR (600 MHz, MeOD) δ 8.64 (d, J = 4.7 Hz, 1H), 8.50 (d, J = 1.4 Hz, 1H), 8.14 (s, 1H), 8.03 (s, 1H), 7.93 – 7.89 (m, 2H), 7.39 (ddd, J = 10.6, 7.0, 3.9 Hz, 1H), 4.38 (d, J = 5.9 Hz, 1H), 3.73 (ddt, J = 12.6, 8.6, 4.3 Hz, 1H), 3.69 – 3.63 (m, 1H), 3.63 – 3.57 (m, 1H), 2.44 – 2.35 (m, 1H), 2.21 – 2.13 (m, 1H), 1.50 (s, 9H). 13C NMR (151 MHz, MeOD) δ (176.2, 175.9), (169.6, 169.5), (156.1, 155.8), 150.5, (144.0, 143.5), (141.2, 139.1), (139.0, 138.9), 133.6, (131.3, 131.2), (130.0, 129.9), (129.8, 129.2), (128.2, 128.1), 126.3, (122.6, 122.6), (81.9, 81.5), (67.6, 67.0), (51.2, 50.1), (47.4, 47.1), (33.9, 33.4), (28.8, 28.6).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-3-yl)phenyl)pyrrolidine-2-carboxylic acid
General procedure C. White solid (28%). Rotamer 1: 1H NMR (600 MHz, MeOD) δ 8.84 (s, 1H), 8.55 (d, J = 4.3 Hz, 1H), 8.20 – 8.16 (m, 1H), 8.13 (s, 1H), 8.02 (s, 1H), 7.83 (s, 1H), 7.54 (dd, J = 7.7, 5.0 Hz, 1H), 4.28 (d, J = 7.0 Hz, 1H), 3.75 – 3.68 (m, 1H), 3.65 (dd, J = 15.0, 7.1 Hz, 1H), 3.59 (dd, J = 17.5, 8.5 Hz, 1H), 2.44 – 2.34 (m, 1H), 2.20 – 2.11 (m, 1H), 1.44 (s, 9H). Rotamer 2: 1H NMR (600 MHz, MeOD) δ 8.84 (s, 1H), 8.55 (d, J = 4.3 Hz, 1H), 8.20 – 8.16 (m, 1H), 8.15 – 8.13 (m, 1H), 8.01 (s, 1H), 7.81 (s, 1H), 7.54 (dd, J = 7.7, 5.0 Hz, 1H), 4.36 (d, J = 5.9 Hz, 1H), 3.75 – 3.68 (m, 1H), 3.65 (dd, J = 15.0, 7.1 Hz, 1H), 3.59 (dd, J = 17.5, 8.5 Hz, 1H), 2.44 – 2.34 (m, 1H), 2.20 – 2.11 (m, 1H), 1.49 (s, 9H). 13C NMR (151 MHz, MeOD) δ (176.7, 176.3), (169.9, 169.8), (156.1, 155.8), (149.2, 148.4), (144.6, 143.9), (139.3, 137.7), (136.8 136.7), 134.48, (131.3, 131.2), (129.9, 129.2), (129.2, 129.1), (128.1, 128.0), 126.3, 125.5, (81.8, 81.5), (67.9, 67.3), (51.1, 50.0), (47.3, 47.1), (33.8, 33.3), (28.8, 28.6).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-4-yl)phenyl)pyrrolidine-2-carboxylic acid
General procedure C. Yellow syrup (35%). Rotamer 1: 1H NMR (600 MHz, MeOD) δ 8.61 (d, J = 4.4 Hz, 2H), 8.26 (d, J = 1.3 Hz, 1H), 8.06 (s, 1H), 7.92 (s, 1H), 7.76 (d, J = 5.9 Hz, 2H), 4.28 (d, J = 7.1 Hz, 1H), 3.75 – 3.68 (m, 1H), 3.66 (dd, J = 15.3, 7.0 Hz, 1H), 3.59 (dt, J = 10.2, 7.8 Hz, 1H), 2.43 – 2.35 (m, 1H), 2.20 – 2.12 (m, 1H), 1.44 (s, 9H). Rotamer 2: 1H NMR (600 MHz, MeOD) δ 8.61 (d, J = 4.4 Hz, 2H), 8.26 (d, J = 1.3 Hz, 1H), 8.05 (s, 1H), 7.90 (s, 1H), 7.76 (d, J = 5.9 Hz, 2H), 4.36 (d, J = 6.0 Hz, 1H), 3.75 – 3.68 (m, 1H), 3.66 (dd, J = 15.3, 7.0 Hz, 1H), 3.59 (dt, J = 10.2, 7.8 Hz, 1H), 2.43 – 2.35 (m, 1H), 2.20 – 2.12 (m, 1H), 1.49 (s, 9H). 13C NMR (151 MHz, MeOD) δ (176.5, 176.1), (169.6, 169.5), (156.1, 155.8), (150.6, 149.8), (144.6, 144.0), 139.6, 134.5, (131.3, 131.1), (130.2, 130.1), (128.1, 128.0), 123.3, (81.9, 81.5), (67.8, 67.2), (51.1, 50.0), (47.4, 47.1), (33.8, 33.3), (28.8, 28.6).
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid
General procedure C. Used in the next step without purification.
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(2-methoxypyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid
General procedure C. Used in the next step without purification.
(2S,3R)-3-(3-Carboxy-5-(pyridin-2-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5e)
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-2-yl)phenyl)pyrrolidine-2-carboxylic acid (37 mg, 90 μmol, 1equiv.) was dissolved in anhydrous DCM (1.8 mL) and cooled to 0 °C. After 5 minutes TFA (1.8 mL) was added dropwise to the stirred solution. The solution was allowed to warm to rt and stirred 2h at ambient temperature. The volatiles were removed under reduced pressure and the residue was dissolved in 1M HCl and concentrated in vacuo. This procedure was repeated times followed by lyophilization to afford the title compound as an off-white solid (30.6 mg, 98%). 1H NMR (400 MHz, D2O) δ 8.84 (ddd, J = 6.0, 1.6, 0.7 Hz, 1H), 8.70 (td, J = 8.0, 1.6 Hz, 1H), 8.41 (t, J = 1.6 Hz, 1H), 8.35 (dd, J = 8.1, 1.0 Hz, 1H), 8.32 (t, J = 1.6 Hz, 1H), 8.17 (t, J = 1.8 Hz, 1H), 8.09 (ddd, J = 7.4, 5.9, 1.2 Hz, 1H), 4.55 (d, J = 10.0 Hz, 1H), 3.91 (td, J = 10.3, 7.4 Hz, 1H), 3.77 (ddd, J = 11.6, 8.2, 3.2 Hz, 1H), 3.61 (ddd, J = 11.8, 10.2, 6.9 Hz, 1H), 2.68 (dtd, J = 13.9, 7.0, 3.2 Hz, 1H), 2.38 (dtd, J = 13.3, 10.4, 8.2 Hz, 1H). 13C NMR (151 MHz, D2O) δ 170.8, 168.6, 150.9, 147.4, 141.5, 141.0, 132.2, 132.1, 132.0, 131.9, 128.6, 126.5, 126.1, 64.9, 47.5, 45.8, 33.0. LC-MS (m/z) calcd for C17H17N2O4 [M+H]+, 313.2, found 313.3
(2S,3R)-3-(3-Carboxy-5-(pyridin-3-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5f)
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-3-yl)phenyl)pyrrolidine-2-carboxylic acid (54 mg, 131 μmol, 1 equiv.) was dissolved in anhydrous DCM (2.6 mL) and cooled to 0 °C. After 5 min TFA (2.6 mL) was added dropwise. The solution was allowed to warm up to rt and was stirred for 2h at ambient temperature. The volatiles were removed under reduced pressure and the residue was dissolved in 1M HCl and concentrated in vacuo. The brown solid was purified by preparative HPLC with subsequent triplicate evaporation from 5 mL 1M HCl followed by lyophilization to afford title compound as a white solid (18 mg, 39%). 1H NMR (600 MHz, D2O) δ 9.11 (d, J = 2.1 Hz, 1H), 8.89 (dt, J = 8.3, 1.7 Hz, 1H), 8.83 (d, J = 5.8 Hz, 1H), 8.28 (t, J = 1.6 Hz, 1H), 8.21 – 8.17 (m, 2H), 8.04 (t, J = 1.6 Hz, 1H), 4.56 (d, J = 10.0 Hz, 1H), 3.88 (td, J = 10.3, 7.4 Hz, 1H), 3.76 (ddd, J = 11.6, 8.3, 3.2 Hz, 1H), 3.60 (ddd, J = 11.9, 10.2, 6.9 Hz, 1H), 2.66 (dtd, J = 14.0, 7.1, 3.2 Hz, 1H), 2.42 – 2.32 (m, 1H). 13C NMR (151 MHz, D2O) δ 170.7, 169.1, 145.0, 140.5, 140.0, 139.4, 139.1, 134.7, 131.8, 131.7, 129.9, 128.0, 127.6, 64.8, 47.6, 45.8, 33.0. LC-MS (m/z) calcd for C17H17N2O4 [M+H]+, 313.2, found 313.3.
(2S,3R)-3-(3-Carboxy-5-(pyridin-4-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5g)
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyridin-4-yl)phenyl)pyrrolidine-2-carboxylic acid) (75 mg, 182 μmol) was dissolved in anhydrous DCM (3.6 mL) and cooled to 0 °C. After 5 minutes TFA (3.6 mL) was added dropwise to the stirring solution. The solution was allowed to warm to rt and was stirred 2h at ambient temperature. The volatiles were removed under reduced pressure and the residue was dissolved in 1M HCl and concentrated three times. The brown solid was purified by preparative HPLC with subsequent triplicate evaporation from 5 mL 1M HCl followed by lyophilization to afford the title compound as a white solid (18 mg, 28%). 1H NMR (400 MHz, D2O) δ 8.87 (d, J = 6.8 Hz, 2H), 8.46 (s, 1H), 8.39 (d, J = 6.9 Hz, 2H), 8.29 (s, 1H), 8.20 (s, 1H), 4.54 (d, J = 9.9 Hz, 1H), 3.90 (td, J = 10.3, 7.4 Hz, 1H), 3.78 (ddd, J = 11.5, 8.2, 3.1 Hz, 1H), 3.62 (ddd, J = 11.6, 10.3, 6.9 Hz, 1H), 2.68 (dtd, J = 10.2, 7.0, 3.1 Hz, 1H), 2.46 – 2.34 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.0, 168.8, 156.6, 141.3, 140.8, 135.6, 132.2, 131.9, 131.4, 128.5, 124.7, 65.0, 47.6, 45.8, 33.0. LC-MS (m/z) calcd for C17H17N2O4 [M+H]+, 313.2, found 313.3.
(2S,3R)-3-(3-Carboxy-5-(pyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5h)
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(pyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid was dissolved in anhydrous DCM (6 mL) and was cooled to 0 °C. After 5 minutes TFA (4 mL) was added dropwise to the stirring solution. The solution was allowed to warm to rt and stirred 2.5h at ambient temperature. The volatiles were removed under reduced pressure and the residue was purified by preparative HPLC with subsequent triplicate evaporation from 5 mL 1M HCl followed by lyophilization to afford title compound as a white solid (16 mg, 4% from 20d). 1H NMR (600 MHz, D2O) δ 9.32 (s, 1H), 9.22 (d, J = 1.4 Hz, 2H), 8.00 – 7.98 (m, 2H), 7.89 (t, J = 1.7 Hz, 1H), 4.47 (d, J = 9.9 Hz, 1H), 3.76 (td, J = 10.3, 7.5 Hz, 1H), 3.66 (ddd, J = 11.7, 8.2, 3.2 Hz, 1H), 3.50 (ddd, J = 12.0, 10.3, 6.8 Hz, 1H), 2.54 (dtd, J = 13.9, 7.1, 3.1 Hz, 1H), 2.23 (dtd, J = 13.5, 10.4, 8.2 Hz, 1H). 13C NMR (151 MHz, D2O) δ 170.2, 168.5, 154.9, 152.3, 140.5, 134.0, 132.3, 131.7, 131.7, 130.1, 127.6, 64.4, 47.2, 45.8, 32.9. LC-MS (m/z) calcd for C16H16N3O4 [M+H]+, 314.4, found 314.3.
(2S,3R)-3-(3-Carboxy-5-(2-hydroxypyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5i)
(2S,3R)-1-(tert-Butoxycarbonyl)-3-(3-carboxy-5-(2-methoxypyrimidin-5-yl)phenyl)pyrrolidine-2-carboxylic acid) was suspended in anhydrous DCM (5 mL). To the solution was added dropwise BBr3 (5 mL) maintaining rt. The mixture was stirred at rt for 2h and quenched with H2O. The resulting aqueous layer was concentrated under reduced pressure and purified by preparative HPLC with subsequent triplicate evaporation from 5 mL 1M HCl followed by lyophilization. The obtained product was recrystallized from H2O/acetone to afford the title compound as a white solid (18 mg, 4% from 20e). 1H NMR (600 MHz, D2O) δ 8.91 (s, 2H), 8.18 (t, J = 1.7 Hz, 1H), 8.14 (t, J = 1.5 Hz, 1H), 7.87 (t, J = 1.6 Hz, 1H), 4.41 (d, J = 9.7 Hz, 1H), 3.82 – 3.77 (m, 1H), 3.74 (ddd, J = 11.6, 8.1, 3.2 Hz, 1H), 3.59 (ddd, J = 12.3, 10.3, 6.8 Hz, 1H), 2.64 (dtd, J = 13.8, 7.0, 3.1 Hz, 1H), 2.36 (dtd, J = 13.5, 10.3, 8.1 Hz, 1H). 13C NMR (151 MHz, D2O) δ 171.6, 169.5, 156.5, 153.9, 153.2, 140.6, 132.8, 131.7, 130.5, 128.4, 126.7, 65.5, 47.9, 45.7, 33.0. LC-MS (m/z) calcd for C16H16N3O5 [M+H]+, 330.4, found 330.3.
Synthesis of Analogues (±)-5j-m
3-Methoxycarbonyl-5-nitrophenylboronic acid (23)
3-Borono-5-nitrobenzoic acid (1 g, 4.74 mmol, 1 equiv.) was loaded in a flask and 10 mL MeOH (0.5 M) were added. The clear solution was cooled on ice and SOCl2 (0.41 mL, 5.67 mmol, 1.2 equiv.) was added dropwise over 10 minutes. After 10 minutes the ice bath was removed and the reaction was allowed to stir at room temperature overnight. The reaction became a thick white suspension. Water (60 mL) was added, followed by 60 mL EtOAc. The layers were separated and the aqueous layer was extracted with EtOAc (3 × 60 mL). The combined organic layers were dried over Mg2SO4, filtered and concentrated in vacuo. The crude product was pure enough and not further purified. The title compound was obtained as a white solid (0.95 g, 89%). 1H NMR (600 MHz, DMSO) δ 8.84 (dd, J = 2.2, 1.1 Hz, 1H), 8.76 (dd, J = 1.4, 1.1 Hz, 1H), 8.66 (dd, J = 2.2, 1.4 Hz, 1H), 3.94 (s, 4H). 13C NMR (151 MHz, DMSO) δ 164.8, 147.6, 140.4, 132.4, 130.5, 125.0, 52.7.
1-Benzyl 2-methyl 4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (21)
Synthesis was performed according to known literature procedure already reported.47
1-Benzyl-2-methyl-3-bromo-4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (22)
A flame-dried round-bottom flask with addition funnel was charged with 1-benzyl 2-methyl 4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (21) (0.5 g, 1.91 mmol, 1 equiv.). To this was added 4 mL anhydrous MeCN, followed by DABCO (0.43 g, 3.82 mmol, 2 equiv.) and the reaction mixture was cooled to −15 °C. A solution of NBS in 6 mL anhydrous MeCN was added slowly over 30 minutes and the reaction mixture was allowed to slowly warm up to room temperature. After 16h the resulting mixture was concentrated in vacuo to a volume of 1–2 mL and directly loaded on the column. The crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc 4:1) to provide the title compound as colorless oil (0.52 g, 80%). 1H NMR (600 MHz, CDCl3) δ 7.39 – 7.28 (m, 5H), 5.13 (s, 2H), 4.00 (dd, J = 9.7, 8.7 Hz, 2H), 3.77 – 3.54 (m, 3H), 2.94 (dd, J = 9.7, 8.7 Hz, 2H).13C NMR (151 MHz, CDCl3) δ 161.8, 152.5, 135.6, 128.7, 128.6, 128.5, 100.1, 68.3, 52.6, 47.5, 36.4. LC-MS (m/z) calcd for C14H14BrNO4 [M+H]+ 340.0 and 342.0, found 340.0 and 342.0.
1-Benzyl 2-methyl 3-(3-(methoxycarbonyl)-5-nitrophenyl)-4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (24)
To a round-bottom flask equipped with reflux condenser was added 1-benzyl 2-methyl 3-bromo-4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (22) (0.29 g, 0.84 mmol, 1 equiv.), Ar-B(OH)2 (23, 0.28 g, 1.3 mmol, 1.5 equiv.), Cs2CO3 (0.39 mg, 1.17 mmol, 1.4 equiv.) and PdCl2dppf·CH2Cl2 (68 mg, 0.08 mmol, 0.10 equiv.). The flask was evacuated and refilled with argon three times. Then THF (16 mL) and water (16 mL) were added and the reaction mixture was stirred at 85 °C until full consumption of the starting material (~3h). The reaction mixture was allowed to cool to down to room temperature and the organic layer was removed under reduced pressure. The aqueous layer was extracted with was EtOAc (3× 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (heptane:EtOAc 3:1) to afford the title compound as yellow solid (312 mg, 85 %). 1H NMR (600 MHz, CDCl3) δ 8.69 (dd, J = 2.1, 1.4 Hz, 1H), 8.30 (t, J = 2.0 Hz, 1H), 8.23 (t, J = 1.6 Hz, 1H), 7.41 – 7.32 (m, 5H), 5.19 (s, 2H), 4.09 (t, J = 9.4 Hz, 2H), 3.98 (s, 3H), 3.91 – 3.63 (bs, 3H), 3.14 (t, J = 9.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 164.7, 163.4, 152.2, 148.5, 135.7, 132.5, 132.0, 128.6, 128.6, 128.5, 124.5, 122.8, 68.3, 52.9, 46.4, 31.5. LC-MS (m/z) calcd for C22H20N2O8 [M+H]+ 441.1, found 441.1.
1-(tert-Butyl) 2-methyl (2S,3S)-3-(3-amino-5-(methoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxylate ((±)-cis-25)
To a flame-dried round-bottom flask containing 1-benzyl 2-methyl 3-(3-(methoxycarbonyl)-5-nitrophenyl)-4,5-dihydro-1H-pyrrole-1,2-dicarboxylate (24) (0.68 g, 1.53 mmol, 1 equiv.) was added Pd/C (68 mg, 10 wt%). The flask was evaporated and refilled with hydrogen gas three times. Then anhydrous MeOH (5.5 ml) was added and the reaction mixture was stirred at rt overnight. The reaction mixture was filtrated over celite rinsed with MeOH (100 mL) and concentrated to dryness to afford the free di amine as a yellow oil (411 mg, 95%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 7.24 – 7.23 (m, 1H), 7.20 (d, J = 1.9 Hz, 1H), 6.68 (t, J = 1.9 Hz, 1H), 4.03 (d, J = 8.5 Hz, 1H), 3.88 (d, J = 2.6 Hz, 4H), 3.74 (s, 2H), 3.56 (q, J = 8.2 Hz, 1H), 3.31 (s, 3H), 3.03 (ddd, J = 10.9, 8.5, 7.4 Hz, 1H), 2.24 – 2.17 (m, 1H), 2.14 – 2.07 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 173.5, 167.3, 146.6, 142.2, 131.1, 119.6, 118.8, 114.6, 66.0, 52.2, 51.6, 48.9, 46.9, 32.6.
The crude di amine intermediate (0.41 g, 1.48 mmol, 1 equiv.) was dissolved in 12 mL anhydrous DCM. To this was added Boc2O (0.325 g, 1.48 mmol, 1 equiv) and the reaction mixture was allowed to stir at rt overnight. The mixture was concentrated to dryness under reduced pressure and directly subjected to purification. The crude residue was purified by flash column chromatography on silica gel (heptane: EtOAc 1:1) to afford the title product as a white solid (0.47 g, 84%). 1H NMR (600 MHz, CDCl3) δ 7.26 – 7.19 (m, 2H), 6.69 (dt, J = 3.7, 2.1 Hz, 1H), 4.51 (d, J = 8.6 Hz, 0.26H), 4.43 (d, J = 8.7 Hz, 0.42H), 4.35 (d, J = 6.1 Hz, 0.11H), 4.21 (d, J = 6.8 Hz, 0.21H), 3.91 – 3.76 (m, 5H), 3.73 (m, 0.5H), 3.68 (s, 1H), 3.57 (m, 1.5H), 3.42 (m, 0.73H), 3.36 – 3.29 (m, 2.27H), 2.58 – 2.43 (m, 0.73H), 2.25 (m, 0.27H), 2.08 (dq, J = 12.0, 5.9 Hz, 0.73H), 2.04 – 1.95 (m, 0.27H), 1.45 (m, 3H), 1.38 (m, 6H). 13C NMR (151 MHz, CDCl3) δ 173.2, 171.8, 171.7, 167.2, 167.1, 154.4, 153.7, 147.1, 146.8, 146.7, 142.1, 138.4, 138.3, 131.7, 131.3, 131.2, 119.1, 119.1, 118.9, 118.7, 118.3, 117.9, 115.0, 114.9, 114.8, 80.4, 80.2, 80.2, 65.7, 65.1, 64.2, 63.8, 52.3, 52.2, 52.1, 51.6, 51.5, 49.9, 48.7, 47.9 47.0, 46.3, 46.2, 46.1, 45.8, 33.1, 32.4, 28.5, 28.4, 27.6. LC-MS (m/z) calcd for C19H26N2O6 [M+H]+ 379.2, found 379.5 (-Boc).
1-(tert-Butyl) 2-methyl (2S,3S)-3-(3-(methoxycarbonyl)-5-((4-methylphenyl)sulfonamido)phenyl)pyrrolidine-1,2-dicarboxylate ((±)-cis-26b)
A flame-dried vial was loaded with (±)-cis-25 (150 mg, 0.4 mmol, 1 equiv.) and TsCl (0.91 mg, 0.48 mmol, 1.2 equiv.) Then pyridine (0.5 mL) was added and the reaction mixture was stirred at rt overnight. The resulting mixture was diluted in EtOAc (10ml) and washed with water (3 × 15 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (heptane-EtOAc 2:1) to provide the title compound as a white solid (188 mg, 92%, mixture of rotamers). 1H NMR (400 MHz, DMSO) δ 10.54 (s, 1H), 7.72 – 7.67 (m, 3H), 7.54 (s, 1H), 7.42 – 7.40 (m, 2H), 7.27 (m, 1H), 4.38 (m, 1H), 3.82 (s, 3H), 3.81 – 3.75 (m, 1H), 3.70 – 3.62 (m, 1H), 3.59 – 3.58 (m, 1H), 3.36 – 2.89 (m, 1H), 3.05 – 2.98 (m, 2H), 2.32 (s, 3H), 2.24 – 2.14 (m, 1H), 2.05 – 2.00 (0.72H), 1.93 – 1.87 (m, 0.28H), 1.43–1.41 (m, 3H), 1.34 – 1.31 (m, 6H). LC-MS (m/z) for C26H32N2O8S [M+H]+ 533.2, found 433.1 (-Boc).
(2S,3R)-3-(3-Carboxy-5-(pyridine-2-sulfonamido)phenyl)pyrrolidine-2-carboxylic acid ((±)-cis-26c)
A flame-dried vial was loaded with (±)-cis-25 (150 mg, 0.4 mmol, 1 equiv.) and pyridine-2-sulfonyl chloride (0.11 g, 0.59 mmol, 1.5 equiv.). To this was added pyridine (0.48 mL) and the reaction mixture was stirred at rt overnight. The reaction mixture was diluted with EtOAc (10 mL) and washed with water (3 × 15 mL). The combined organic layers was dried over MgSO4, filtered and concentrated in vacuo. The crude material was purified by flash column chromatography on silica gel (heptane-EtOAc 2:1) to afford the title compounds as white solid (174 mg, 85%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 4.8 Hz, 1H), 7.95 (m, 1H), 7.86 (td, J = 7.7, 1.7 Hz, 1H), 7.69 – 7.64 (m, 2H), 7.49 (m, 1H), 7.32 (s, 1H), 7.16 (d, J = 9.3 Hz, 1H), 4.49 (d, J = 8.6 Hz, 0.4H), 4.43 (d, J = 8.6 Hz, 1H), 3.86 (s, 3H), 3.85 – 3.78 (m, 1H), 3.67 – 3.56 (m, 1H), 3.48 – 3.39 (m, 1H), 3.14 (m, 3H), 2.49 (m, 1H), 2.13 – 2.03 (m, 1H), 1.45 (m, 4H), 1.38 (m, 5H).
1-(tert-Butyl) 2-methyl (2S,3S)-3-(3-benzamido-5-(methoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxylate ((±)-cis-26d)
To a flame-dried vial was added (±)-cis-25 (150 mg, 0.4 mmol, 1 equiv.), followed by pyridine (0.48 mL) and benzoyl chloride (51 μl, 0.44 mmol, 1.1 equiv.). The reaction mixture was stirred at room temperature overnight and then diluted with EtOAc (40 mL). The organic layer was washed with water (3× 15 mL) then dried over MgSO4, filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (heptane-EtOAc 2:1) to provide the title compound as a white solid (175 mg, 92%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 8.14 – 8.07 (m, 1H), 7.94 – 7.87 (m, 3H), 7.72 – 7.69 (m, 1H), 7.60 – 7.57 (m, 1H), 7.54 – 7.50 (m, 2H), 4.60 (d, J = 8.7 Hz, 0.26H), 4.53 (d, J = 8.6 Hz, 0.46H), 4.41 (d, J = 7.2 Hz, 0.1H), 4.28 (d, J = 7.2 Hz, 0.16H), 3.93 (s, 3H), 3.73 (s, 1H), 3.93–3.59 (m, 2H), 3.55 – 3.45 (m, 1H), 3.38 – 3.34 (m, 2H), 2.66 – 2.57 (m, 0.7H), 2.38 – 2.33 (m, 0.3H), 2.22 – 2.09 (m, 1H), 1.48 (s, 3H), 1.41 (m, 6H). LC-MS (m/z) for C26H30N2O7 [M+H]+ 483.2, found 383.1 (-Boc).
(±)-2,3-trans-(3-Amino-5-carboxyphenyl)pyrrolidine-2-carboxylic acid dihydrochloride (5j)
Aniline (±)-cis-25 (50 mg, 0.1 mmol, 1 equiv.) was dissolved in THF (1 mL, 0.1 M) and cooled to −78 °C. After 10 minutes LDA (0.2 mL, 0.2 mmol, 1M) was added dropwise, followed by stirring at 0 °C for 10 min. Saturated NH4Cl was added to quench the reaction. 10 mL water was added and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (heptane-EtOAc 1:1) to afford the title compound as white foam (25 mg, 50%, d.r. 1.3:1 (trans:cis)).
The intermediate was dissolved in dioxane (1.9 mL) and to the solution was added 4N HCl (1.9 mL). The reaction mixture was refluxed for 20 hours and then concentrated to dryness under reduced pressure. The crude material was purified by preparative HPLC to afford the title compound as an off-white solid (15 mg, 35%). Rt: 4.15 min; flow: 20 mL min−1; gradient 0–25 % B (45 min); λ = 210 nm. 1H NMR (400 MHz, D2O) δ 8.14 (s, 1H), 7.96 (s, 1H), 7.66 (s, 1H), 4.24 (m, 1H), 3.73 (m, 2H), 3.58 (m, 1H), 2.62 (m, 1H), 2.35 (m, 1H). 13C NMR (151 MHz, D2O) δ 172.0, 168.7, 142.0, 132.8, 131.3, 128.7, 127.3, 123.2, 65.9, 47.8, 45.6, 32.9. LC-MS (m/z) for C12H14N2O4 [M+H]+ 251.1, found 251.0
(±)-2,3-trans-(3-Carboxy-5-((4-methylphenyl)sulfonamido)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5k)
To a flame-dried 2 mL vial containing (±)-cis-26b (50 mg, 0.1 mmol, 1 equiv.) was added 25wt% NaOMe in MeOH solution (20 μL) and MeOH (80 μl). The reaction mixture was stirred at 65 ºC for 24h. To the resulting mixture was added acetic acid and the solution was concentrated in vacuo. The obtained residue was directly used in the next step without further purification. The crude residue was dissolved in dioxane (1.9 mL) and to the solution was added 4N HCl (1.9 mL). The reaction mixture was refluxed for 20h and then concentrated to dryness under reduced pressure. The crude material was purified by preparative HPLC to afford the title compound (30 mg, 74%, d.r. 10:1). Rt: 18.82 min; flow: 20 mL min−1; gradient 0–100 % B (45 min); λ = 210 nm. 1H NMR (400 MHz, MeOD) δ 7.76 (s, 1H), 7.65 (m, 2H), 7.52 (t, J = 1.7 Hz, 1H), 7.51 (t, J = 1.9 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 4.33 (d, J = 9.6 Hz, 1H), 3.68 – 3.60 (m, 2H), 3.50 – 3.43 (m, 1H), 2.56 – 2.49 (m, 1H), 2.36 (s, 3H), 2.16 (m, 1H). 13C NMR (600 MHz, MeOD) δ 170.5, 168.7, 145.5, 141.9, 140.3, 137.9, 133.9, 130.8, 130.8, 128.4, 125.7, 125.7, 122.4, 66.0, 47.1, 34.6, 31.0, 21.6. LC-MS (m/z) for C19H20N2O6S [M+H]+ 405.10, found 405.1.
(±)-2,3-trans-3-(3-Carboxy-5-(pyridine-2-sulfonamido)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5l)
To a flame-dried 2 mL vial containing (±)-cis-26c (66 mg, 0.13 mmol, 1 equiv.) was added 25wt% NaOMe in MeOH solution (26 μL) and MeOH (100 μL) and the reaction mixture was stirred at 65 ºC for 24h. To the reaction mixture was added acetic acid (pH 4) and the solution was concentrated under reduced pressure. The crude material was used in the next step without further purification. The crude residue was dissolved in dioxane (1.4 mL) and 4M HCl (1.4 mL). The reaction mixture was stirred at reflux overnight and after cooling to rt it was concentrated to dryness in vacuo. The crude material was purified by preparative HPLC to provide the title compound as white solid (8 mg, 16%, d.r. 38:1). Rt: 26.25 min; flow: 20 mL min−1; gradient 0–20 % B (45 min); λ = 210 nm. 1H NMR (400 MHz, MeOD) δ 8.59 (dt, J = 4.7, 1.4 Hz, 1H), 7.94 – 7.88 (m, 2H), 7.69 (t, J = 1.6 Hz, 1H), 7.63 (dd, J = 2.1, 1.4 Hz, 1H), 7.53 (t, J = 1.9 Hz, 1H), 7.55 – 7.47 (m, 2H), 4.28 (d, J = 9.4 Hz, 1H), 3.63 – 3.52 (m, 2H), 3.46 – 3.36 (m, 1H), 2.47 (dtd, J = 14.1, 7.2, 3.3 Hz, 1H), 2.20 – 2.07 (m, 1H). 13C NMR (600 MHz, MeOD) δ 170.5, 168.7, 158.8, 151.3, 141.8, 140.0, 139.9, 133.8, 128.7, 125.8, 125.7, 124.0, 122.6, 66.0, 49.7, 47.1, 34.6. LC-MS (m/z) C17H17N3O6S [M-H]− 390.1, found 390.1.
(±)-2,3-trans-3-(3-Benzamido-5-carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (5m)
To a flame-dried 2ml vial was added (±)-cis-26d (50 mg, 0.10 mmol) followed by 25wt% NaOMe in MeOH solution (26 μL) and MeOH (100 μL). The reaction mixture was stirred at 65 ºC for 24h and subsequently concentrated under reduced pressure. The crude material was used in the next step without further purification. The crude intermediate (50 mg, 0.10 mmol, 1 equiv.) was added anhydrous MeOH (1.8 mL) and water (1.28 mL). The reaction was cooled to 0 °C and LiOH•H2O (44 mg, 1 mmol, 10 equiv.) was added in portions. The reaction was allowed to stir at room temperature until all starting material was consumed. The reaction mixture was concentrated to dryness and the residue was re-dissolved in water (15 mL). The aqueous layer was washed with DCM and then acidified to pH 3 with 1N HCl. The aqueous layer was extracted with EtOAc (3× 15 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The residue (26mg) was used in the next step without further purification. The crude residue was dissolved in anhydrous DCM (810 μL). To the solution was added TFA (650 μL) and the reaction was allowed to stir for 8 h at room temperature. The resulting mixture was concentrated to dryness and the residue was purified by preparative HPLC to afford the title compound as a single diastereomer as a white solid (8 mg, 20%). Rt: 19.58 min; flow: 20 mL min−1; gradient 0–70% B (50 min); λ = 210 nm. 1H NMR (400 MHz, MeOD) δ 8.15 (dt, J = 5.2, 2.0 Hz, 2H), 7.93 – 7.89 (m, 2H), 7.81 (s, 1H), 7.58 – 7.52 (m, 1H), 7.51 – 7.45 (m, 2H), 4.41 (d, J = 9.3 Hz, 1H), 3.76 – 3.66 (m, 1H), 3.64 – 3.55 (m, 1H), 3.51 – 3.40 (m, 1H), 2.61 – 2.51 (m, 1H), 2.32 – 2.19 (m, 1H). 13C NMR (151 MHz, MeOD) δ 170.7, 169.3, 169.2, 141.5, 141.1, 136.1, 133.6, 133.4, 129.9, 129.9, 128.9, 128.9, 125.8, 125.7, 122.9, 66.2, 49.8, 47.2, 34.7. LC-MS (m/z) for C19H18N2O5 [M+H]+ 355.1, found 355.0.
Synthesis key intermediate 28a
Methyl 3-bromo-5-iodobenzoate (31)
3-Bromo-5-iodobenzoic acid (3 g, 9.17 mmol, 1 equiv.) was dissolved in anhydrous MeOH (30 mL, 0.3 M) and cooled on an ice bath. After 10 minutes, SOCl2 (0.8 mL, 11.0 mmol, 1.2 equiv.) was added dropwise over 10 minutes. After stirring the mixture for another 10 minutes the ice bath was removed and the mixture was heated to reflux for 16h. The mixture was allowed to cool down to rt and subsequently concentrated to dryness. The residue was resuspended in EtOAc and washed three times with 1N NaOH. The organic layer was dried over MgSO4, filtered and concentrated to give the title compound as a white solid (2.85 g, 91%). The product was used in the next step without further purification. 1H NMR (600 MHz, DMSO) δ: 8.27 (t, J = 1.7 Hz, 1H), 8.20 (t, J = 1.5 Hz, 1H), 8.04 (t, J = 1.6 Hz, 1H), 3.86 (s, 3H). 13C NMR (151 MHz, DMSO-) δ 163.7, 143.3, 136.5, 133.1, 131.0, 122.7, 960.1, 52.8.
(R)-tert-Butyl 2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (30)
(R)-1-(tert-Butoxycarbonyl)pyrrolidine-2-carboxylic acid (1 g, 4.64 mmol, 1 equiv.) was dissolved in anhydrous DCM (19 mL, 0.25 M) in a flame-dried flask with argon atmosphere. To this was added HOBt (0.75g, 5.57 mmol, 1.2 equiv.) and 8-aminoquinoline (0.80 g, 5.57 mmol, 1.2 equiv.) in one portion each. After stirring the mixture for 5 minutes at rt EDC·HCl (1.07 g, 5.57 mmol, 1.2 equiv.) was added in portion. The reaction mixture became brownish and was stirred for 24h at rt. The reaction mixture was diluted with DCM (50 mL) and saturated NaHCO3 (50 mL) was added. The aqueous layer was extracted with DCM (3 × 50 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The title compound was purified by flash column chromatography on silica gel (heptane-EtOAc 7:1 → 5:1) to afford the product as a light-yellow solid (1.35 g, 85%, mixture of rotamers). 1H NMR (400 MHz, DMSO) δ 10.42 (s, 0.45H), 10.27 (s, 0.55H), 8.64 (dd, J = 1.4 Hz, 1H), 8.42 dd, J = 8.3, 1.6 Hz, 1H), 7.70 – 7.67 (dd, J = 8.3, 1Hz, 1H), 7.64 (dd, J = 8.3, 4.2 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 4.50 (dd, J = 8.5, 4.1 Hz, 1H), 3.54 – 3.40 (m, 2H), 2.31 – 2.17 (bm, 1H), 2.12 – 1.99 (bm, 1H), 1.93 – 1.83 (m, 1H), 1.45 (s, 4 H), 1.23 (s, 5H); 13C NMR (151 MHz, DMSO) δ 171.5, 170.9, 154.2, 153.4, 148.9, 137.9, 136.6, 134.0, 127.8, 126.9, 122.2, 121.9, 116.2, 115.9, 79.3, 78.9, 61.2, 60.9, 46.9, 46.7, 30.9, 29.7, 28.1, 27.8, 24.0, 23.4. LC-MS (m/z) calcd for C19H23N3O3 [M+H]+ 342.2, found 342.5.
(2R,3R)-tert-Butyl 3-(3-bromo-5-(methoxycarbonyl)phenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (29)
To a sealed tube equipped with a stir bar and argon atmosphere was loaded 30 (0.6 g, 1.76 mmol, 1 equiv.), 31 (1.2 g, 3.52 mmol, 2 equiv.), Pd(OAc)2 (40 mg, 0.17 mmol, 0.1 equiv.), Ag2CO3 (0.49 g, 1.76 mmol, 1equiv.) and PivOH (54 mg, 0.53 mmol, 0.3 equiv.). To this was added anhydrous toluene (6 mL, 0.3 M) in one portion. The reaction mixture was placed in a preheated oil bath at 110 ˚C and stirred for 3d. The reaction mixture became dark brown. The reaction mixture was allowed to cool down to rt, filtered over a silica-celite pad and rinsed with 1:1 EtOAc-heptane. The filtrate was concentrated in vacuo. The title compound was obtained after flash column chromatography (heptane-EtOAc 5:1) as a light-yellow foam (0.43 g, 44% (yield range 40–48%), mixture of rotamers). 1H NMR (600 MHz, DMSO) δ 9.75 (s, 0.4H), 9.73 (s, 0.6H), 8.76 (s, 1H), 8.3 (d, J = 8.25 Hz, 1H), 8.26 (d, J = 7.3 Hz, 1H), 7.85 (s, 0.6H), 7.82 (s, 0.4H), 7.79 (s, 0.6H), 7.76 (s, 0.4H), 7.60 – 7.57 (m, 2H), 7.51 (s, 1H), 7.48 – 7.45 (t, J = 7.9 Hz, 1H), 5.00 (d, J = 8.0 Hz, 0.6 Hz), 4.95 (d, J = 8.0 Hz, 0.4H), 3.93 – 3.86 (m, 1H), 3.81 – 3.78 (m, 1H), 3.70 (s, 3H), 3.49 – 3.39 (bm, 1H), 2.46 (m, 1H), 2.14 (m, 1H), 1.44 (s, 3H), 1.26 (s, 6H); 13C NMR (151 MHz, DMSO) δ 169.9, 165.0, 153.5, 149.0, 141.3, 138.3, 136.9, 136.2, 134.0, 131.6, 130.4, 128.2, 128.1, 127.1, 122.4, 121.9, 117.0, 79.4, 65.2, 52.8, 47.2, 46.2, 40.6, 31.7, 28.8, 28.6, 28.4, 27.7, 22.6. LC-MS (m/z) calcd for C27H28BrN3O5 [M+H]+ 554.1 and 556.1, found 554.6 and 556.6.
(2S,3R)-3-(3-Bromo-5-carboxyphenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (32)
Intermediate 29 (1 g, 1.80 mmol, 1 equiv.) was dissolved in 18 mL (0.1 M) EtOH. To the yellow solution was added NaOH (0.72 g, 18.0 mmol, 10 equiv.). The reaction mixture was placed in a preheated oil bath at 100 ˚C and stirred at reflux for 24h. The reaction mixture became slurry and brown. The mixture was allowed to cool down to rt and then concentrated to dryness in vacuo. The residue was resuspended in EtOAc and water. The aqueous layer was extracted three times with EtOAc to remove 8-aminoquinoline. Then the aqueous layer was acidified with 1N HCl to pH 2–3 and was again extracted with EtOAc. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give the title compound as light-brown foam (0.67 g, 90%, mixture of rotamers). The synthesis was continued without further purification. 1H NMR (600 MHz, DMSO) δ 13.38 (bs, 1H), 12.66 (bs, 1H), 7.93 (s, 1H), 7.86 – 7.83 (m, 2H), 4.10 (d, J =7.7 Hz, 0.36 H), 4.07 (d, J = 7.7 Hz, 0.64H), 3.60 – 3.54 (m, 1H), 3.53 – 3.48 (m, 1H), 3.42 – 3.35 (m, 1H), 2.26 – 2.19 (m, 1H), 2.07 – 2.00 (m, 1H), 1.43 (s, 3 H), 1.36 (s, 6H). LC-MS (m/z) calcd for C17H20BrNO6 [M+H]+ 414.1 and 416.1, found 314.3 and 316.3 (-Boc).
(2S,3R)-Di-tert-butyl 3-(3-bromo-5-(tert-butoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxylate (28a)
In a flame-dried vial with argon atmosphere was loaded 32 (0.5 g, 1.21 mmol, 1 equiv.), followed by anhydrous DCM (2.4 mL, 0.5 M). To the brown solution was added TBTA (1.32 g, 6 mmol, 5 equiv.) in one portion. The vial was capped and the reaction was stirred at rt for 24h. During the reaction time a white precipitate was formed. After 24h the reaction mixture was concentrated to dryness in vacuo. The title compound was purified by flash column chromatography on silica gel (heptane-EtOAc 7:1) to afford the title compound as a white solid (0.48 g, 73%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.80 (s, 1H), 7.54 (s, 1H), 4.18 (d, J = 6.6 Hz, 0.3H), 4.08 (d, J = 6.6 Hz, 0.7H), 3.80 – 3.75 (m, 0.7H), 3.68 – 3.62 (m, 0.3H), 3.59 – 3.53 (m, 1H), 3.40 – 3.35 (m, 1H), 2.33 – 2.24 (m, 1H), 2.07 – 1.98 (m, 1H), 1.59 (s, 9H), 1.49–1.43 (m, 18H); 13C NMR (151 MHz, DMSO) δ 171.2, 164.3, 153.9, 143.6, 134.3, 131.4, 127.2, 122.7, 82.1, 81.7, 80.4, 66.2, 49.9, 48.5, 46.1, 32.1, 28.5, 28.3, 28.2, 22.8. LC-MS (m/z) calcd for C25H36Br NO6 [M+H]+ 526.2 and 528.2, found 314.3 and 316.3 (-Boc, −2 x tert-Bu).
Synthesis of 4a and 2,3-cis-4a tert-Butyl (2R,3R)-3-(3-(methoxycarbonyl)phenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (33)
To a seal tube equipped with a stir bar and argon atmosphere was loaded 30 (0.5 g, 1.46 mmol, 1 equiv.), methyl-3-iodobenzoate (0.77 g, 2.92 mmol, 2 equiv.), Pd(OAc)2 (32 mg, 0.15 mmol, 0.1 equiv.), Ag2CO3 (0.48 g, 1.75 mmol, 1.2 equiv.) and PivOH (45 mg, 0.44 mmol, 0.3 equiv.). To this was added anhydrous toluene (4.9 mL, 0.3 M) in one portion. The reaction mixture was placed in a preheated oil bath at 110 ˚C and stirred for 3d. The reaction mixture became dark brown. The reaction mixture was allowed to cool down to rt, filtered over a silica-celite pad and rinsed with 1:1 EtOAc-heptane. The filtrate was concentrated in vacuo. The title compound was obtained after flash column chromatography (heptane-EtOAc 5:1) as a white foam (0.43 g, 62% (mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 9.62 (bs, 1H), 8.66 (bs, 1H), 8.45 (s, 1H), 8.08 (bs, 1H), 7.96 (t, J = 1.8 Hz, 1H), 7.60 (m, 1H), 7.49 – 7.34 (m, 4H), 7.14 (m, 1H), 4.81 – 4.58 (m, 1H), 4.13 – 3.93 (m, 1H), 3.86 – 3.77 (m, 4H), 3.62 (m, 1H), 2.75 (m, 1H), 2.31 – 2.18 (m, 1H), 1.42 (m, 9H). 13C NMR (151 MHz, CDCl3) δ 168.9, 166.9, 154.3, 148.2, 138.4, 137.5, 136.2, 133.8, 132.5, 130.3, 129.6, 128.6, 128.5, 127.8, 127.3, 121.6, 116.5, 80.7, 66.7, 66.1, 52.1, 48.6, 46.3, 28.6, 28.4, 28.2. LC-MS (m/z) calcd for C27H29N3O5 [M+H]+ 476.2, found 476.6.
(2S,3R)-3-(3-Carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (4a)
Intermediate 33 (25 mg, 0.05 mmol, 1 equiv.) was dissolved in EtOH (0.27 mL, 0.2 M). To this was added NaOH (65 mg, 1.63 mmol, 30 equiv.) and the vial was sealed. The reaction mixture was placed in a preheated oil bath at 100 ˚C and stirred for 24h. The reaction mixture became slurry and brown. The mixture was allowed to cool down to room temperature and then concentrated to dryness in vacuo. The residue was resuspended in EtOAc and 1N NaOH. The aqueous layer was extracted with 3 x EtOAc to remove 8-aminoquinoline. Then the aqueous layer was acidified with 1N HCl to pH 2–3 and extracted three times with EtOAc. The organic layers of the second extraction were combined, dried over MgSO4 and concentrated in vacuo to give the di acid intermediate as a light-brown foam (10 mg, 55%). The obtained product was not further purified and used as crude in the next step. The intermediate was dissolved in DCM (1 mL) and TFA (1 mL) was added. The reaction mixture was stirred for 5h at rt and then concentrated to dryness. The crude residue was purified by preparative HPLC to afford a white solid. The solid was dissolved in 2 mL 1N HCl and concentrated to dryness under reduced pressure. This was repeated two more times and after lyophilization the title compound was obtained as a white solid (10 mg, 74%). Rt: 10.2 min (flow: 20 mL min−1; gradient 0–50% B (25 min); 1H NMR (600 MHz, D2O) δ 8.07 (t, J = 1.7 Hz, 1H), 8.02 (dt, J = 7.9, 1.3 Hz, 1H), 7.71 (dt, J = 7.9, 1.5 Hz, 1H), 7.60 (dd, J = 8.3, 7.1 Hz, 1H), 4.27 (d, J = 9.4 Hz, 1H), 3.73 – 3.67 (m, 2H), 3.57 (ddd, J = 11.5, 10.2, 6.9 Hz, 1H), 2.58 (dtd, J = 13.8, 7.1, 3.4 Hz, 1H), 2.38 – 2.24 (m, 1H). Analytical data in agreement with reported ones.36
(2R,3R)-3-(3-Carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (2,3-cis-4a)
Intermediate 33 (0.05 g, 0.1 mmol) was loaded in a vial to which was added 3 mL 40% aqueous H2SO4. The reaction was heated to 120 °C and stirred for 36h. After cooling down to rt 4N NaOH was added to reach pH 12. The aqueous layer was washes with DCM three times. After this the aqueous layer was acidified to pH4 and concentrated to dryness under reduced pressure. The crude residue was purified by preparative HPLC. Concentration afforded a white solid which was dissolved in 2 mL 1N HCl and concentrated to dryness under reduced pressure. This was repeated two more times and after lyophilization the title compound was obtained as a white solid (7 mg, 27%). Rt 21.4 min (flow: 20 mL min−1; gradient 0–50% B (45 min). 1H NMR (400 MHz, D2O) δ 8.00 (dt, J = 7.6, 1.5 Hz, 1H), 7.96 (t, J = 1.8 Hz, 1H), 7.61 (dt, J = 7.8, 1.5 Hz, 1H), 7.58 – 7.52 (m, 1H), 4.53 (d, J = 9.4 Hz, 1H), 4.07 (td, J = 9.4, 7.3 Hz, 1H), 3.86 (ddd, J = 11.9, 7.9, 4.0 Hz, 1H), 3.52 (ddd, J = 11.9, 9.4, 7.6 Hz, 1H), 2.62 – 2.53 (m, 1H), 2.44 (dtd, J = 13.4, 9.4, 8.0 Hz, 1H). 13C NMR (151 MHz, D2O) δ 171.0, 170.3, 137.9, 133.3, 123.0, 129.1, 129.0, 129.0, 64.8, 45.6, 45.3, 29.9.
Synthesis of analogs 4g-i, 5b-d and 6a,b
Methyl 2-bromo-5-iodobenzoate (36)
2-Bromo-5-iodobenzoic acid (2.5 g, 7.65 mmol, 1 equiv.) was dissolved in anhydrous MeOH (26 mL, 0.3 M) and cooled on an ice bath. After 10 minutes, SOCl2 (0.7 mL, 9.2 mmol, 1.2 equiv.) was added dropwise over 10 minutes. After stirring the mixture for another 10 minutes the ice bath was removed and the mixture was heated to reflux for 16h. The mixture was allowed to cool down to room temperature and subsequently concentrated to dryness. The residue was resuspended in EtOAc and washed three times with 1N NaOH. The organic layer was dried over MgSO4, filtered and concentrated to give the title compound as a white solid (1.69 g, 66%). The product was used in the next step without further purification. 1H NMR (600 MHz, DMSO) δ 8.06 (d, J = 2.2 Hz, 1H), 7.81 (dd, J = 8.4, 2.2 Hz, 1H), 7.53 (d, J = 8.4 Hz, 1H), 3.85 (s, 3H). 13C NMR (151 MHz, DMSO δ 164.8, 141.5, 138.8, 135.6, 134.2, 119.9, 93.3, 52.8.
Dimethyl 4-nitrophthalate
4-Nitrophthalate 37 (1 g, 4.73 mmol, 1 equiv.) was dissolved in anhydrous MeOH (6.3 mL, 0.75 M) and cooled on an ice bath. After 10 minutes, SOCl2 (0.8 mL, 10.6 mmol, 2.2 equiv.) was added dropwise over 10 minutes. After stirring the mixture for another 10 minutes the ice bath was removed and the mixture was heated to reflux for 16h. The mixture was allowed to cool down to rt and subsequently concentrated to dryness. The residue was re-suspended in EtOAc and washed three times with 1N NaOH. The organic layer was dried over MgSO4, filtered and concentrated to give the title compound as a yellow solid (0.78 g, 69%). The product was used in the next step without further purification. 1H NMR (600 MHz, CDCl3) δ 8.63 (d, J = 2.3 Hz, 1H), 8.39 (dd, J = 8.4, 2.3 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 3.97 (s, 3H), 3.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 166.8, 165.5, 148.9, 138.2, 132.8, 130.1, 126.2, 124.6, 53.4.
Dimethyl 4-aminophthalate
Dimethyl 4-nitrophthalate (0.44 g, 1.85 mmol, 1 equiv.) was dissolved in anhydrous MeOH (6 mL, 0.3 M). To this was added 10wt% Pd/C (0.044 g), followed by hydrogen gas balloon. The reaction was stirred vigorously at rt overnight. The mixture was filtered over a celite-silica plug and rinsed with EtOAc. The filtrate was concentrated in vacuo to yield the title compound as a sticky, colorless oil which crystalized upon standing (0.37 g, 95%). 1H NMR (600 MHz, CDCl3) δ: 7.72 (d, J = 8.4 Hz, 1H), 6.73 (d, J = 2.4 Hz, 1H), 6.68 (dd, J = 8.4, 2.4 Hz, 1H), 4.10 (bs, 2H), 3.90 (s, 3H), 3.84 (s, 3H). 13C NMR (151 MHz, CDCl3) δ: 169.8, 166.7, 149.9, 136.63, 132.1, 118.6, 115.2, 113.5, 52.8, 52.2. LC-MS (m/z) calcd for C10H11NO4 [M+H]+ 210.1 found 210.5.
Dimethyl 4-iodophthalate (38)
Dimethyl 4-aminophthalate (0.63 g, 3 mmol, 1 equiv.) was dissolved in 1 mL anhydrous acetonitrile and 19 mL anhydrous toluene. To the solution was added iodine (0.34 g, 3.28 mmol, 1.1 equiv.) and the dark red solution was cooled to 0 °C. After 15 minutes, tBuNO2 (0.53 mL, 1.79 mmol, 1.1 equiv.) was added dropwise over 15 minutes. The reaction mixture was stirred at 0 °C for 2 hours and then left stirring while slowly reaching rt overnight. After 24h the reaction mixture was concentrated to dryness and resuspended in EtOAc. The organic layer was washed twice with basic Na2S2O3 solution, followed by a wash with brine. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by flash column chromatography (heptane-EtOAc 10:1) to give the title compound as a yellow oil (440 mg, 46%). 1H NMR (600 MHz, CDCl3) δ 8.04 (s, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 8.0 Hz, 2H), 3.91 (s, 3H), 3.90 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 167.2, 166.6, 140.0, 137.6, 133.7, 130.9, 130.4, 52.9, 52.8. LC-MS (m/z) calcd for C10H9IO4 [M+H]+ 321.0 found 321.3.
3-Bromo-2-chloro-5-iodobenzoic acid
3-Bromo-2-chlorobenzoic acid 43 (2g, 8.55 mmol, 1 equiv.) was dissolved in 7 mL TfOH and then cooled on an ice bath. NIS (2.31 g, 10.3 mmol, 1.2 equiv.) was added in portions over 10 minutes and then stirred at rt overnight. To the reaction mixture was added EtOAc, followed by water and saturated Na2S2O3 solution. The water layer was extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The title compound was obtained as a white solid (3.05 g, 99%). The product was used in the next step without further purification. 1H NMR (600 MHz, DMSO) δ 8.29 (d, J = 2.0 Hz, 1H), 8.02 (d, J = 2.0 Hz, 1H). 13C NMR (151 MHz, DMSO) δ 165.1, 142.9, 137.2, 135.9, 130.8, 124.7, 93.3.
Methyl 3-bromo-2-chloro-5-iodobenzoate (44)
3-Bromo-2-chloro-5-iodobenzoic acid (2 g, 5.53 mmol, 1 equiv.) was dissolved in anhydrous MeOH (7.5 mL, 0.75 M) and cooled on an ice bath. After 10 minutes, SOCl2 (0.5 mL, 6.64 mmol, 1.2 equiv.) was added dropwise over 10 minutes. After stirring the mixture for another 10 minutes the ice bath was removed and the mixture was heated to reflux for 16h. The mixture was allowed to cool down to rt and subsequently concentrated to dryness. The residue was resuspended in EtOAc and washed three times with 1N NaOH. The organic layer was dried over MgSO4, filtered and concentrated to give the title compound as an off-white solid (1.46 g, 70%). The product was used in the next step without further purification. 1H NMR (600 MHz, DMSO) δ 8.35 (d, J = 2.1 Hz, 1H), 8.07 (d, J = 2.0 Hz, 1H), 3.86 (s, 3H). 13C NMR (151 MHz, DMSO) δ 163.9, 143.6, 137.7, 134.2, 131.1, 124.8, 93.4, 53.1.
C-H- activation-arylation – General Procedure D
To a seal tube equipped with a stir bar and argon atmosphere was added 30 (1 equiv.), ArI (2 equiv.), Pd(OAc)2 (0.1 equiv.), Ag2CO3 (1 equiv.) and PivOH (0.3 equiv.). To this was added anhydrous toluene (0.3M) in one portion. The reaction mixture was placed in a preheated oil bath at 110 ˚C and stirred for 3d. The reaction mixture became dark brown. The reaction mixture was allowed to cool down to room temperature and was filtered over a silica-celite pad and rinsed with EtOAc. The filtrate was concentrated in vacuo. The crude material was purified as stated below.
tert-Butyl (2R,3R)-3-(4-bromo-3-(methoxycarbonyl)phenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (34a)
General procedure D. The reaction was performed with 0.6 g (1.76 mmol) of 30 and aryl iodide 36. The title compound was obtained after flash column chromatography (heptane-EtOAc 5:1) as a light-yellow foam (0.6 g, 61%). 1H NMR (400 MHz, DMSO) δ 9.80– 9.74 (m, 1H), 8.79 (s, 1H), 8.35 (d, J = 7.2 Hz, 1H), 8.26 (dd, J = 7.7 Hz, 1.3Hz, 1H), 7.72 – 7.68 (m, 1H), 7.62 – 7.57 (m, 2H), 7.50 – 7.50 (m, 2H), 7.39 – 7.36 (m, 1H), 4.94 – 4.89 (m, 1H), 3.87 – 3.77 (m, 2H), 3.69 (s, 3H), 3.53 – 3.38 (m, 1H), 2.47 – 2.41 (m, 1H), 2.18 – 2.10 (m, 1H), 1.43 (s, 3H), 1.24 (s, 6H). 13C NMR (151 MHz, DMSO) δ 174.6, 174.1, 171.0, 158.7, 158.4, 153.9, 143.1, 143.0, 142.7, 142.6, 141.6, 138.7, 138.7, 138.1, 137.9, 136.8, 136.7, 136.0, 135.9, 132.8, 131.9, 127.2, 123.8, 121.8, 121.6, 84.4, 84.2, 69.8, 69.6, 57.5, 51.78 51.3, 51.0, 51.0, 33.6, 33.3, 33.1, 32.6. LC-MS (m/z) calcd for C27H28N3O5 [M+H]+ 554.1 and 556.01, found 554.6 and 556.1.
Dimethyl 4-((2R,3R)-1-(tert-butoxycarbonyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidin-3-yl)phthalate (34c)
General procedure D. The reaction was performed with 0.235 g (0.69 mmol) of 30 and aryl iodide 38. The title compound was obtained after flash column chromatography (heptane-EtOAc 1:1) as a light-yellow foam (0.13 g, 37%). 1H NMR (600 MHz, CDCl3) δ 9.86–9.80 (m, 1H), 8.78 (m, 1H), 8.33 (d, J = 8.1 Hz, 1H), 8.26 (d, J = 7.5 Hz),7.69 – 7.64 (m, 1H), 7.61 – 7.57 (m, 3H), 7.46 (m 2H), 4.99 – 4.93 (m, 1H) 3.94 – 3.88 (m, 1H), 3.82 – 3.79 (m, 1H), 3.66 (s, 3H), 3.65 (s, 3H), 3.54–3.41 (m, 1H), 2.23 – 2.14 (m, 1H), 1.43 (s, 3H), 1.24 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 169.2, 167.2, 166.8, 153.1, 148.7, 141.6, 137.9, 136.3, 133.5, 131.4, 131.16, 130.9, 129.6, 128.6, 128.65, 127.6, 126.6, 121.9, 116.5, 79.2, 79.0, 64.6, 52.3, 46.93, 45.8, 28.4, 28.1, 27.9, 27.3. LC-MS (m/z) calcd for C29H31lN3O7 [M+H]+ 534.2, found 534.7.
tert-Butyl (2R,3R)-3-(3-bromophenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate
General procedure D. The reaction was performed with 0.6 g (1.76 mmol) of 30 and 3-bromoiodobenzene. The title compound was obtained after flash column chromatography (heptane-EtOAc 5:1) as inseparable mixture of the title compound and 30 (0.44 g, 28% of 30)). The mixture was not further purified and used as such in the next step. 1H NMR (600 MHz, DMSO) δ 9.76–9.73 (m, 1H), 8.84 (s, 1H), 8.34 (d, J = 8.2 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 7.59 (m, 3H), 7.48 (t, J = 8.0 Hz, 1H), 7.29 (d, J = 7.8 Hz, 1H), 7.12 (d, 7.8 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 4.93 – 4.88 (m, 1H), 3.51 – 3.37 (m, 2H), 2.15 – 2.08 (m, 1H), 1.90 – 1.85 (m, 1H), 1.43 (3H), 1.23 (s, 6H). LC-MS (m/z) calcd for C25H26BrN3O3 [M+H]+ 496.1and 498.1 found 496.6 and 498.6 (as well as 30 (342.6)).
tert-Butyl (2R,3R)-3-(3-bromo-4-chloro-5-(methoxycarbonyl)phenyl)-2-(quinolin-8-ylcarba-moyl)-pyrrolidine-1-carboxylate (45a)
General procedure D. The reaction was performed with 0.6 g (1.76 mmol) of 30 and aryl iodide 44. The title compound was obtained after flash column chromatography (heptane-EtOAc, gradient 3:1 → 2:1) as a light-yellow foam (0.32 g, 31%). 1H NMR (600 MHz, CDCl3) δ 9.53 (s, 1H), 8.69 (s, 1H), 8.48 (d, J = 6.9 Hz, 1H), 8.12 (s, 1H), 7.65 (s, 1H), 7.57 (s, 1H), 7.49 – 7.42 (m, 3H), 4.73 – 4.59 (m, 1H), 4.09 – 3.93 (m, 1H), 3.72 (m, 4H), 3.59 (bs, 1H), 2.65 – 2.55 (m, 1H), 2.23 – 2.19 (m, 1H), 1.51 (s, 3H), 1.37 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 168.6, 165.4, 154.1, 148.4, 138.4, 137.3, 136.3, 136.1, 133.4, 132.4, 132.3, 129.6, 127.9, 127.2, 125.1, 122.1, 121.8, 116.9, 80.9, 66.5, 52.7, 47.9, 46.2, 28.6, 28.4. LC-MS (m/z) calcd for C27H27BrClN3O5 [M+H]+ 588.1 and 590.1, found 588.6 and 590.6.
Epimerization and Cleavage of directing group – General procedure E
The product of the C-H activation (1 equiv.) was dissolved in EtOH (0.1 M). To this was added NaOH (10 equiv.) and the vial was sealed. The reaction mixture was placed in a preheated oil bath at 100 ˚C and stirred for 24h. The reaction mixture became slurry and brown. The mixture was allowed to cool down to rt and then concentrated to dryness in vacuo. The residue was resuspended in EtOAc and 1N NaOH. The aqueous layer was extracted 3x with EtOAc to remove 8-aminoquinoline. Then the aqueous layer was acidified with 1N HCl to pH 2–3 and extracted three times with EtOAc. The organic layers of the second extraction were combined, dried over MgSO4 and concentrated in vacuo to give the title compound as a light-brown foam. The obtained product was not further purified.
(2S,3R)-3-(4-Bromo-3-carboxyphenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (35a)
General procedure E. The reaction was performed with 0.1 g (0.18 mmol) of 34a. Brown foam (64 mg, 86%, mixture of rotamers). 1H NMR (600 MHz, MeOD) δ 7.73 (m, 1H), 7.67 (d, J = 8.3 Hz, 1H), 7.38–7.34 (m, 1H), 4.27 (d, J = 6.1 Hz, 0.35 Hz), 4.25 (d, J = 7.0 Hz, 0.65 H), 3.71–3.64 (m, 1H), 3.58–3.48 (m, 2H), 2.37–2.29 (m, 1H), 2.12–2.02 (m,1H), 1.49 (s, 3H), 1.44 (s, 9H). LC-MS (m/z) calcd for C17H20BrNO6 [M-H]-, 414.1 and 416.1, found 314.3 and 316.3 (-Boc).
4-((2S,3R)-1-(tert-butoxycarbonyl)-2-carboxypyrrolidin-3-yl)phthalic acid (35c)
General procedure E. The reaction was performed with 0.13 g (0.25 mmol) of 34b. Brown foam (50 mg, 53%, mixture of rotamers). 1H NMR (600 MHz, MeOD) δ 7.77–7.76 (m, 1H), 7.68 – 7.66 (m, 1H), 7.56 – 7.53 (m, 1H), 4.31 (d, J = 6.0 Hz, 0.3H), 4.24 (d, J = 7.0 Hz, 0.7H), 3.72 – 3.67 (m, 1H), 3.61 – 3.54 (m, 1H), 2.40 – 2.33 (m, 1H), 2.14 – 2.08 (m, 1H), 1.49 (s, 3H), 1.44 (s, 6H). LC-MS (m/z) calcd for C18H21NO8 [M-H]- 380.1, found 280.3 (-Boc).
(2S,3R)-3-(3-Bromophenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid
General procedure E. The reaction was performed with 0.44 g (0.89 mmol) of tert-butyl (2R,3R)-3-(3-bromophenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate. Brown foam (0.24 g, 68%, mixture of rotamers). Product was obtained with 32% impurity. 1H NMR (600 MHz, MeOD) δ 7.50 – 7.48 (m, 1H), 7.45 – 7.44 (m, 1H), 7.30 – 7.26 (m, 2H), 4.27 – 4.18 (m, 1H (1.49H)), 3.73 – 3.65 (m, 1H (1.43H)), 3.59 – 3.39 (m, 2H (4.12H)), 2.41 – 2.29 (m, 1.45H), 2.14 – 1.88 (m, 1H (3.24H)), 1.51–1.44 (m, 9H (13 H)). LC-MS (m/z) calcd for C16H20BrNO4 [M-H]- 370.1 and 372.1, found 270.3 and 272.3 (-Boc).
(2S,3R)-3-(3-Bromo-5-carboxy-4-chlorophenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid
General procedure E. The reaction was performed with 0.3 g (0.51 mmol) of 45. Brown foam (0.2 g, 88%, mixture of rotamers). 1H NMR (600 MHz, MeOD) δ 7.95 (d, J = 2.3 Hz, 0.7 Hz),7.93 (d, J = 2.2 Hz, 0.3 H), 7.8 (d, J = 2.3 Hz, 0.7H), 7.78 (d, J = 2.2 Hz, 0.3H), 4.38 (d, J = 6.1 Hz, 0.H), 4.30 (d, J = 7.3 Hz, 0.7H), 3.83 – 3.77 (m, 1H), 3.67 (m, 2H), 2.48 – 2.42 (m, 1H), 2.23 – 2.17 (m, 1H), 1.61 (s, 3H), 1.56 (s, 6H). LC-MS (m/z) calcd for C17H19BrClNO6 [M-H]- 446.0 and 448.0; found 348.4 and 350.2 (-Boc).
Di-tert-butyl (2S,3R)-3-(3-bromo-5-(tert-butoxycarbonyl)-4-chlorophenyl)pyrrolidine-1,2-dicarboxylate (46a)
To a flame-dried vial with argon atmosphere was added 0.2 g of (2S,3R)-3-(3-Bromo-5-carboxy-4-chlorophenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carbo-xylic acid followed by anhydrous DCM (0.3–0.5 M). To the brown solution was added TBTA (5 equiv.) in one portion. The vial was capped and the reaction was stirred at rt for 24h. During the reaction time a white precipitate was formed. After 24h the reaction mixture was concentrated to dryness in vacuo. The title compound was purified by column chromatography on silica gel (heptane-EtOAc 5:1). The product was obtained as white foam (0.13 g, 52%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 2.2 Hz, 1H), 7.43 (d, J = 2.2 Hz, 1H), 4.18 (d, J = 6.2 Hz, 0.3H), 4.05 (d. J = 6.4 Hz, 0.7H), 3.78 – 3.74 (m, 0.7H), 3.65 – 3.61 (m, 0.3H), 3.58 – 3.51 (m, 1H), 3.36 – 3.32 (m, 1H), 2.33 – 2.26 (m, 1H), 2.00 – 1.96 (m, 1H), 1.60 (s, 9H), 1.56 – 1.44 (m, 18H). 13C NMR (151 MHz, DMSO) δ 171.1, 164.9, 153.8, 141.4, 135.29, 134.3, 134.3, 131.3, 128.1, 127.9, 124.9, 83.6, 83.5, 81.9, 80.6, 80.3, 66.1, 65.7, 49.2, 47.8, 46.1, 46.0, 32.7, 31.9, 28.6, 28.5, 28.3, 28.2, 28.1. LC-MS (m/z) calcd for C25H35BrClNO6 [M+H]+ 560.1 and 562.1, found 348.3 and 350.2 (-Boc, −2 x tert-butyl).
(2S,3R)-3-(4-Amino-3-carboxyphenyl)-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (35b)
35a (0.1 g, 0.24 mmol, 1 equiv.), NaN3 (78 mg, 1.2 mmol, 5 equiv.) Cs2CO3 (0.39 g, 1.2 mmol, 5 equiv.) and CuI (9.2 mg, 0.05 mmol, 0.2 equiv.) were loaded in a flame dried vial with argon atmosphere. To this was added degassed and anhydrous EtOH (1.2 mL, 0.2 M). The vial was capped and the reaction was stirred at 95 °C for 3d. The reaction was allowed to cool down to rt and concentrated to dryness. Water was added and the aqueous layer was carefully acidified to pH 2–3. Subsequently, the aqueous layer was extracted with EtOAc three times and the combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude reaction mixture was not further purified and used as such in the deprotection step. The reaction yielded a 0.7:1 mixture of 35a to 35b (based on crude NMR). LC-MS (m/z) calcd for C17H22N2O6 [M+H]+350.15, found 351.1 and 251.1 (-Boc).
3-((2S,3R)-1,2-Bis(tert-butoxycarbonyl)pyrrolidin-3-yl)-5-(tert-butoxycarbonyl)benzoic acid (28b)
HCOOH (0.2 mL, 5.32 mmol, 2 equiv.) and Ac2O (0.5 mL, 5.32 mmol, 2 equiv.) were mixed and stirred in a vial for 1.5h at rt. (2S,3R)-di-tert-butyl 3-(3-bromo-5-(tert-butoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxylate (1.4 g, 2.66 mmol, 1 equiv.), Pd(OAc)2 (30 mg, 0.13 mmol, 0.05 equiv.) and Xantphos (77 mg, 0.13 mmol, 0.05 equiv.) were loaded into a vial equipped with a septa containing screw-cap. The vial was evaporated and backfilled with argon three times. Then degassed, anhydrous toluene (5.31 mL, 0.5 M) was added, followed by anhydrous tert-BuOH (0.5 mL, 5.32 mmol, 2 equiv.). To the orange reaction mixture was added the mixture of HCOOH and Ac2O in a dropwise manner. During the addition the color changed to dark-green. Then Et3N (1.8 mL, 13.3 mmol, 5 equiv.) was added dropwise. Instantly gas evolution was observed and a color change of the reaction mixture to dark red (careful: overpressure!). After the addition was completed, the reaction was placed in a preheated oil bath at 80 ˚C and the reaction was stirred overnight at 80 ˚C. The reaction mixture became black and was allowed to cool down to room temperature. The pressure was released carefully, followed by the addition of water and EtOAc. The aqueous layer was carefully acidified to pH 2 with 1N HCl. The aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (2:1 heptane:EtOAc +1 vol% AcOH) to afford the title compound as white solid (1.18 g, 90%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 8.59 (s, 0.7H), 8.58 (s, 0.3H), 8.17 (s, 0.3 H), 8.16 (s, 0.7H), 8.12 (s, 0.7H), 8.11 (s, 0.3H), 4.26 (d, J = 7.0 Hz, 0.3H), 4.13 (d, J = 7.0 Hz, 0.7H), 3.85 – 3.81 (m, 0.7H), 3.73 – 3.69 (m, 0.3H), 3.61 – 3.55 (m, 1H), 3.50 – 3.47 (m, 1H), 2.35 – 2.29 (m, 1H), 2.13 – 2.07 (m, 1H), 1.62 (s, 9H), 1.50 – 1.42 (m, 18 H). 13C NMR (151 MHz, CDCl3) δ 171.1, 169.7, 164.5, 153.8, 141.9, 133.5, 133.1, 132.6, 130.0, 129.9, 82.0, 81.5, 80.4, 66.2, 49.9, 46.1, 32.2 31.9, 29.0, 28.4, 28.2, 28.0, 22.7, 14.1.; LC-MS (m/z) calcd for C26H37NO8 [M+H]+ 491.2, found 280.4 (-Boc, −2 x tert-butyl).
Di-tert-butyl (2S,3R)-3-(3-(tert-butoxycarbonyl)-5-((trimethylsilyl)ethynyl)phenyl)pyrrolidine-1,2-dicarboxylate
To a flame-dried vial with argon atmosphere was added arylbromide 39a (0.5g, 0.95 mmol, 1 equiv.), PdCl2(PPh3)2 (0.1 equiv.), and CuI (0.1 eq). The vial was evaporated and refilled with argon three times. DMF (0.3 M) was added, followed by TMSA (1.5 eq) and Et3N (0.45 M). The reaction was placed into a preheated oil bath at 60 ºC and stirred for 12h. The reaction was allowed to cool down to rt before EtOAc was added. The organic layer was washed twice with water and once with brine. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc 10:1) to afford the title compound as a white foam (0.4 g, 77%). 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.80 (s, 1H), 7.50 (s, 1H), 4.21 (d, J = 6.2 Hz, 0.3H), 4.09 (d, J = 6.5 Hz, 0.7H), 3.79 – 3.75 (m, 0.7H), 3.66 – 3.63 (m, 0.3H), 3.59 – 3.54 (m, 1H), 3.40 – 3.37 (m, 1H), 2.31–2.24 (m, 1H), 2.06 – 2.00 (m, 1H), 1.59 (s, 9H), 1.49 (s, 3H), 1.45 (s, 6H), 1.43 (s, 9H), 0.26 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.4, 164.9, 153.9, 141.7, 134.3, 132.7, 131.8, 128.7, 123.9, 104.1, 95.6, 81.8, 81.7, 81.6, 81.6, 80.4, 80.0, 66.2, 50.0, 46.2, 32.0, 28.6, 28.5, 28.3, 28.3, 28.2, 28.1, 0.03. LC-MS (m/z) calcd for C30H45NO4Si [M+H]+ 544.3, found 444.8 (-Boc).
Di-tert-butyl (2S,3R)-3-(3-(tert-butoxycarbonyl)-5-ethynylphenyl)pyrrolidine-1,2-dicarboxylate (41a)
TMS-acetylene intermediate (0.39 g, 0.72 mmol, 1 equiv.) was loaded in a flame-dried vial with argon atmosphere. To this was added anhydrous THF (0.3 M) and the solution was cooled on an ice bath. After 15 minutes, a 1M solution of TBAF in THF (1.1 eq) was added dropwise. After 5 minutes the ice bath was removed and the reaction was stirred at room temperature for 30 minutes. The reaction was quenched with saturated NH4Cl and subsequently the aqueous layer was extracted with EtOAc three times. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The title compound was purified by flash column chromatography on silica gel (heptane-EtOAc 10:1). The product was obtained as a light-yellow foam (0.28 g, 82%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.85 (s, 1H), 7.52 (s, 1H), 4.20 (d, J = 6.2 Hz, 0.3H), 4.09 (d, J = 6.6 Hz, 0.7H), 3.79 – 3.76 (m, 0.7H), 3.67 – 3.62 (m, 0.3H), 3.59 – 3.53 (m, 1H), 3.41 – 3.37 (m, 1H), 3.12–3.10 (m, 1H), 2.33 – 2.25 (m, 1H), 2.06 – 2.00 (m, 1H), 1.59 (s, 9H), 1.49 (s, 3H), 1.45 (s, 6H), 1.42 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.3, 164.8, 154.2, 153.9, 142.0, 134.6, 132.9, 132.0, 132.0, 128.9, 128.8, 122.9, 122.8, 82.8, 81.8, 81.6, 80.4, 80.1, 66.3, 66.1, 50.0, 48.6, 46.3, 46.2, 32.7, 32.1, 28.6, 28.5, 28.3, 28.2, 28.1. LC-MS (m/z) calcd for C27H37NO6 [M+H]+ 472.3, found 372.8 (-Boc) and 260.5 (-Boc, −2 x tert-Bu).
CuAAC reaction - General procedure I
Acetylene (1 equiv.) and CuI (0.05 equiv) were loaded in a flame-dried flask with argon atmosphere. To this was added DMF (0.16 M), MeOH (1.3 M), and TMSN3 (1.5 equiv.) in one portion each. The vial was capped and stirred at 100 ºC for 16 hours. After the reaction was allowed to cool to rt, the mixture was filtered over celite, rinsed with EtOAc and concentrated to dryness. Further purification is stated below.
Di-tert-butyl (2S,3R)-3-(3-(tert-butoxycarbonyl)-5-(1H-1,2,3-triazol-5-yl)phenyl)pyrrolidine-1,2-dicarboxylate (42a)
General procedure I. The reaction was performed with 0.15 g (0.32 mmol) of 41a. The title compound was purified by flash column chromatography on silica gel (heptane-EtOAc 1:1). The product was obtained as a white foam (71 mg, 65%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 11.73 (bs, 1H), 8.28 (s, 1H), 8.03 (s, 1H), 7.93 (s, 1H), 7.87 (s, 1H), 4.29 (d, J = 6.5 Hz,0.31H), 4.16 (d, J = 6.8 Hz, 0.7H), 3.84 – 3.81 (m, 0.7H), 3.72 – 3.68 (m, 0.3H), 3.62 – 3.57 (m, 1H), 3.51 – 3.47 (m, 1H), 2.38 – 2.30 (m, 1H), 2.16 – 2.09 (m, 1H), 1.63 (s, 9H), 1.50 (s, 3H), 1.45 (s, 6H), 1.42 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.3, 165.2, 153.8, 142.2, 133.1, 130.7, 128.7, 128.6, 128.5, 125.9, 125.9, 81.7, 81.4, 81.4, 80.2, 79.9, 66.3, 66.0, 50.2, 48.8, 46.3, 46.1, 32.8, 32.1, 29.7, 28.5, 28.4, 28.2, 28.0. LC-MS (m/z) calcd for C27H38N4O6 [M+H]+ 515.3, found 515.6 and 303.1 (-Boc, −2 x tert-Bu).
5-((2S,3R)-1,2-bis(tert-butoxycarbonyl)pyrrolidin-3-yl)-3-(tert-butoxycarbonyl)-2-chlorobenzoic acid (46b)
Formic acid (21 μL, 0.54 mmol, 2 equiv.) and Ac2O (52 μL, 0.54 mmol, 2 equiv.) were stirred in a vial for 1.5h at rt. Then, 45a (0.15 g, 0.27 mmol, 1equiv.), Pd(OAc)2 (6 mg, 0.03 mmol, 0.1 equiv.) and Xantphos (16 mg, 0.03 mmol, 0.1 equiv.) were loaded in a flask equipped with screw cap containing a septa. The flask was evaporated and backfilled with argon three times. Then anhydrous toluene (0.55 mL, 0.5 M) was added, followed by anhydrous tBuOH (52 μL, 0.54 mmol, 2 equiv.). To the orange reaction mixture was added the mixture of HCOOH and Ac2O in a dropwise manner. During the addition the color changed to dark-green. Then Et3N (190 μL, 1.35 mmol, 5 equiv.) was added dropwise. Instantly, gas evolution was observed and a color change of the reaction mixture to dark red. (Overpressure!) After the addition was completed, the reaction was placed in a preheated oil bath at 80 ˚C and the reaction was stirred overnight at 80 ˚C. The reaction mixture became black and was allowed to cool down to rt. The pressure was released carefully, followed by the addition of water and EtOAc. The aqueous layer was carefully acidified to pH 2 with 1N HCl. The aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (heptane-EtOAc 2:1 with 1 vol% AcOH) to afford the title compound as a white foam (89 mg, 63%, mixture of rotamers). 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 2.2 Hz, 1H), 7.43 (d, J = 2.2Hz, 1H), 4.18 (d, J = 6.2 Hz, 0.35H), 4.05 (d, J = 6.3 Hz, 0.65H), 3.79 – 3.73 (m, 0.65H), 3.66 – 3.61 (m, 0.35H), 3.57 – 3.51 (m, 1H), 3.37 – 3.31 (m, 1H), 2.33 – 2.25 (m, 1H), 2.03 – 1.94 (m, 1H), 1.60 (s, 9H), 1.48 – 1.44 (m, 9H). 13C NMR (151 MHz, CDCl3) δ 171.1, 165.0, 153.8, 141.4, 135.3, 134.3, 131.3, 128.1, 124.9, 83.6, 81.9, 80.6, 66.1, 49.2, 47.8, 46.0, 31.9, 28.6, 28.5, 28.3, 28.2, 28.1. LC-MS (m/z) calcd for C26H36ClNO8 [M+H]+ 526.2, found 314.4 (-Boc, −2 x tert-Bu).
Final compounds for pharmacological characterization
Deprotection – General procedure J
The protected compound was dissolved in 1 mL anhydrous DCM. To this was added dropwise 1mL TFA. The reaction mixture was stirred at room temperature overnight and then concentrated in vacuo. The crude mixture was purified by preparative HPLC if necessary. The obtained TFA salt was treated with 2 mL 1N HCl and concentrated to dryness in vavcuo. This procedure was repeated three times. After lyophilization the final product was obtained as HCl salt if not otherwise stated-
(2S,3R)-3-(4-Bromo-3-carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (4g)
General procedure J. The reaction was performed with 50 mg (0.09 mmol) of 35a. The crude product was purified by preparative HPLC. Rt 12.8 min (flow: 20 mL min−1; gradient 0–50% B (25 min); λ = 210 nm). The solid was treated with 1M HCl (2 mL) and the volatiles were evaporated under reduced pressure to afford the hydrochloride salt as a white solid (12.3 mg, 39%). 1H NMR (600 MHz, D2O) δ 7.76 (d, J = 8.3 Hz, 1H), 7.72 (d, J = 2.3 Hz, 1H), 7.45 (dd, J = 8.3, 2.3 Hz, 1H), 4.27 (d, J = 9.4 Hz, 1H), 3.70 – 3.63 (m, 2H), 3.55 (ddd, J = 11.8, 10.2, 6.8 Hz, 1H), 2.57 (dtd, J = 13.9, 7.1, 3.4 Hz, 1H), 2.30 – 2.23 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.4, 171.1, 138.5, 134.4, 134.0, 131.7, 129.2, 118.8, 65.3, 47.4, 45.7, 32.8. LC-MS (m/z) calcd for C12H12BrNO4 [M+H]+ 314.0 and 316.0; found 314.4 and 316.5.
(2S,3R)-3-(4-Amino-3-carboxyphenyl)pyrrolidine-2-carboxylic acid dihydrochloride (4h)
General procedure J. The crude product was purified by preparative HPLC. Rt 3.72 min (flow: 20 mL min−1; gradient 0–25% B (30 min); λ = 210 nm). The solid was treated with 1M HCl (2 mL) and the volatiles were evaporated under reduced pressure to afford the hydrochloride salt as a white solid (10 mg, 14% over 2 steps). 1H NMR (600 MHz, D2O) δ 8.09, 7.64 (d, J = 8.4 Hz, 1H), 7.33 (dd, J = 8.4, 2.6 Hz, 1H), 4.23 (dd, J = 9.7, 2.6 Hz, 1H), 3.70 – 3.63 (m, 2H), 3.57 – 3.53 −8m, 1H), 2.56 (ddt, J = 13.1, 6.3, 3.3 Hz, 1H), 2.28 (dtd, J = 13.5, 10.2, 7.9 Hz, 1H). 13C NMR (151 MHz, D2O) δ 172.3, 170.1, 136.0, 133.4, 130.7, 122.8, 121.0, 66.1, 47.7, 45.6, 32.4. LC-MS (m/z) calcd for C12H14N2O4 [M+H]+ 251.1, found 251.5.
4-((2S,3R)-2-Carboxypyrrolidin-3-yl)phthalic acid tri sodium salt (4i)
General procedure J. The reaction was performed with 30 mg (0.08 mmol) of 35c. The crude product was purified by preparative HPLC. Rt 8.72 min (flow: 20 mL min−1; gradient 0–50% B (25 min); λ = 210 nm). After lyophilization the solid was treated with 1M NaOH (4 equiv.) and the volatiles were evaporated under reduced pressure to afford the tri sodium salt as a white solid (20 mg, 72%) 1H NMR (600 MHz, D2O) δ 7.79 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 1.9 Hz, 1H), 7.58 (dd, J = 8.0, 1.9 Hz, 1H), 4.21 (d, J = 8.8 Hz, 1H), 3.69–3.65 (m, 2H), 3.57–3.52 (m, 1H), 2.58–2.53 (m, 1H), 2.32–2.25 (m, 1H). 13C NMR (151 MHz, D2O) δ 173.9, 173.4, 173.0, 142.5, 135.0, 132.4, 129.8, 129.6, 127.5, 66.5, 48.1, 45.6, 32.9. LC-MS (m/z) calcd for C13H13NO6 [M+H]+ 280.1, found 280.5.
(2S,3R)-3-(3-Bromo-5-carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (5b)
General procedure J. The reaction was performed with 50 mg (0.09 mmol) of 28a. No further purification was required. The title compound was treated with 1N HCl (2 mL) and all volatiles were removed under reduced pressure to afford the hydrochloride salt as a white solid (25.7 mg, 77%). 1H NMR (600 MHz, D2O) δ 8.17 (s, 1H), 8.01 (s, 1H), 7.89 (s, 1H), 4.23 (d, J = 9.3 Hz, 1H), 3.70 – 3.64 (m, 2H), 3.59 – 3.48 (m, 1H), 2.58 (dtd, J = 13.8, 7.0, 3.4 Hz, 1H), 2.33 – 2.21 (m, 1H). 13C NMR (151 MHz, D2O) δ 172.3, 169.1, 141.8, 135.4, 132.6, 131.7, 127.4, 122.3, 66.1, 47.8, 45.5, 32.9. LC-MS (m/z) calcd for C12H12BrNO4 [M+H]+, 314.0, 316.0 found 314.3, 316.3.
5-((2S,3R)-2-Carboxypyrrolidin-3-yl)isophthalic acid hydrochloride (5c)
General procedure J. The reaction was performed with 52 mg (0.1 mmol) of 28b. No further purification was required. The title compound was treated with 1N HCl (2 mL) and all volatiles were removed under reduced pressure to afford the hydrochloride salt as a white solid (23.6 mg, 75%). 1H NMR (600 MHz, D2O) δ 8.54 (s, 1H), 8.29 (s, 2H), 4.43 (d, J = 9.6 Hz, 1H), 3.74 (ddd, J = 9.6 8.1, 3.3 Hz, 1H), 3.59 (td, J = 10.8, 6.8 Hz, 1H), 2.64 (dtd, J = 13.9, 7.0, 3.3 Hz, 1H), 2.35 (dtd, J = 13.9, 10.8, 8.1 Hz, 1H).; 13C NMR (151 MHz, D2O) δ 171.4, 169.1, 139.9, 133.4, 131.1, 130.1, 65.3, 47.6, 45.7, 32.9. LC-MS (m/z) calcd for C13H13NO6 [M+H]+, 280.1 found 280.3.
(2S,3R)-3-(3-Carboxy-5-(1H-1,2,3-triazol-5-yl)phenyl)pyrrolidine-2-carboxylic acid hydrochloride (5d)
General procedure J. The reaction was performed with 70 mg (0.14 mmol) of 42a. No further purification was required. The title compound was treated with 1N HCl (2 mL) and all volatiles were removed under reduced pressure to afford the hydrochloride salt as a yellow solid (44 mg, 92%). 1H NMR (600 MHz, D2O) δ 8.26–8.25 (m, 2H), 8.04 (s, 1H), 8.02 (s, 1H), 4.46 (d, J = 9.4 Hz, 1H), 3.82–3.73 (m, 2H), 3.62–3.58 (m, 1H), 2.66–2.61 (m, 1H), 2.38–2.31 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.3, 169.4, 144.5, 140.1, 131.2, 130.3, 129.8, 128.6, 126.3, 125.0, 65.2, 47.7, 45.8, 32.9. LC-MS (m/z) calcd for C14H14N4O4 [M+H]+ 303.0, found 303.5.
(2S,3R)-3-(3-Bromo-5-carboxy-4-chlorophenyl)pyrrolidine-2-carboxylic acid hydrochloride (6a)
General procedure J. The reaction was performed with 50 mg (0.09 mmol) of 46a. No further purification required. The title compound was treated with 1N HCl (2 mL) and all volatiles were removed under reduced pressure to afford the hydrochloride salt as a white solid (32 mg, 96%). 1H NMR (600 MHz, D2O) δ 7.93 (d, J = 2.2 Hz, 1H), 7.71 (d, J = 2.2 Hz, 1H), 4.35 (d, J = 9.6 Hz, 1H), 3.71 – 3.66 (m, 2H), 3.58 – 3.53 (m 1H), 2.62 – 2.57 (m 1H), 2.30 – 2.23 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.2, 170.4, 139.2, 135.0, 135.0, 130.2, 127.7, 124.1, 65.1, 46.9, 45.6, 32.7. LC-MS (m/z) calcd for C12H11BrClNO4 [M+H]+ 348.0 and 350.0, found 348.3 and 350.3.
5-((2S,3R)-2-Carboxypyrrolidin-3-yl)-2-chloroisophthalic acid hydrochloride (6b)
General procedure J. The reaction was performed with 89 mg (0.17 mmol) of 46a. No further purification required. The title compound was treated with 1N HCl (2 mL) and all volatiles were removed under reduced pressure to afford the hydrochloride salt as a white solid (55 mg, 93%). 1H NMR (600 MHz, D2O) δ 7.84 (s, 1H), 4.38 (d, J = 9.6 Hz, 1H), 3.77 – 3.68 (m, 2H), 3.59 – 3.54 (m, 1H), 2.63 – 2.58 (m, 1H), 2.33 – 2.26 (m, 1H). 13C NMR (151 MHz, D2O) δ: 171.3, 170.6, 138.3, 134.8, 130.5, 127.3, 65.1, 47.0, 45.6, 32.7. LC-MS (m/z) calcd for C13H12ClNO6 [M+H]+ 314.0, found 314.3.
Synthesis Analogs 9b-e
1-(tert-Butyl)-2-methyl (2R,4S)-4-allyl-5-oxopyrrolidine-1,2-dicarboxylate (47)
A solution of 1-(tert-Butyl)-2-methyl (R)-5-oxopyrrolidine-1,2-dicarboxylate (3.9 g, 16.3 mmol, 1.0 equiv.) in anhydrous THF (160 mL) was cooled to −78 °C and 1 M LiHMDS in THF (19.3 mL, 19.3 mmol, 1.2 equiv.) was added dropwise over 45 min. The mixture was left to stir at −78 °C for 30 min after which a solution of 3-bromoprop-1-ene (4.2 mL, 5.8 g, 48.1 mmol, 3 equiv.) with DMPU (21.5 mL, 160 mmol, 10 equiv.) in anhydrous THF (240 mL) was added dropwise over 1.5 h. After the addition was completed the reaction mixture was stirred for another 2 h at −78 ºC. The mixture was quenched with saturated NH4Cl solution and allowed to reach rt. The mixture was transferred to a separation funnel and the aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was filtered over a short silica pad and rinsed with 1:1 EtOAc-heptane. The filtrate was concentrated and the crude mixture was purified by flash column chromatography (heptane-EtOAc 10:1) to afford the title compound as a colorless oil (3.25 g, 36%). 1H-NMR (600 MHz, CDCl3) δ 5.76 – 5.69 (m, 1H), 5.11 – 5.06 (m, 2H), 4.56 (dd, J = 9.7, 1.7 Hz, 1H), 3.77 (s, 3H), 2.76 (m, 1H), 2.64 – 2.59 (m, 1H), 2.21 – 2.15 (m, 2H), 2.03– 1.97 (m, 1H), 1.49 (s, 9H). 13C-NMR (151 MHz, CDCl3) δ 174.2, 171.8, 149.4, 134.3, 117.7, 83.59, 56.9, 52.5, 41.2, 34.4, 27.9, 27.7. LC-MS (m/z) calcd for C14H21NO5 [M+H]+ 284.1, found 284.4, 184.5 (-Boc). Analytical data is in agreement with literature. Nakache et al., JOC 2006, 71 (7), 2760–2778.
1-(tert-Butyl) 2-methyl (2R)-4-allylpyrrolidine-1,2-dicarboxylate (48)
Lactam 47 (3.1 g, 11 mmol, 1.0 equiv.) was dissolved in anhydrous THF (57 mL), and cooled to −78 °C. After 10 minutes a 1 M solution of LiEt3BH (11 mL, 11 mmol, 1 equiv.) was added dropwise over 20 min. The mixture was left to stir at −78 °C for 20 minutes. Then the reaction was quenched with saturated NaHCO3 (6.5 mL) and allowed to warm up to 0 °C. 50% H2O2 (5.6 mL) was added dropwise and the reaction stirred for another 20 min. The organic solvent was evaporated and the aqueous layer was extracted with DCM. The organic layers were combined, dried over MgSO4 and concentrated in vacuo. The resulting residue was used for the next reaction without further purification. The flask with the residue was evaporated and refilled with argon. The residue was dissolved in anhydrous DCM (56 mL) and cooled to −78 °C. To the stirring solution, triethylsilane (3.7 mL, 23 mmol, 2.1 equiv.) and boron trifluoride etherate (2.85 mL, 23 mmol, 2.1 eq) were added dropwise. The reaction mixture was left to stir at −78 °C for 3h and then quenched with saturated NaHCO3 solution. The resulting mixture was extracted with DCM and the organic layers were combined, dried over MgSO4 and concentrated in vacuo. The crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc 10:1), to afford the title compound a colorless oil (2.1 g, 72%, mixture of rotamers). 1H-NMR (600 MHz, CDCl3) δ 5.80 – 5.67 (m, 1H), 5.07 – 5 (m, 2H), 4.37 (dd, J = 2.5, 8.9 Hz, 0.36H), 4.27 (dd, J = 3.1, 8.9 Hz, 0.54H), 3.71 – 3.63 (m, 4H), 3.07 – 2.96 (m, 1H), 2.45–5.32 (m, 1H), 2.14 – 2.03 (m, 3H), 1.94–1.80 (m, 1H), 1.46 (s, 4H), 1.40 (s, 5H). 13C-NMR (151 MHz, CDCl3) δ 173.6, 173.4, 154.4, 153.7, 135.8, 116.5, 79.9, 58.9, 58.6, 52.1, 51.9, 51.5, 51.2, 37.1, 36.9, 36.1, 35.3, 28.4, 28.3. LC-MS (m/z) calcd for C14H23NO4 [M+H] + 270.2, found 170.5 (-Boc).
(2R,4S)-4-Allyl-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid
Intermediate 48 (2.1 g, 7.8 mmol, 1.0 equiv.) was dissolved in THF (20 mL) and MeOH (4 mL). A 1 M solution of aq. LiOH (12 mL, 12 mmol, 1.5 equiv.) was added and the reaction was stirred at rt overnight. The reaction mixture was concentrated to dryness in vacuo and then water and DCM were added. The aqueous layer was acidified with 1N HCl to pH 3 and extracted with DCM. The combined organic layers were dried over MgSO4 and concentrated in vacuo to give the title compound as a white solid (1.8 g, 90%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 5.76 (m, 1H), 5.08 – 5.02 (m, 2H), 4.39 (d, J = 8.3 Hz, 0.7H), 4.29 (bs, 0.3H), 3.72 (bs, 0.3H), 3.51 (m, 0.7H), 3.05 (bs, 0.3H), 2.97 – 2.94 (m, 7H), 2.49 – 2.47 (m, 0.7H), 2.37 (m, 1H), 2.18 (m, 2.7H), 1.99 – 1.92 (m, 0.3H), 1.72–1.64 (m, 0.7H), 1.49 – 1.42 (m, 9H). 13 C NMR (151 MHz, CDCl3) δ 177.9, 173.5 157.2, 153.7, 135.7, 116.6, 82.1, 80.3, 59.4, 58.7, 51.9, 51.3, 37.1, 36.8, 36.0, 33.4, 28.4. LC-MS (m/z) calcd for C13H21NO4 [M+H] + 256.2, found 156.5 (-Boc).
tert-Butyl-(2R,4S)-4-allyl-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate
(2R,4S)-4-Allyl-1-(tert-butoxycarbonyl)pyrrolidine-2-carboxylic acid (1.8 g, 7.1 mmol, 1.0 equiv.) and 8-aminoquinoline (1.2 g, 8.5 mmol, 1.2 equiv.) were dissolved in anhydrous DCM (14 mL, 0.5 M). EDC·HCl (1.6 g, 8.5 mmol, 1.2 equiv.) and HOBt (1.14 g, 8.5 mmol, 1.2 equiv) were added to the solution and the reaction was left to stir at rt for 24h. To the reaction was added saturated NaHCO3 solution and the aqueous layer was extracted three times with DCM. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc, gradient 7:1 to 6:1), to afford the title compound as a white solid (2.1 g, 80%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 10.65 (s, 0.4H), 10.40 (s, 0.6H), 8.79 (m, 2H), 8.15 (bs, 1H), 7.53 (bs, 2H), 7.44 (bs, 1H), 5.77 (ddt, J = 17.1, 10.2, 7.0 Hz, 1H), 5.06 (dd, J = 17.1, 1.6 Hz, 1H), 5.01 (dd, J = 10.2, 1.6 Hz, 1H), 4.64 (s, 0.4 H), 4.49 (s, 0.6H), 3.83 (m, 0.6H), 3.7 (m, 0.4H), 3.23 (m, 0.6H), 3.09 (m, 0.4H), 2.47 (m, 0.4H), 2.43 (m, 1.6H), 2.18 (m, 2H), 1.96 (m, 0.6H), 1.83 (m, 0.4H), 1.55 (s, 4H), 1.37 (s, 5H). 13C NMR (151 MHz, CDCl3) δ 171.4, 170.9, 155.4, 154.6, 148.5, 148.2, 138.8, 138.6, 136.2, 136.0, 135.8, 134.5, 134.0, 127.9, 127.3, 121.8, 121.7, 121.5, 116.6, 116.4, 80.7, 80.4, 62.3, 61.6, 52.2, 51.9, 37.4, 37.1, 36.6, 36.5, 35.1, 29.7, 28.5, 28.2. LC-MS (m/z) calcd for C22H27N3O3 [M+H] + 382.2, found 382.6. Mp: 130.3–134.3 °C.
tert-Butyl-(2R,3R,4S)-4-allyl-3-(3-(methoxycarbonyl)phenyl)-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (49)
tert-Butyl-(2R,4S)-4-allyl-2-(quinolin-8-ylcarbamoyl)pyrrolidine-1-carboxylate (0.5 g, 1.3 mmol, 1.0 equiv.), methyl 3-iodobenzoate (686 mg, 2.6 mmol, 2.0 equiv), Palladium acetate (30 mg, 0.13 mmol, 0.1 equiv.), pivalic acid (44 mg, 0.4 mmol, 0.3 equiv.) and silver carbonate (361 mg, 1.3 mmol, 1.0 equiv.) were placed in a flame-dried, argon-filled microwave vial and dissolved in anhydrous toluene (6.5 mL, 0.2 M). The microwave vial was sealed and heated to 110 °C under vigorous stirring. The reaction was left to stir at 110 ºC for 48 h. After the reaction was allowed to cool down to rt, the mixture was filtered through a celite-silica pad and rinsed with EtOAc. The resulting filtrate was concentrated in vacuo and then purified by column chromatography on silica gel (heptane-EtOAc, gradient 10:1→5:1→4:1), to afford the title compound as a white foam (244 mg, 37%, 57% brsm, mixture of rotamers). However, the product could not be obtained in 100% purity. Nonetheless, the product was used in the next step. 1H NMR (600 MHz, DMSO) (Mixture of conformers) δ 9.65 (s, 0.4H), 9.59 (s,0.6H), 8.71 (m, 1H), 8.31 (s, 2H), 7.91 (m, 1H), 7.58 – 7.55 (m, 2H), 7.51 – 7.45 (m, 3H) 7.20 (td (J = 7.7, 1.8 Hz, 1H), 5.75 (m, 1H), 5.01 – 4.93 (m, 2H), 3.91 – 3.89 (m, 1H), 3.72 (s, 3H), 3.61 – 3.55 (m, 1H), 3.19 – 3.10 (m, 1H), 2.96 (m, 1H), 2.08 (m, 1H), 1.97 – 1.92 (m, 1H), 1.43 (s, 3H), 1.22 (s, 6H). 13C NMR (151 MHz, DMSO) δ 169.9, 169.3, 166.4, 153.9, 153.5, 149.4, 148.9, 138.2, 137.2, 136.8, 136.4, 133.9, 133.6, 130.0, 129.9, 128.9, 128.2, 128.0, 127.1, 122.4, 122.9, 116.9, 116.8, 79.6, 79.4, 66.4, 66.1, 52.7, 52.4, 51.3, 39.2, 35.2, 31.7, 28.6, 28.3, 22.6, 14.4. LC-MS (m/z) calcd for C30H33N3O5 [M+H] + 516.2, found 516.8.
(2S,3R,4S)-4-Allyl-1-(tert-butoxycarbonyl)-3-(3-carboxyphenyl)pyrrolidine-2-carboxylic acid
Intermediate 49 (0.34 g, 0.67 mmol, 1.0 equiv.) was dissolved in EtOH (3.4 mL), in a microwave vial. NaOH pellets (0.27 mg, 6.7 mmol, 10.0 equiv.) were added and the vial was sealed and the reaction mixture was heated to 100 °C and stirred for 20h. The reaction mixture became brown and slurry over time. The reaction was allowed to cool down to rt and then concentrated to dryness in vacuo. The residue was dissolved in 1N NaOH and then extracted three times with EtOAc to remove 8-aminoquinoline. The aqueous layer was acidified with 1N HCl to pH 2–3 and extracted with EtOAc three times. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The resulting brown-ish foam (0.15 g, 60–90%) was used in the next step without further purification (mixture of rotamers). 1H NMR (400 MHz, DMSO) δ 12.45 (bs, 2H), 7.86 – 7.84 (m, 2H), 7.62 (d, J = 7.9 Hz, 1H), 7.48 (m, 1H), 5.72 – 5.62 (m, 1H), 4.98 (d, J = 15.8 Hz, 1H), 4.94 (d, J = 10.3Hz, 1H), 4.08 (d, J = 9.3 Hz, 1H), 3.77 – 3.72 (m, 1H), 3.11 – 2.97 (m, 1H), 1.99 – 1.97 (m, 2H), 1.42 (s,13H), 1.34 (s, 6H). 13C NMR (151 MHz, DMSO) δ 173.3, 172.7, 172.0, 167.3, 167.2, 153.2, 153.0, 152.7, 139.6, 139.4, 135.8, 135.8, 132.5, 132.4, 131.2, 130.1, 130.1, 129.7, 129.0, 128.9, 128.3, 127.8, 126.7, 116.6, 116.5, 79.2, 79.1, 66.5, 66.3, 58.6, 55.0, 54.2, 51.4, 51.2, 44.7, 43.9, 34.3, 34.2, 27.9, 27.9, 21.1.
Di-tert-butyl-(2S,3R,4S)-4-allyl-3-(3-(tert-butoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxlate
(2S,3R,4S)-4-Allyl-1-(tert-butoxycarbonyl)-3-(3-carboxyphenyl)pyrrolidine-2-carboxylic acid (561 mg, 1.5 mmol, 1.0 equiv.) was loaded in a vial and dissolved in anhydrous DCM (3 mL, 0.5 M). To this was added in one portion TBTA (1.6 g, 7.5 mmol, 5 equiv.). The reaction was stirred for 24h at rt. The brownish suspension was concentrated to dryness and the crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc 10:1) to afford the title compound as a yellow oil (0.42 g, 57%, mixture of rotamers) with small impurities (~10%). 1H NMR (600 MHz, CDCl3) δ 7.90 (m, 1H), 7.84 (s, 1H) 7.42 – 7.36 (m, 2H), 5.69 – 5.59 (m, 1H), 5.01 – 4.94 (m, 2H), 4.18 – 4.11 (m, 0.4H), 4.08 – 4.06 (d, J = 9.1Hz, 0.75H), 3.99 (m, 0.6H), 3.86 – 3.82 (m, 0.25H), 3.21 – 3.16 (m, 1H), 2.97 – 2.92 (dd, J = 11.0, 9.0 Hz, 1H), 2.46 – 2.37 (m, 1H), 2.19 – 2.10 (m,1H), 1.99 – 1.90 (m,1H), 1.60 (s, 9H), 1.43 (s, 9H), 1.34 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.1, 170.9, 165.6, 153.9, 153.6, 139.5, 135.4, 135.2, 132.4, 131.9, 131.8, 129.1, 128.6, 128.5, 128.1, 116.8, 81,3, 81.2, 81.1, 80.2, 79.9, 67.3, 60.4, 59.8 59.7, 56.5, 55.3, 51.9, 51.6, 45.4, 44.7, 36.3, 35.2, 35.1, 28.5, 28.4, 28.3, 28.3, 28.2, 28.0, 27.9,21.0, 14.2. LC-MS (m/z) calcd for C28H41NO6 [M+H]+ 488.2, found 388.6 (-Boc) and 276.6 (-Boc, −2 x tert-butyl).
Di-tert-butyl-(2S,3R,4S)-3-(3-(tert-butoxycarbonyl)phenyl)-4-(2-oxoethyl)pyrrolidine-1,2- dicarboxylate (50)
Di-tert-butyl-(2S,3R,4S)-4-allyl-3-(3-(tert-butoxycarbonyl)phenyl)pyrrolidine-1,2-dicarboxlate (0.42 g, 0.86 mmol, 1 equiv.) was dissolved in 8.5 ml 1:1 THF/H2O. To the colorless solution was added OsO4 (75 μL, 0.01 equiv., 4wt% in H2O). The mixture was stirred at rt for 15 minutes and became black. Then NaIO4 (732 mg, 3.4 mmol, 4 equiv.) was added in one portion and stirred for another 4 h at rt. The reaction mixture became a white suspension over time. To the mixture was added water and the organic layer was extracted three with EtOAc. The combines organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by flash column chromatography on silica gel (heptane-EtOAc 3:1) to yield the tile compound as a white foam (0.22 g, 53%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 9.62 (s, 0.35H), 9.59 (s, 0.65H), 7.90 (d, J = 6.6Hz, 1H), 7.82 (s, 1H), 7.38 (s, 1H), 4.20 – 4.06 (m, 2H), 3.16 – 3.09 (m, 1H), 2.98 – 2.91 (m, 1H), 2.80 – 2.75 (m, 1H), 2.50 – 2.40 (m, 2H), 1.58 (s, 9H) 1.46 (s, 3H), 1.42 (s, 6H), 1.32 (s, 9H). 13C-NMR (151 MHz, CDCl3) δ 199.8, 199.4, 170.9, 165.3, 153.4, 138.3, 132.6, 131.7, 129.0, 128.9, 128.8, 81.4, 80.3, 80.1, 66.5, 56.4, 55.2, 51.9, 51.5, 45.3, 45.0, 39.9, 39.3, 28.4, 28.3, 28.1, 27.9, 27.8. LC-MS (m/z) calcd for C27H39NO7 [M+H]+ 489.3, found 278.5 (-Boc, −2 x tert-butyl).
Di-tert-butyl-(2S,3R,4S)-3-(3-(tert-butoxycarbonyl)phenyl)-4-(2-(methylamino)ethyl)pyrrolidine-1,2-dicarboxylate (51a)
Aldehyde 50 (20 mg, 0.04 mmol, 1 equiv.) was dissolved in anhydrous MeOH (0.8 mL, 0.05 M). To this was added MeH2N·HCl (14 mg, 0.2 mmol, 5 equiv.) in one portion, followed by NaBH3CN (13 mg, 0.2 mmol, 5 equiv.). The white suspension was stirred at rt overnight and then concentrated to dryness. To the crude mixture was added 1N NaOH and the aqueous layer was extracted three times with EtOAc. The combined organic layer was dried over MgSO4, filtered and concentrated in vacuo. The crude product was used in the next step without further purification (crude yield: 10 mg, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 7.88 (bs, 1H), 7.80 (bs, 1H), 7.35 (s, 2H), 4.08 – 3.94 (m, 1.75H), 3.84 – 3.79 (m, 0.25H), 3.12 – 3.05 (m, 1H), 2.90 – 2.85 (m, 1H), 2.33 – 2.24 (m, 1H), 2.16 – 2.01 (m, 2H), 1.92 (s, 1H), 1.60 (s, 9H), 1.42 (s, 9H), 1.33 (s, 9H), 1.25 (s, 3H, over integrates to 6H) LC-MS (m/z) calcd for C28H44N2O6 [M+H] + 505.3, found 505.8.
Di-tert-butyl-(2S,3R,4S)-4-(2-(benzylamino)ethyl)-3-(3-(tert-butoxycarbonyl)phenyl)pyrolidine-1,2-dicarboxylate (51b)
Aldehyde 50 (50 mg, 0.1 mmol, 1 equiv.) was dissolved in anhydrous MeOH (1 mL, 0.1 M). To this was added BnH2N·HCl (44 mg, 0.3 mmol, 3 equiv.) in one portion, followed by NaBH3CN (10 mg, 0.15 mmol, 1.5 equiv.). The colorless mixture was stirred at rt overnight and then concentrated to dryness. The crude mixture was purified by flash column chromatography (Et3N:MeOH:EtOAc:heptane 0.05:0.05:1:4). To afford the title compound as white foam (40mg, 70%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 7.88 (m, 1H), 7.83 (m, 1H), 7.37 (s, 2H), 7.28 – 7.26 (m, 2H), 7.22 – 7.19 (m, 3H), 4.12 – 4.01 (m, 1.8H), 3.90 – 3.87 (m, 0.2H), 3.64 (m, 2H), 3.17 – 3.13 (m, 1H), 2.93 – 2.89 (m, 1H), 2.55 – 2.38 (m, 3H), 1.60 (s, 9H), 1.43 (s, 9H), 1.32 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.1, 171.0, 165.5, 153.8, 153.5, 139.3, 132.4, 131.9, 131.7, 129.2, 129.1, 128.6, 128.6, 128.5, 128.4, 128.2, 127.1, 81.2, 81.1, 80.2, 79.9, 67.2, 67.1, 61.1, 57.3, 56.2, 53.6, 53.5, 52.4, 51.9, 47.4, 43.9, 43.3, 42.2 34.0, 31.4, 29.7, 28.4, 28.3, 28.2, 27.9, 27.9. LC-MS (m/z) calcd for C34H48N2O6 [M+H] + 581.4, found 581.9.
Di-tert-butyl-(2S,3R,4S)-3-(3-(tert-butoxycarbonyl)phenyl)-4-(2-hydroxyethyl)pyrrolidine-1,2-dicarboxylate (51c)
Aldehyde 50 (20 mg, 0.04 mmol, 1 equiv.) was dissolved in anhydrous MeOH (0.4 mL, 0.01 M). To this was added NaBH4 (1.5 mg, 0.04 mmol, 1 equiv.) in one portion. The reaction was stirred at rt for 30 minutes after which saturated NH4Cl solution was added. The reaction mixture was concentrated to dryness. The residue was taken up in EtOAc and water. The aqueous layer was extracted with EtOAc three times. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by column chromatography on silica gel (heptane-EtOAc, gradient 2:1 → 1:1) to afford the title compound as a colorless sticky oil (16.5 mg, 82%, mixture of rotamers). 1H NMR (600 MHz, CDCl3) δ 7.89 (s, 1H), 7.84 – 7.82 (m, 1H), 7.40 (s,1H), 4.11 – 4.03 (m, 1.75H), 3.96 – 3.93 (m, 0.25H), 3.54 – 3.50 (m, 1H), 3.20 – 3.16 (m, 1H), 2.95 – 2.92 (m, 1H), 2.52 – 2.43 (m, 1H), 1.59 (s, 9H) 1.42 (s, 9H), 1.33 (s, 9H). 13C NMR (151 MHz, CDCl3) δ 171.1, 165.5, 153.6, 139.4, 139.3, 132.4, 131.8, 131.7, 129.2, 128.7, 128.6, 81.3, 81.2, 81.2, 81.1, 80.2, 79.9, 67.2, 61.2, 61.1, 57.3, 56.1, 52.4, 52.0 42.9, 42.2, 34.1, 33.9, 29.7, 28.5, 28.4, 28.3, 28.2, 28.0, 27.9. LC-MS (m/z) calcd for C27H41NO7 [M+H] + 492.3; found 280.5 (-Boc, −2 x tert-butyl).
2-(3S,4R,5S)-1,5-bis(tert-butoxycarbonyl)-4-(3-(tert-butoxycarbonyl)phenyl)pyrrolidin-3-yl)acetic acid (51d)
Aldehyde 50 (30 mg, 0.06 mmol, 1 equiv.) was dissolved in a 1:1:1 mixture of THF/tBuOH/H2O (1.22 mL, 0.05M). To the colorless solution was added NaH2PO4 (7.4 mg, 0.06 mmol, 1 equiv.) and 2-methyl-2-butene (28 μL, 0.06 mmol, 4 equiv.). Then, NaO2Cl (11 mg, 0.12 mmol, 2 equiv.) was added and the reaction was allowed to stir at rt. The reaction became light yellow. After 1 h the reaction was colorless and water was added. The aqueous layer was acidified with 1N HCl to pH 3 and extracted with EtOAc three times. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The crude mixture was purified by column chromatography on silica gel (heptane-EtOAc 2:1 + 1vol% AcOH) to afford the title compound as a white foam (29 mg, 94%, mixture of rotamers). 1H NMR (400MHz, CDCl3) (Mixture of conformers) δ 7.90 (s, 1H), 7.82 (s, 1H), 7.3 (s, 2H), 4.21 – 4.13 (m, 1H), 4.09 – 4.02 (m, 1H), 3.23 – 3.18 (m, 1H), 2.98 – 2.93 (m, 1H), 2.73 (m, 1H), 2.37 – 2.22 (m, 2H), 1.59 (s, 9H), 1.42 (s, 9H), 1.32 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 176.7, 175.8, 170.8, 165.4, 153.7, 138.2, 132.5, 131.8, 129.2, 128.9, 128.8, 81.4, 81.4, 80.7, 76.7, 66.8, 56.2, 51.7, 41.3, 35.3, 28.4, 28.3, 28.2, 28.0, 27.9, 27.9. LC-MS (m/z) calcd for C27H39NO8 [M+H] + 505.3, found 294.5 (-Boc, −2 x tert-butyl).
Deprotection of 51a-d – General Deprotection M
For the deprotection the starting material was dissolved in 1mL anhydrous DCM. To this was added 1 mL TFA. The reaction mixture was stirred at room temperature overnight and subsequently concentrated to dryness to yield the final compound. Purification was performed as described below.
(2S,3R,4S)-3-(3-Carboxyphenyl)-4-(2-(methylamino)ethyl)pyrrolidine-2-carboxylic acid (9b)
General deprotection M. The crude product was purified by preparative HPLC and concentrated to dryness by lyophilization to afford the title compound as its zwitterion as a white solid (6.3 mg, 53% over two steps). Compound contains small amounts of an unknown impurity (<5%). Rt: 3.68 min; flow: 20 mL min−1; gradient 100% A (10 min); λ = 210 nm. 1H NMR (600MHz, D2O) δ 7.95–7.93 (m, 2H), 7.58 (d, J = 7.7 Hz, 1H), 7.49 (t, J = 7,7 Hz, 1H), 4.25 (d, J = 10.5 Hz, 1H), 3.74 (dd, J = 11.7, 7.3 Hz, 1H), 3.25 – 3.16 (m, 2H), 2.82 – 2.77 (m, 1H), 2.70 – 2.66 (m, 1H), 2.62 – 2.57 (m, 1H), 2.46 (s, 3H), 1.75 – 1.70 (m, 2H). 13C NMR (151 MHz, D2O) δ 171.3, 170.0, 137.3, 133.2, 130.5, 129.5, 129.5, 128.9, 65.9, 53.8, 49.4, 46.7, 43.4, 32.3. 26.31. LC-MS (m/z) calcd for C15H20N2O4 [M+H] + 293.1, found 293.5.
(2S,3R,4S)-4-(2-(Benzylamino)ethyl)-3-(3-carboxyphenyl)pyrrolidine-2-carboxylic acid (9c)
General deprotection M. The crude product was purified by preparative HPLC and concentrated to dryness by lyophilization to afford the title compound as its zwitterion as a white solid (21 mg, 84%). Rt: 10.02 min; flow: 20 mL min−1; gradient 0–50% B (10 min); λ = 210 nm. 1H NMR (600 MHz, D2O) δ 7.91 (d, J = 7.8 Hz, 1H) 7.79 (s, 1H), 7.52 – 7.47 (m, 1H), 7.44 (t, J = 7.7 Hz, 1H)), 7.35 – 7.32 (m, 1H), 7–29 – 7.26 (m, 2H), 7.09 – 7.08 (m, 2H), 4.14 – 4.09 (m, 1H), 4.04 – 3.86 (m, 2H), 3.70 – 3.67 (m, 1H), 3.15 – 3.08 (m, 2H), 2.58 – 2.53 (m, 2H), 2.34 (dt, J = 12.3, 5.2 Hz, 1H), 1.83 – 1.78 (m, 1H), 1.70 – 1.63 (m, 1H). 13C NMR (151 MHz, D2O) δ 171.5, 169.9, 137.6, 132.9, 130.4, 129.6, 129.5, 129.0, 128.8, 66.6, 54.0, 50.4, 49.5, 44.0, 43.4, 26.7. LC-MS (m/z) calcd for C21H24N2O4 [M+H]+ 369.2, found 369.4.
(2S,3R,4S)-3-(3-Carboxyphenyl)-4-(2-hydroxyethyl)pyrrolidine-2-carboxylic acid (9d)
General deprotection M. The crude product was purified by preparative HPLC and concentrated to dryness by lyophilization to afford the title compound as its zwitterion as a white solid (8 mg, 90%). Rt: 10.99 min; flow: 20 mL min−1; gradient 0–20% B (20 min); λ = 210 nm. 1H NMR (600 MHz, D2O) δ 7.92 (s, 1H), 7.9 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H)), 7.47 (t, J = 7.7 Hz, 1H), 4.14 (d, J = 10.5 Hz, 1H), 3.73 (dd, J = 11.7, 7.2 Hz, 1H), 3.39 – 3.31 (m, 2H), 3.17 – 3.09 (m, 1H), 2.59 – 2.52 (m, 1H), 1.57 – 1.48 (m, 2H). 13C NMR (151 MHz, D2O) δ 172.3, 170.3, 138.3, 133.4, 130.5, 129.4, 129.2, 129.1, 66.4, 59.7, 54.5, 50.2, 43.8, 32.3. LC-MS (m/z) calcd for C14H17NO5 [M+H] + 280.1, found 280.5.
(2S,3R,4S)-4-(Carboxymethyl)-3-(3-carboxyphenyl)pyrrolidine-2-carboxylic acid hydrochloride (9e)
General deprotection M. The obtained product was not further purified. The salt was treated with 1M aq HCl (2 mL) and the volatiles were evaporated under reduced pressure, repeating the same procedure three times to afford the title compound as hydrochloride salt as a white solid (18 mg, quantitative). 1H NMR (600 MHz, D2O) δ 7.93 (s, 1H), 7.91 (d, J = 7.7 Hz, 1H), 7.56 (d, J = 7.7 Hz, 1H)), 7.46 (t, J = 7.7 Hz, 1H), 4.29 – 4.25 (m, 1H), 3.81 (dd, J = 11.8, 7.5 Hz, 1H), 3.23 – 3.19 (m, 2H), 2.89 – 2.81 (m, 1H), 2.46 – 2.35 (m, 2H). 13C NMR (151 MHz, D2O) δ 175.3, 171.6, 170.2, 137.1, 133.4, 130.4, 129.5, 129.5, 129.2, 65.8, 53.5, 49.6, 42.5, 34.2. LC-MS (m/z) calcd for C14H15NO6 [M+H] + 294.1, found 294.6. Analytical HPLC: Rt: 9.61 min; flow: 1 mL min−1; gradient 0–25 % B (20 min); λ = 210 nm.
Pharmacological studies
Radio ligand binding
Affinities for native AMPA, KA, and NMDA receptors in rat cortical synaptosomes were determined using 5 nM [3H]AMPA, 5 nM [3H]KA, and [3H]CGP39653, respectively, as previously detailed.56 Rat brain membrane preparations used in these receptor-binding experiments were prepared according to a method previously described.57 Cpm values were fitted by nonlinear regression using GraphPad Prism version 7.02, GraphPad Software, La Jolla, CA, USA.
Ligand binding affinities at recombinant rat homomeric GluA1, GluA2 and GluK1–3 were determined as previously detailed.58,59 Ligands were diluted in assay buffer (GluA1–2: 50 mM Tris-HCl, 0.1M KSCN, 2.5 mM CaCl2 pH 7.2 at 4°C, GluK1–3: 50 mM Tris-HCl pH 7.1 at 4°C), mixed with sf9 insect cell membranes expressing the respective receptors and radioligand (GluA1–2: [3H]-(S)-AMPA (55.7 Ci/mmol; PerkinElmer), GluK1: [3H]-(S)-NF608 (16.3 Ci/mmol) (Alcaide et al.), GluK2 and GluK3: [isopropenyl-3H]-kainic acid (43.6–47.2 Ci/mmol; PerkinElmer) in a total volume of 0.25 mL followed by 2h equilibration at 4°C. GluA1–2 membranes were filtered through 24 mm GF/C-type glass fibre filters on a 12-well Millipore manifold and washed twice with ice-cold assay buffer. Filters were added to pony vials with 3 mL UltimaGold scintillation fluid and DPM determined with a TriCarb 2900 scintillation counter (PerkinElmer). GluK1–3 membranes were filtered through GF/B-type glass fibre filters in microtitre plate format (UniFilter-96, PerkinElmer) and washed twice with ice-cold assay buffer on a FilterMate manifold (PerkinElmer). The filters were dried at 70 °C for 1 h and 50 μL/well Microscint 20 (PerkinElmer) was added. Radioactivity was detected with a TopCounter (PerkinElmer). All competition curves (n ≤ 3 per ligand) were determined in triplicate at 12–16 ligand concentrations. Data were analyzed using GraphPad Prism 6 to determine ligand affinity (Ki) and Hill coefficient (nH) using the one-site Ki and four-parameter logistics equations, respectively.
Two-Electrode Voltage-Clamp electrophysiology
DNA constructs with rat cDNAs encoding GluN1–1a (Genbank accession number U11418 and U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were linearized using restriction enzymes and used as templates to synthesize cRNA using the mMessage mMachine kit (Ambion, Life Technologies, Paisley, UK) for expression in Xenopus oocytes. The cDNA encoding rat GluN2B was modified without changing the amino acid sequence to remove a T7 RNA polymerase termination site located in the C-terminal domain.60 Xenopus oocytes obtained from Rob Weymouth (Xenopus 1, Dexter, MI) were prepared as previously described,61 and injected with cRNAs encoding GluN1 and GluN2 in a 1:2 ratio. The oocytes were maintained at 18°C in Barth’s solution containing (in mM) 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 0.33 Ca(NO3)2, 0.91 CaCl2, 10 HEPES (pH 7.5 with NaOH) supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin and 50 μg/ml gentamycin (Invitrogen, Life Technologies, Paisley, UK). Two-electrode voltage-clamp recordings were performed at room temperature on Xenopus oocytes 2–6 days post-injection with the extracellular recording solution comprised of (in mM) 90 NaCl, 1 KCl, 10 HEPES, 0.5 BaCl2 and 0.01 EDTA (pH 7.4 with NaOH) at a holding potential of −40 mV essentially as previously described.61 NMDA receptor ligands were dissolved in extracellular recording solution and applied to the oocyte by gravity-driven perfusion. Concentration-inhibition data were fitted to the Hill equation to obtain IC50 values for individual oocytes using GraphPad Prism software (GraphPad Software, San Diego, CA, USA) as previously described.61
Supplementary Material
Figure 4. Concentration-inhibition data of 4h and 6b at NMDA receptor subtypes.

A) Representative two-electrode voltage-clamp recordings showing inhibition of GluN1/2A and GluN1/2D by 4h. Receptors were activated by 1 μM glutamate in the continuous presence of 100 μM glycine. B) Concentration-inhibition data for 4h and 6b at different recombinant NMDA receptor subtypes (GluN1/2A-D) measured using two-electrode voltage-clamp recordings. Data are mean ± SEM from 5–6 oocytes for each receptor subtype. The IC50 values were used to estimate Ki values using the Cheng-Prusoff relationship54 (Table 6).
Table 4.
Chemical structures and binding affinities of analogues 9a-s at native iGluRs (rat synaptosomes) and homomeric recombinant iGluRs. All values in μM.a
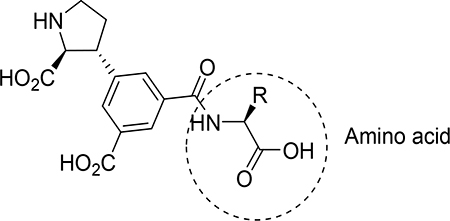 | |||||||
|---|---|---|---|---|---|---|---|
| Native iGluRs (synaptosomes)b | Cloned homomeric iGluRsc | ||||||
| Amino acid | AMPA IC50 | KA IC50 | NMDA Ki | GluK1 Ki | GluK2 Ki | GluK3 Ki | |
| 8a | Gly | 17 [4.76 ± 0.04] | 44 [4.37 ± 0.06] | 0.60 [6.23 ± 0.05] | 25.2 ± 1.8 | ’100 | 22.3 ± 6.0 |
| 8b | L- Ala | 32 [4.52 ± 0.10] | 54 [4.28 ± 0.06] | 0.87 [6.06 ± 0.02] | 13.3 ± 0.5 | ’100 | 17.8 ± 2.1 |
| 8c | L- Val | 27 [4.57 ± 0.02] | 27 [4.57 ± 0.05] | 3.4 [5.47 ± 0.04] | 4.41 ± 0.63 | ’100 | 5.80 ± 0.54 |
| 8d | L- Leu | 33 [4.49 ± 0.07] | 66 [4.20 ± 0.09] | 3.9 [5.41 ± 0.03] | 11.7 ± 1.4 | ’100 | 20.6 ± 3.1 |
| 8e | L- Ile | 10–50 | 10–50 | 5–10 | (68 ± 5)d | (91 ± 6)d | (51 ± 6)d |
| 8f | L- Phe | 10–50 | >50 | 1–5 | (86 ± 8)d | (103 ± 10)d | (66 ± 7)d |
| 8g | L-Tyr | 10 | 10–50 | 0.96 [6.02 ± 0.01] | (62 ± 1)d | (84 ± 9)d | 5.69 ± 0.90 (40 ± 7)d |
| 8h | L- Ser | 10–50 | >50 | 1–5 | (75 ± 2)d | (89 ± 14)d | (63 ± 6)d |
| 8i | L- Thr | 10–50 | 10–50 | 5 | (80 ± 2)d | (104 ± 7)d | (55 ± 10)d |
| 8j | L- Pro | 10–50 | 10–50 | 1.1 [5.98 ± 0.03] | 1.44 ± 0.18 (35 ± 1)d | (86 ± 1)d | 2.41 ± 1.06 (18 ± 4)d |
| 8k | L- Glu | 10–50 | >100 | 0.83 [6.08 ± 0.01] | (91 ± 6)d | (100 ± 14)d | (86 ± 3)d |
| 8l | L- Gln | -- | -- | -- | -- | -- | -- |
| 8m | L- Asp | 5–10 | >100 | 1–5 | (86 ± 7)d | (102 ± 5)d | (83 ± 6)d |
| 8n | L- Asn | -- | -- | -- | -- | -- | -- |
| 8o | L- Lys | 10–50 | 10–50 | 10–50 | 3.27 ± 0.55 (50 ± 5)d | (79 ± 4)d | 5.06 ± 0.03 (37 ± 2)d |
| 8p | L- Cys | 10–50 | 10–50 | 1–5 | (80 ± 2)d | (88 ± 6)d | 10–50 (60 ± 4)d |
| 8q | L- His | 10–50 | 10–50 | 5 | (76 ± 1)d | (66 ± 1)d | 6.23 ± 2.69 (44 ± 3)d |
| 8r | L- Trp | 10 | 10–50 | 1–5 | (72 ± 3)d | (90 ± 6)d | 10–50 (62 ± 5)d |
| 8s | L- Arg | 10 | 10 | 10–50 | 2.65 ± 1.06 (40 ± 1)d | 10–50 (66 ± 1)d | 2.78 ± 1.41 (29 ± 9)d |
--: Analog unstable in buffer - values not determined. Radio ligands used: AMPA, [3H]AMPA; KA, [3H]KA; [3H]CGP-39653; GluK1, [3H]SYM2081; GluK2 and GluK3, [3H]KA
Data are mean values of three to six individual experiments performed in triplicate. For AMPA and KA: pIC50 values with SEM in brackets. For NMDA: pKi values with SEM in brackets.
Mean values ± SEM of at least three experiments conducted in triplicate at 12–16 drug concentrations.
%specific binding mean values ± SD of three experiments performed at 10 μM ligand concentration.
Table 5.
Chemical structures and binding affinities of analogues 9a-e at native iGluRs (rat synaptosomes) and homomeric recombinant iGluRs. All values in μM.a
 | ||||||
|---|---|---|---|---|---|---|
| Native iGluRs (synaptosomes)b | Cloned homomeric iGluRsc | |||||
| AMPA IC50 | KA IC50 | NMDA Ki | GluK1 Ki | GluK2 Ki | GluK3 Ki | |
| 9a | 48 | 22 | 25 | 0.62 | 82 | 2.2 |
| 9b | 69 [4.16 ± 0.02] | >100 | >100 | ∼10 (50 ± 6)d | >100 (99 ± 6)d | >100 (83 ± 6)d |
| 9c | 15 [4.83 ± 0.05] | 45 [4.72 ± 0.03] | >100 | 2.98 ± 0.96 | >100 (86 ± 6)d | 2.46 ± 0.54 |
| 9d | 35 [4.47 ± 0.007] | 13 [4.89 ± 0.3] | 61 [4.22 ± 0.03] | >10 (60 ± 6)d | >100 (93 ± 7)d | >10 (57 ± 2)d |
| 9e | >100 | >100 | >100 | >100 (93 ± 3)d | >100 (97 ± 7)d | >100 (95 ± 3)d |
-,--: Values not determined, Radio ligands used: AMPA, [3H]AMPA; KA, [3H]KA; [3H]CGP-39653; GluK1, [3H]NF608; GluK2 and GluK3, [3H]KA
Data are mean values of three to six individual experiments performed in triplicate. For AMPA and KA: pIC50 values with SEM in brackets. For NMDA: pKi values with SEM in brackets.
Mean values ± SEM of at least three experiments conducted in triplicate at 12–16 drug concentrations.
% specific binding mean values ± SD of three experiments.
Acknowledgement
We would like to thank the Danish Research Council, Health and medical sciences for financial support. This work was funded by the National Institutes of Health (P20GM103546 and R01NS097536).
Footnotes
Supporting information available:
Experimental details for the synthesis of 8a-s.
References
- (1).Meldrum BS Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr 2000, 130 (4), 1007S–1015S. [DOI] [PubMed] [Google Scholar]
- (2).Traynelis S; Wollmuth L; McBain C; Menniti F; Vance K; Ogden K; Hansen KB; Yuan H; Myers S; Dingledine R Glutamate Receptor Ion Channels: Structure, Regulation, and Function Stephen. Pharmacol Rev 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Riaza Bermudo-Soriano C; Perez-Rodriguez MM; Vaquero-Lorenzo C; Baca-Garcia E New Perspectives in Glutamate and Anxiety. Pharmacol. Biochem. Behav 2012, 100 (4), 752–774. [DOI] [PubMed] [Google Scholar]
- (4).Sanacora G; Treccani G; Popoli M Towards a Glutamate Hypothesis of Depression: An Emerging Frontier of Neuropsychopharmacology for Mood Disorders. Neuropharmacology 2012, 62 (1), 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).McCarthy DJ; Alexander R; Smith MA; Pathak S; Kanes S; Lee CM; Sanacora G Glutamate-Based Depression GBD. Med. Hypotheses 2012, 78 (5), 675–681. [DOI] [PubMed] [Google Scholar]
- (6).Vikelis Michail; Mitsikostas Dimos D.. The Role of Glutamate and Its Receptors in Migraine. CNS Neurol. Disord. - Drug Targets 2007, 6 (4), 251–257. [DOI] [PubMed] [Google Scholar]
- (7).Weiss B; Alt A; Ogden AM; Gates M; Dieckman DK; Clemens-smith A; Ho KH; Jarvie K; Rizkalla G; Wright R. a; Calligaro DO; Schoepp D; Mattiuz EL; Stratford RE; Johnson B; Salhoff C; Katofiasc M; Phebus L. a; Schenck K; Cohen M; Filla S. a; Ornstein PL; Johnson KW; Bleakman D Pharmacological Characterization of the Competitive GLU K5 Receptor Antagonist Decahydroisoquinoline LY466195 in Vitro and in Vivo. J. Pharmacol. Exp. Ther 2006, 318 (2), 772–781. [DOI] [PubMed] [Google Scholar]
- (8).Lerma J; Marques JM Kainate Receptors in Health and Disease. Neuron 2013, 80 (2), 292–311. [DOI] [PubMed] [Google Scholar]
- (9).De Bartolomeis A; Sarappa C; Magara S; Iasevoli F Targeting Glutamate System for Novel Antipsychotic Approaches: Relevance for Residual Psychotic Symptoms and Treatment Resistant Schizophrenia. Eur. J. Pharmacol 2012, 682 (1–3), 1–11. [DOI] [PubMed] [Google Scholar]
- (10).Coyle JT; Basu A; Benneyworth M; Balu D; Konopaske G Novel Antischizophrenia Treatments. 2012, 213 (213), 1–27. [Google Scholar]
- (11).Javitt DC Glutamatergic Theories of Schizophrenia. Isr. J. Psychiatry Relat. Sci 2010, 47 (1), 4–16. [PubMed] [Google Scholar]
- (12).Almeida A; Heales SJR; Bolaños JP; Medina JM Glutamate Neurotoxicity Is Associated with Nitric Oxide-Mediated Mitochondrial Dysfunction and Glutathione Depletion. Brain Res. 1998, 790 (1–2), 209–216. [DOI] [PubMed] [Google Scholar]
- (13).Gu B; Nakamichi N; Zhang WS; Nakamura Y; Kambe Y; Fukumori R; Takuma K; Yamada K; Takarada T; Taniura H; Yoneda Y Possible Protection by Notoginsenoside R1 against Glutamate Neurotoxicity Mediated by N-Methyl-D-Aspartate Receptors Composed of an NR1/NR2B Subunit Assembly. J. Neurosci. Res 2009, 87 (9), 2145–2156. [DOI] [PubMed] [Google Scholar]
- (14).Kumar A; Singh RL; Babu GN Cell Death Mechanisms in the Early Stages of Acute Glutamate Neurotoxicity. Neurosci. Res 2010, 66 (3), 271–278. [DOI] [PubMed] [Google Scholar]
- (15).Danysz W; Parsons CG Alzheimer’s Disease, β-Amyloid, Glutamate, NMDA Receptors and Memantine - Searching for the Connections. Br. J. Pharmacol 2012, 167 (2), 324–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Francis PT Glutamatergic Approaches to the Treatment of Cognitive and Behavioural Symptoms of Alzheimer’s Disease. Neurodegener. Dis 2008, 5 (3–4), 241–243. [DOI] [PubMed] [Google Scholar]
- (17).Fan MMY; Raymond LA N-Methyl-d-Aspartate (NMDA) Receptor Function and Excitotoxicity in Huntington’s Disease. Prog. Neurobiol 2007, 81 (5–6), 272–293. [DOI] [PubMed] [Google Scholar]
- (18).Corona JC; Tovar-y-Romo LB; Tapia R Glutamate Excitotoxicity and Therapeutic Targets for Amyotrophic Lateral Sclerosis. Expert Opin. Ther. Targets 2007, 11 (11), 1415–1428. [DOI] [PubMed] [Google Scholar]
- (19).Muir KW Glutamate-Based Therapeutic Approaches: Clinical Trials with NMDA Antagonists. Curr. Opin. Pharmacol 2006, 6 (1 SPEC. ISS.), 53–60. [DOI] [PubMed] [Google Scholar]
- (20).Lee K; Goodman L; Fourie C; Schenk S; Leitch B; Montgomery JM AMPA Receptors as Therapeutic Targets for Neurological Disorders. Adv. Protein Chem. Struct. Biol 2016, 103, 203–261. [DOI] [PubMed] [Google Scholar]
- (21).Foster AC; Fagg GE Taking Apart NMDA Receptors. Nature 1987, 329 (6138), 395–396. [DOI] [PubMed] [Google Scholar]
- (22).Huettner JE; Bean BP Block of N-Methyl-D-Aspartate-Activated Current by the Anticonvulsant MK-801: Selective Binding to Open Channels. Proc. Natl. Acad. Sci 1988, 85 (4), 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Burnell ES; Irvine M; Fang G; Sapkota K; Jane DE; Monaghan DT Positive and Negative Allosteric Modulators of N -Methyl- d -Aspartate (NMDA) Receptors: Structure–Activity Relationships and Mechanisms of Action. J. Med. Chem 2019, 62 (1), 3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Honoré T; Davies SN; Dreier J; Fletcher EJ; Jacobsen P; Lodge D; Nielsen FE Quinoxalinediones: Potent Competitive Non-NMDA Glutamate Receptor Antagonists. Science (80-.). 1988, 241 (4866), 701–703. [DOI] [PubMed] [Google Scholar]
- (25).Löscher W A New Pyrrolyl-Quinoxalinedione Series of Non-NMDA Glutamate Receptor Antagonists: Pharmacological Characterization and Comparison with NBQX and Valproate in the Kindling Model of Epilepsy. Eur. J. Neurosci 1999, 11 (1), 250–262. [DOI] [PubMed] [Google Scholar]
- (26).Demmer CSCS; Rombach D; Liu N; Nielsen B; Pickering DSDS; Bunch L Revisiting the Quinoxalinedione Scaffold in the Construction of New Ligands for the Ionotropic Glutamate Receptors. ACS Chem. Neurosci 2017, 8 (11), 2477–2495. [DOI] [PubMed] [Google Scholar]
- (27).Stensbøl TB; Madsen U; Krogsgaard-larsen P The AMPA Receptor Binding Site: Focus on Agonists and Competitive Antagonists. Curr. Pharm. Des 2002, 8, 857–872. [DOI] [PubMed] [Google Scholar]
- (28).Dolman NP; More JCA; Alt A; Knauss JL; Troop HM; Bleakman D; Collingridge GL; Jane DE Structure-Activity Relationship Studies on N3-Substituted Willardiine Derivatives Acting as AMPA or Kainate Receptor Antagonists. J. Med. Chem 2006, 49 (8), 2579–2592. [DOI] [PubMed] [Google Scholar]
- (29).Jane DE; Hoo K; Kamboj R; Deverill M; Bleakman D; Mandelzys A Synthesis of Willardiine and 6-Azawillardiine Analogs: Pharmacological Characterization on Cloned Homomeric Human AMPA and Kainate Receptor Subtypes. J. Med. Chem 1997, 40 (22), 3645–3650. [DOI] [PubMed] [Google Scholar]
- (30).Gmelin R Die Freien Aminosäuren Der Samen von Acacia Willardiana. 1959, 316, 164–169. [DOI] [PubMed] [Google Scholar]
- (31).Ornstein PL; Schoepp DD; Arnold MB; Augenstein NK; Lodge D; Millar JD; Chambers J; Campbell J; Paschal JW 6-Substituted Decahydroisoquinoline-3-Carboxylic Acids as Potent and Selective Conformationally Constrained NMDA Receptor Antagonists. J. Med. Chem 1992, 35 (19), 3547–3560. [DOI] [PubMed] [Google Scholar]
- (32).Isaacson J; Nicoll R Aniracetam Reduces Glutamate Receptor Desensitization and Slows the Decay of Fast Excitatory Synaptic Currents in the Hippocampus. Proc. Natl. Acad. Sci 1991, 88, 10936–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ornstein PL; Schoepp DD; Arnold MB; Augenstein NK; Lodge D; Millar JD; Chambers J; Campbell J; Paschal JW; Zimmerman DM; Leander JD 6-Substituted Decahydroisoquinoline −3-Carboxylic Acids as Potent and Selective Conformationally Constrained NMDA Receptor Antagonists. J. Med. Chem 1992, 35 (19), 3547–3560. [DOI] [PubMed] [Google Scholar]
- (34).O’Neill MJ; Bond A; Ornstein PL; Ward MA; Hicks CA; Hoo K; Bleakman D; Lodge D Decahydroisoquinolines: Novel Competitive AMPA/Kainate Antagonists with Neuroprotective Effects in Global Cerebral Ischaemia. Neuropharmacology 1998, 37 (10–11), 1211–1222. [DOI] [PubMed] [Google Scholar]
- (35).Krogsgaard-Larsen N; Delgar CG; Koch K; Brown PMGE; Møller C; Han L; Huynh THV; Hansen SW; Nielsen B; Bowie D; Pickering DS; Kastrup JS; Frydenvang K; Bunch L Design and Synthesis of a Series of L-Trans-4-Substituted Prolines as Selective Antagonists for the Ionotropic Glutamate Receptors Including Functional and X-Ray Crystallographic Studies of New Subtype Selective Kainic Acid Receptor Subtype 1 (GluK1) Anta. J. Med. Chem 2017, 60 (1), 441–457. [DOI] [PubMed] [Google Scholar]
- (36).Larsen AM; Venskutonytė R; Valadés EA; Nielsen B; Pickering DS; Bunch L Discovery of a New Class of Ionotropic Glutamate Receptor Antagonists by the Rational Design of (2S,3R)-3-(3-Carboxyphenyl)-Pyrrolidine-2-Carboxylic Acid. ACS Chem. Neurosci 2011, 2 (2), 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dolman NP; More JCA; Alt A; Knauss JL; Pentikäinen OT; Glasser CR; Bleakman D; Mayer ML; Collingridge GL; Jane DE Synthesis and Pharmacological Characterization of N 3 -Substituted Willardiine Derivatives: Role of the Substituent at the 5-Position of the Uracil Ring in the Development of Highly Potent and Selective GLU K5 Kainate Receptor Antagonists. J. Med. Chem 2007, 50 (7), 1558–1570. [DOI] [PubMed] [Google Scholar]
- (38).Krogsgaard-Larsen N; Storgaard M; Møller C; Demmer CS; Hansen J; Han L; Monrad RN; Nielsen B; Tapken D; Pickering DS; Kastrup JS; Frydenvang K; Bunch L Structure-Activity Relationship Study of Ionotropic Glutamate Receptor Antagonist (2S,3R)-3-(3-Carboxyphenyl)Pyrrolidine-2-Carboxylic Acid. J. Med. Chem 2015, 58 (15), 6131–6150. [DOI] [PubMed] [Google Scholar]
- (39).Gynther M; Proietti Silvestri I; Hansen JC; Hansen KBKB; Malm T; Ishchenko Y; Larsen Y; Han L; Kayser S; Auriola S; Petsalo A; Nielsen B; Pickering DSDS; Bunch L Augmentation of Anticancer Drug Efficacy in Murine Hepatocellular Carcinoma Cells by a Peripherally Acting Competitive N-Methyl- d -Aspartate (NMDA) Receptor Antagonist. J. Med. Chem 2017, 60 (23), 9885–9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nitta I; Watase H; Tomiie Y Structure of Kainic Acid and Its Isomer, Allokainic Acid [6]. Nature 1958, 181 (4611), 761–762. [DOI] [PubMed] [Google Scholar]
- (41).Takemoto T; Daigo K Constituents of Chondra Armata. Chem. Pharm. Bull 1958, 6 (5), 578–580. [Google Scholar]
- (42).Takemoto T; Daigo K Ueber Die Inhaltsstoffe von Chondria Armata Und Ihre Pharmakologische Wirkung. Arch. Pharm. (Weinheim) 1960, 293 (6), 627–633. [Google Scholar]
- (43).Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev 2010, 62 (3), 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Venskutonyte R; Frydenvang K; Gajhede M; Bunch L; Pickering DS; Kastrup JS Binding Site and Interlobe Interactions of the Ionotropic Glutamate Receptor GluK3 Ligand Binding Domain Revealed by High Resolution Crystal Structure in Complex with (S)-Glutamate. J. Struct. Biol 2013, 176 (2), 381. [DOI] [PubMed] [Google Scholar]
- (45).Frydenvang K; Lash LL; Naur P; Postila PA; Pickering DS; Smith CM; Gajhede M; Sasaki M; Sakai R; Pentik??nen OT; Swanson GT; Kastrup JS Full Domain Closure of the Ligand-Binding Core of the Ionotropic Glutamate Receptor IGluR5 Induced by the High Affinity Agonist Dysiherbaine and the Functional Antagonist 8,9-Dideoxyneodysiherbaine. J. Biol. Chem 2009, 284 (21), 14219–14229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Swanson GT; Gereau IV RW; Green T; Heinemann SF Identification of Amino Acid Residues That Control Functional Behavior in GluR5 and GluR6 Kainate Receptors. Neuron 1997, 19 (4), 913–926. [DOI] [PubMed] [Google Scholar]
- (47).Huy P; Neudörfl J-M; Schmalz H-GG; Neudörfl JM; Schmalz H-GG A Practical Synthesis of Trans −3-Substituted Proline Derivatives through 1,4-Addition. Org. Lett 2011, 13 (2), 216–219. [DOI] [PubMed] [Google Scholar]
- (48).Affron DP; Davis OA; Bull JA Regio- and Stereospecific Synthesis of C-3 Functionalized Proline Derivatives by Palladium Catalyzed Directed C(Sp 3)–H Arylation. Org. Lett 2014, 16 (18), 4956–4959. [DOI] [PubMed] [Google Scholar]
- (49).Affron DP; Bull JA Palladium-Catalyzed Directed C(Sp 3)-H Arylation of Saturated Heterocycles at C-3 Using a Concise Optimization Approach. European J. Org. Chem 2016, 2016 (1), 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fujiwara Y; Kawauchi T; Taniguchi H Palladium-Promoted One-Step Carboxylation of Aromatic Compounds with Carbon Monoxide. J. Chem. Soc. Chem. Commun 1980, No. 5, 220–221. [Google Scholar]
- (51).Sandmeyer T Ueber Die Ersetzung Der Amid-Gruppe Durch Chlor, Brom Und Cyan in Den Aromatischen Substanzen. Berichte der Dtsch. Chem. Gesellschaft 1884, 17 (2), 2650–2653. [Google Scholar]
- (52).Sonogashira K Development of Pd-Cu Catalyzed Cross-Coupling of Terminal Acetylenes with Sp2-Carbon Halides. J. Organomet. Chem 2002, 653 (1–2), 46–49. [Google Scholar]
- (53).Rostovtsev VV; Green LG; Fokin VV; Sharpless KBA Stepwise Huisgen Cycloaddition Process: Copper(i)-Catalyzed Regioselective TMLigation∫ of Azides and Terminal Alkynes. Angew. Chemie - Int. Ed 2002, 41 (14), 2596–2599. [DOI] [PubMed] [Google Scholar]
- (54).Yung-Chi C; Prusoff WH Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol 1973, 22 (23), 3099–3108. [DOI] [PubMed] [Google Scholar]
- (55).Lind GE; Mou T-C; Tamborini L; Pomper MG; De Micheli C; Conti P; Pinto A; Hansen KB Structural Basis of Subunit Selectivity for Competitive NMDA Receptor Antagonists with Preference for GluN2A over GluN2B Subunits. Proc. Natl. Acad. Sci 2017, 114 (33), E6942–E6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Assaf Z; Larsen AP; Venskutonytě R; Han L; Abrahamsen B; Nielsen B; Gajhede M; Kastrup JS; Jensen AA; Pickering DS; Frydenvang K; Gefflaut T; Bunch L; Venskutonytė R; Han L; Abrahamsen B; Nielsen B; Gajhede M; Kastrup JS; Jensen AA; Pickering DS; Frydenvang K; Gefflaut T; Bunch L Chemoenzymatic Synthesis of New 2,4-Syn-Functionalized (S)-Glutamate Analogues and Structure-Activity Relationship Studies at Ionotropic Glutamate Receptors and Excitatory Amino Acid Transporters. J. Med. Chem 2013, 56 (4), 1614–1628. [DOI] [PubMed] [Google Scholar]
- (57).Ransom RW; Stec NL Cooperative Modulation of [ 3 H]MK-801 Binding to the N-Methyl-d-Aspartate Receptor-Ion Channel Complex by l-Glutamate, Glycine, and Polyamines. J. Neurochem 1988, 51 (3), 830–836. [DOI] [PubMed] [Google Scholar]
- (58).Alcaide A; Marconi L; Marek A; Haym I; Nielsen B; Møllerud S; Jensen M; Conti P; Pickering DS; Bunch L Synthesis and Pharmacological Characterization of the Selective GluK1 Radioligand (S)-2-Amino-3-(6-[ 3 H]-2,4-Dioxo-3,4-Dihydrothieno[3,2-d]Pyrimidin-1(2H)-Yl)Propanoic Acid ([ 3 H]-NF608). Medchemcomm 2016, 7 (11), 2136–2144. [Google Scholar]
- (59).Sagot E; Pickering DS; Pu X; Umberti M; Stensbøl TB; Nielsen B; Chapelet M; Bolte J; Gefflaut T; Bunch L; Stensboel TB; Nielsen B; Chapelet M; Bolte J; Gefflaut T; Bunch L Chemo-Enzymatic Synthesis of a Series of 2,4-Syn-Functionalized (S)-Glutamate Analogues: New Insight into the Structure-Activity Relation of Ionotropic Glutamate Receptor Subtypes 5, 6, and 7. J. Med. Chem 2008, 51 (14), 4093–4103. [DOI] [PubMed] [Google Scholar]
- (60).Hansen KB; Ogden KK; Yuan H; Traynelis SF Distinct Functional and Pharmacological Properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA Receptors. Neuron 2014, 81 (5), 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Hansen KB; Tajima N; Risgaard R; Perszyk RE; Jørgensen L; Vance KM; Ogden KK; Clausen RP; Furukawa H; Traynelis SF Structural Determinants of Agonist Efficacy at the Glutamate Binding Site of N-Methyl-D-Aspartate Receptors. Mol. Pharmacol. 2013, 84 (1), 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.