

Abstract
Background:
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. In this study we assessed the effect of antisense oligonucleotide eluforsen on CFTR biological activity measured by Nasal Potential Difference (NPD) in patients with the most common mutation, F508del-CFTR.
Methods:
This multi-centre, exploratory, open-label study recruited adults with CF homozygous or compound heterozygous for the F508del-CFTR mutation. Subjects received intranasal eluforsen three times weekly for 4 weeks. The primary endpoint was the within-subject change from baseline in total chloride transport (Cl-free+iso), as assessed by NPD. Secondary endpoints included within-subject change from baseline in sodium transport.
Results:
In the homozygous cohort (n = 7; per-protocol population), mean change (90% confidence interval) in Cl-free+iso was −3.0 mV (−6.6; 0.6) at day 15, −4.1 mV (−7.8; −0.4, p = .04) at day 26 (end of treatment) and − 3.7 mV (−8.0; 0.6) at day 47. This was supported by improved sodium transport as assessed by an increase in average basal potential difference at day 26 of +9.4 mV (1.1; 17.7, p = .04). The compound heterozygous cohort (n = 7) did not show improved chloride or sodium transport NPD values. Eluforsen was well tolerated with a favourable safety profile.
Conclusions:
In F508del-CFTR homozygous subjects, repeated intranasal administration of eluforsen improved CFTR activity as measured by NPD, an encouraging indicator of biological activity.
Keywords: Antisense Oligonucleotide, Clinical Trial, Cystic Fibrosis Transmembrane Conductance, Regulator delta F508, Nasal Potential Difference, Pulmonary Medicine
1. Introduction
Cystic fibrosis (CF) is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene [1] leading to absence or dysfunction of the CFTR protein. Features of CF lung disease include mucus dehydration, chronic inflammation and impaired clearance of bacterial pathogens [2]. The most common CF-causing mutation is a deletion of three nucleotides leading to the loss of a phenylalanine at position 508 of the protein (p.Phe508del [F508del hereafter]). Nearly 90% of patients with CF have an F508del-encoding mutation (F508del-CFTR hereafter), and over half of these individuals are homozygous for the mutation [3,4].
CFTR is a chloride and bicarbonate channel that is expressed at the apical membrane of epithelial cells in various organs including the upper and lower respiratory tracts [5]. CFTR has additional regulatory roles including inhibition of sodium transport through the epithelial sodium channel (ENaC), activation of other epithelial transporters, and pH regulation [6–10]. Dysfunction of CFTR therefore leads to ion transport defects, which can be detected in the upper airways by measuring the Nasal Potential Difference (NPD). NPD is a direct and sensitive method of evaluation of chloride and sodium transport across the nasal epithelium by assessment of transepithelial bioelectric properties, and with the appropriate reagents is specific for CFTR-dependent chloride transport [11–15]. The nasal epithelium shares histological and ion transport features with the bronchial epithelium [12]. NPD is used to diagnose CF in patients with equivocal results from sweat and genetic testing, and has been used as an endpoint for investigating CFTR-targeted interventions such as CFTR modulators and gene transfer [14,16–18]. In several studies evaluating CFTR-directed correctors and potentiators, NPD responses towards normal were associated with improved sweat chloride and clinical improvements in later phase studies [17,19–21].
Substantial progress has been made in the development of disease-modifying therapies using small molecules that directly target CFTR [16,17]. However CFTR-enhancing therapies with broader applicability and greater efficacy are still needed. Recently, molecular therapies that target RNA have been successfully advanced as a novel treatment option to target specific mutations in diseases such as Duchenne’s muscular dystrophy and spinal muscular atrophy [22,23]. Eluforsen,2 previously known as QR-010, is an antisense 33-mer RNA oligonucleotide with a phosphorothioate backbone and full 2’-O-methylation, designed to bind specifically to the mRNA region around the F508-encoding deletion and to restore CFTR protein function in the airway epithelium. Pre-clinical data showed that eluforsen improved CFTR function as measured by in vitro functional assays using primary bronchial epithelial cultures from F508del-CFTR homozygous donors and NPD in F508del-CFTR CF mice [24]. Based on these data, eluforsen is being developed as a novel RNA-based therapy for patients with CF carrying the F508del-CFTR mutation. The aim of this exploratory study was to assess the ability of topical, intranasal administration of eluforsen to improve CFTR biological activity in respiratory epithelium as assessed by NPD in subjects with CF who are homozygous or compound heterozygous for the F508del-CFTR mutation.
2. Methods
Further details of the methods are provided in the online supplementary material.
2.1. Study design and participants
This was an exploratory, open-label study conducted at five hospitals in the USA and Europe. The trial is registered with clinicaltrials.gov (NCT02564354). The protocol and informed consent form were approved by the institutional review board or independent ethics committee at each investigational site prior to study initiation, and the study was conducted in accordance with the ethical principles of Good Clinical Practice and the Declaration of Helsinki. Each subject provided written informed consent at screening, prior to undergoing any protocol-related procedures. Eligible participants were aged ≥18 years, had a confirmed diagnosis of CF (sweat chloride N60 mmol/L), with confirmation of the F508del-CFTR gene mutation (homozygous or compound heterozygous), and a total chloride transport response consistent with CF (i.e. more positive than −6.6 mV) as measured by NPD at baseline. The assessment of the NPD for eligibility included a quantitative analysis of the total chloride response that was based on real time NPD review performed by the enrolling investigator. This was necessary, as the central NPD analysis laboratory could not provide real time quantitative analysis for the pre-dose NPD tracings and simultaneously preserve blinding to site and study timing. A total chloride transport response (i.e. a response to chloride-free plus isoproterenol infusion [Cl-free+iso]) more positive than −6.6 mV has a 95% sensitivity to discriminate between CF and non-CF subjects, as derived from the Therapeutic Development Network (TDN) database [25]. Further eligibility criteria are shown (supplementary Table S1 in the online supplement. Use of ivacaftor or lumacaftor was prohibited from 30 days prior to screening through to the end of the study, and ancillary therapy regimens and airway clearance techniques remained consistent throughout the study period. Subjects were grouped into an F508del CFTR homozygous cohort and a compound heterozygous cohort.
2.2. Analysis populations
The safety population included all subjects who received at least one dose of eluforsen. The full analysis set included all subjects who received at least one dose of eluforsen and had one or more pre- and one or more post-baseline NPD assessments. The per-protocol population included all subjects in the full analysis set with an average baseline Cl-free +iso NPD response more positive than −6.6 mV.
2.3. Procedures
Subjects received 10 mg eluforsen (5 mg/250 μL saline per nostril) via bilateral intranasal administration three times a week for 4 weeks (unless an intolerable adverse event [AE] was experienced), for a total of 12 doses. The dosing regimen used was based on preclinical data from NPD studies of eluforsen in mice [24]. Eluforsen was administered via the LMA Mucosal Atomization Device 300 (Teleflex, Wayne, PA, USA) by a trained healthcare provider. NPD and nasal assessments (via the Nasal Examination Rating Scale [NERS]) were conducted according to the standard operating procedure used by US Cystic Fibrosis Foundation TDN and European Cystic Fibrosis Society Clinical Trials Network (CTN; SOP 528.01). NPD solutions were shipped to all sites by a central provider to avoid variability in the preparation. In brief, the procedure for NPD assessment involved recording voltage tracings from the left and right nostrils while solutions were sequentially perfused through a nasal catheter. Solutions comprised Ringer’s solution to assess the basal potential difference (PD), amiloride to inhibit ENaC, chloride-free solution to induce an electrogenic chloride gradient, isoproterenol to stimulate cyclic adenosine monophosphate-dependent chloride secretion across the epithelium and adenosine triphosphate to stimulate calcium-activated chloride conductance (serving as a positive control for epithelial membrane integrity) [11–14,19]. The final quality review and scoring were performed by a single independent central reader who received data in a randomised batched manner, and was blinded to genotype, subject ID and time point. Assessment of nasal symptoms for safety monitoring was conducted using the Sino-Nasal Outcome Test (SNOT-22).
2.4. Outcomes and measurements
The primary endpoint was the within-subject change from baseline in total chloride transport (Cl-free+iso), measured by NPD. Other chloride transport parameters that were assessed included change in PD following Cl-free perfusion (delta Cl-free) and change in PD following isoproterenol perfusion (delta iso). The sodium transport parameters that were assessed included the average basal PD (from five sites in the inferior meatus) prior to infusion of any solution, the maximum basal PD prior to infusion of any solution, PD following perfusion of Ringer’s solution (Ringer’s PD) and change in PD following perfusion of amiloride (amiloride PD). This selection of NPD parameters was based on those recommended by Rowe et al. [19]. The primary data analysis was based on the average measurements of both nostrils. To provide baseline stability, baseline was defined as the average of the two most recent pre-dose values, where each pre-dose value was the average of two nostrils.
2.5. Statistical analysis
All NPD data were assessed for normality using graphical and diagnostic statistics. NPD analyses were performed in each cohort. In accordance with the predefined statistical analysis plan, changes from baseline to day 15, day 26 and day 47 for each cohort were tested by one-sided paired t-tests at the 5% alpha level. The primary NPD response (i.e. a response to Cl-free+iso) was pre-defined as a decrease, which is, by definition, a one-sided response. As such, to be consistent with the primary objective of the trial (i.e. to demonstrate a decrease in Cl-free +iso), the statistical test was pre-planned as one- sided. Summary statistics, p values and corresponding 90% confidence intervals (CIs) are provided.
3. Results
3.1. Subject disposition and demographics
Subjects were recruited between 19 October 2015 and 14 July 2016. As shown in Fig. 1, 18 eligible subjects with CF were enrolled in this study. Three subjects in the homozygous cohort and one subject in the compound heterozygous cohort did not meet the NPD entry criterion (Cl-free+iso value was more negative than −6.6 mV at baseline) when assessed by the independent central reader and so were excluded from the per-protocol population (n = 7 per cohort; supplementary Table S2). A summary of baseline characteristics (Table 1) reveals typical clinical features of the classical CF phenotype. All subjects in the compound heterozygous cohort had a different mutation on the second allele. Of the NPD tracings, 4/204 (homozygous) and 10/164 (compound heterozygous) could not be used due to uninterpretable readings, as judged by a blinded interpreter. Subjects in the compound heterozygous cohort were older than those in the homozygous cohort, and showed a more negative baseline Cl-free+iso value (Table 1), but this was within the range of expected CF values.
Fig. 1.
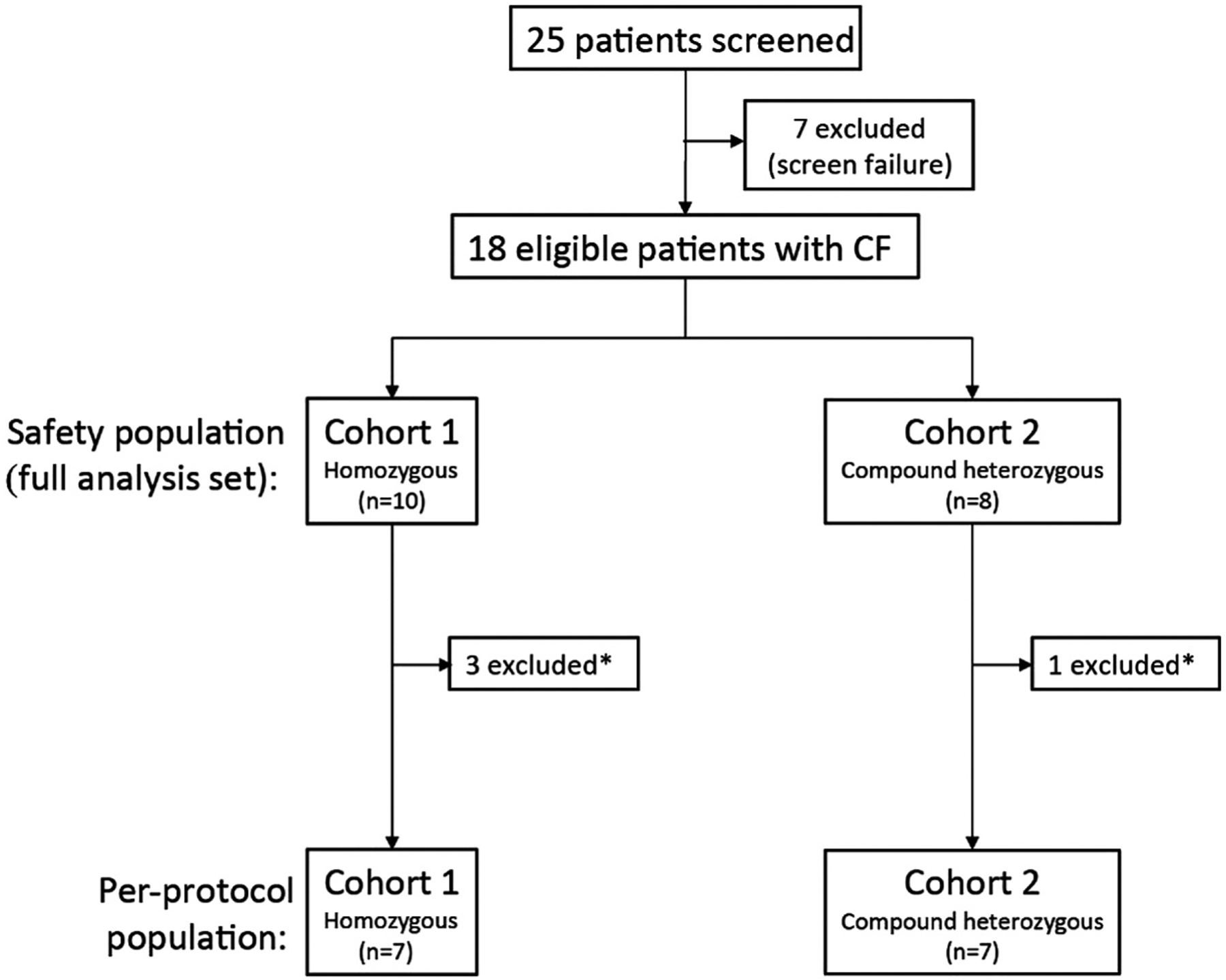
Trial profile. Patient numbers differ between safety population (full analysis set) and per-protocol population due to subjects not meeting NPD inclusion criterion (−6.6 mV cut-off established by TDN to discriminate between non-CF and CF values) when interpreted by a central reader, as described in the Methods and Discussion sections. *Did not meet the NPD inclusion criterion (CI-free+isoproterenol value was more negative than −6.6 mV at baseline). CF, cystic fibrosis; NPD, Nasal Potential Difference; TDN, Therapeutic Development Network.
Table 1.
Baseline and demographic characteristics.
| Homozygous cohort (n = 7) | Compound heterozygous cohort (n = 7) | |
|---|---|---|
| Age (years) | 27 (20; 36) | 36 (18; 63) |
| Sex, n (%) | ||
| Male | 5(71) | 3(43) |
| Female | 2 (29) | 4(57) |
| BMI (kg/m2) | 22 (20; 26) | 23 (20; 28) |
| History of sinus surgery, n (%) | 0 (0) | 2 (29) |
| History of nasal polyps, n (%) | 0 (0) | 3(43) |
| Predicted FEV1 (%) | 72.7 (45.2; 108.8) | 72.7 (52.3; 93.8) |
| Sweat chloride (mmol/L) | 104.3 (80.0; 117.5) | 104.6 (86.0; 134.0) |
| SNOT−22 total score | 15 (8.0; 23.0) | 21 (8.0; 59.0) |
| Cl-free + iso (mV) | +1.9 (−4.0; 6.4) | −0.8 (−5.0; 6.3) |
| Delta Cl-free (mV) | +1.3 (−5.1; 4.5) | −0.4 (−4.9; 6.8) |
| Delta isoproterenol (mV) | +0.6 (−1.7; 1.9) | −0.3 (−3.7; 4.8) |
| Average basal PD (mV) | −31.9 (−44.3; −16.4) | −27.0 (−45.0; −15.8) |
| Maximum basal PD (mV) | −39.5 (−49.9; −19.7) | −34.4 (−53.8; −18.1) |
| Ringer’s PD (mV) | −42.7 (−72.7; −20.9) | −32.8 (−60.5; −18.0) |
Data are mean (range) unless otherwise specified. Each subject in the compound heterozygous cohort had a different mutation in the second allele: p.Gln493, splicing (c.489 + 1 G N T), p.Asn1303Lys, p.Ile336Lys, splicing (c.2657 + 5 G N A), p.Tyr1092 [STOP] and splicing (c.1585–1 G N A).
BMI: body mass index; Cl-free+iso: within-subject change from baseline in total chloride transport; FEV1: forced expiratory volume in 1 s; PD: potential difference; SNOT-22: Sino-Nasal Outcome Test.
3.2. Chloride transport
Eluforsen was associated with increased total chloride transport as assessed by NPD from baseline to end of treatment (EOT) and at post-treatment follow-up for subjects in the homozygous cohort (Fig. 2a and b, and Table 2). This was observed as an increase in polarisation of the nasal epithelium (i.e. more negative values) following Cl-free+iso on day 15 (mean change −3.0 mV [90% CI –6.6; 0.6], p = .08), which was significantly increased by EOT on day 26 (mean change −4.1 mV [90% CI –7.8; −0.4], p = .04) and sustained at the final post-treatment study visit on day 47 (mean change −3.7 mV [90% CI –8.0; 0.6], p = .07). Sensitivity analyses of the Cl-free+iso data of the homozygous cohort showed that the different analysis methods were consistent with the results from the primary analysis method (i.e. the average of both nostrils method; supplementary Fig. S1). The relative contributions of Cl-free response versus isoproterenol response to the total chloride transport were similar in magnitude (delta Cl-free versus delta iso; Table 2 and supplementary Fig. S2). Individual subject data demonstrate a favourable distribution of responders with active treatment (supplementary Figs. S4 and S5a). Eluforsen did not increase mean total chloride transport in the compound heterozygous cohort (Fig. 2; mean change: day 15 + 2.4 mV [90% CI –1.0; 50.8], day 26 + 2.2 mV [90% CI –2.1; 6.4] and day 47 + 3.0 mV [90% CI –1.1; 7.2]). Sensitivity analyses of the Cl-free+iso data for the compound heterozygous cohort are shown in supplementary Fig. S3; individual subject data for the compound heterozygous cohorts are shown in supplementary Figs. S4 and S5b.
Fig. 2.
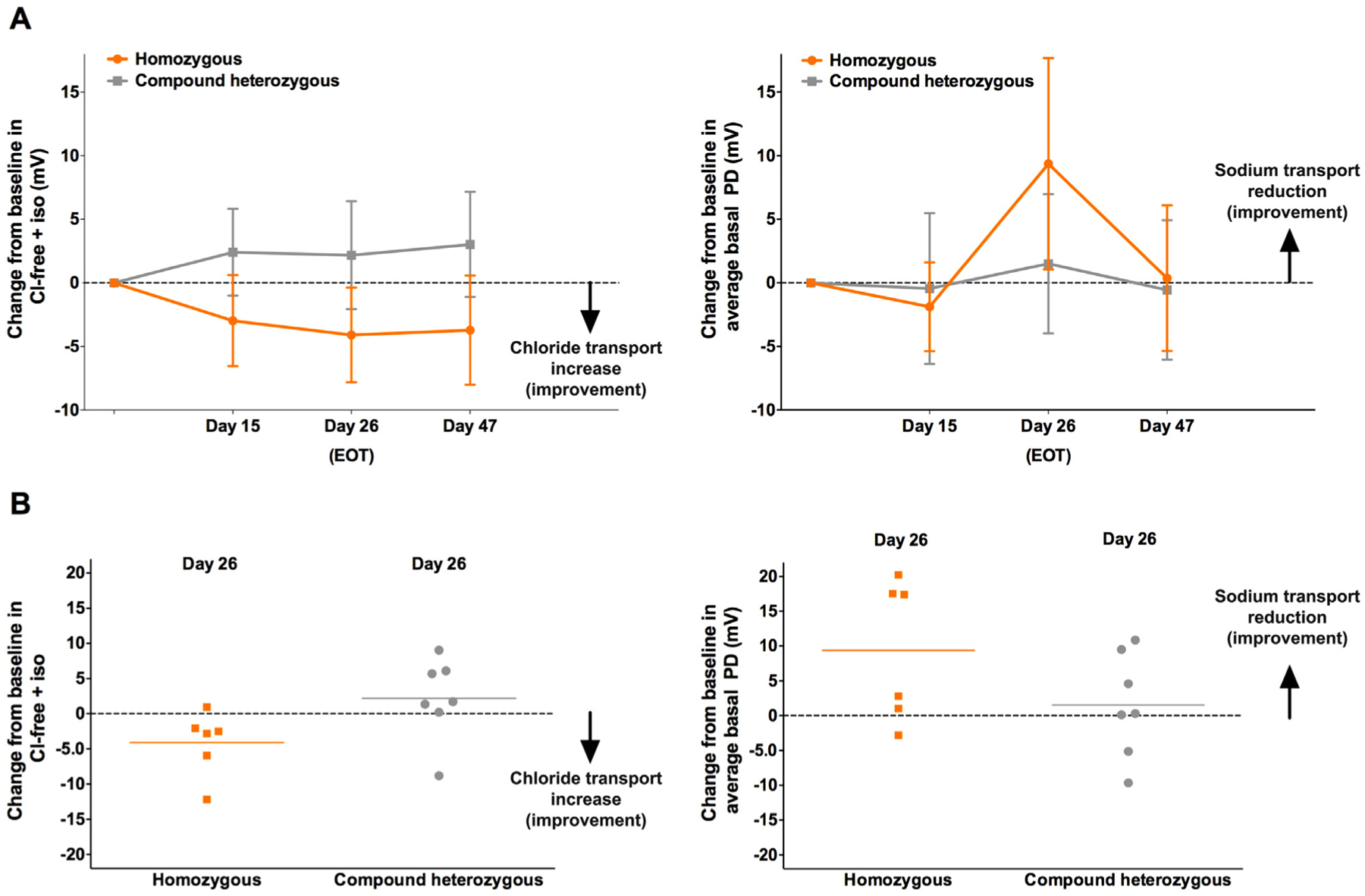
Parameters of NPD. Error bars show 90% CI of the mean. Change from baseline in total chloride transport (Cl-free+iso; left) and average basal PD (right) presented for both homozygous and compound heterozygous cohorts (per-protocol population), showing mean (and CI) for all time points (a) and individual data on day 26 (b). Mean total chloride transport and average basal PD values at day 0 are presented in Table 1. CI, confidence interval; Cl-free+iso, within-subject change from baseline in total chloride transport; EOT, end of treatment; NPD, Nasal Potential Difference; PD, potential difference.
Table 2.
Mean change from baseline values for the different chloride and sodium transport parameters as assessed by NPD for the homozygous cohort (per-protocol population).
| Day 15 | Day 26 (EOT) | Day 47 (EOS) | ||||
|---|---|---|---|---|---|---|
| Mean change from baseline (SD) [90% CI] | p value | Mean change from baseline (SD) [90% CI] | p value | Mean change from baseline (SD) [90% CI] | p value | |
| Chloride transport parameters (mV) | ||||||
| Cl-free+iso | −3.0 (4.9) [−6.6; 0.6] | 0.080 | −4.1 (4.5) [−7.8; −0.4] | 0.039 | −3.7 (5.9) [−8.0; 0.6] | 0.072 |
| Delta Cl-free | −0.9 (3.4) [−3.4; 1.6] | 0.249 | −2.7 (2.0) [−4.3; −1.1] | 0.011 | −1.4 (3.2) [−3.8; 1.0] | 0.152 |
| Delta isoproterenol | −2.1 (1.9) [−3.5; −0.6] | 0.015 | −1.4 (2.7) [−3.7; 0.8] | 0.131 | −2.3 (3.9) [−5.2; 0.5] | 0.082 |
| Sodium transport parameters (mV) | ||||||
| Average basal PD | −1.9 (4.8) [−5.4; 1.6] | 0.832 | +9.4 (10.1) [1.1; 17.7] | 0.036 | +0.4 (7.8) [−5.4; 6.1] | 0.453 |
| Maximum basal PD | −0.8 (4.2) [−3.9; 2.3] | 0.681 | +6.8 (9.4) [−0.9; 14.5] | 0.068 | −1.0 (9.6) [−8.0; 6.0] | 0.604 |
| Ringer’s PD | +0.8 (6.4) [−3.9; 5.5] | 0.377 | −1.0 (11.1) [−10.2; 8.2] | 0.582 | +0.9 (16.0) [−10.9; 12.7] | 0.444 |
| Amiloride PD | +5.8 (3.7) [3.1; 8.6] | 0.997 | 3.4 (8.7) [−3.8; 10.5] | 0.808 | −0.01 (13.4) [−9.8; 9.8] | 0.499 |
SD, 90% CI and corresponding p value (paired t-test, one-sided alpha = 0.05) are shown. CI: confidence interval; EOS: end of study; EOT: end of treatment; iso: isoproterenol; NPD: nasal potential difference; PD: potential difference; SD: standard deviation.
3.3. Sodium transport
Several different measures of sodium transport (average basal PD, maximum basal PD, Ringer’s PD and amiloride-sensitive PD) were evaluated to assess the effect of eluforsen on ENaC activity. Treatment with eluforsen resulted in decreased (more positive) average basal PD at day 26 only (mean change +9.4 mV [90% CI 1.1; 17.7], p = .04) in the homozygous cohort (Figs. 2a and b and S6a, and Table 2). Sensitivity analyses of the average basal PD data for the homozygous cohort showed that the different analysis methods were consistent with results from the primary analysis method (Fig. S1). Maximum basal PD was also improved, but no meaningful change in Ringer’s PD or amiloride-sensitive PD was observed (supplementary Fig. S7), possibly due to the small number of subjects [19].
3.4. Safety
Eluforsen was well tolerated during the study. No serious AEs were reported. Mild-to-moderate AEs were reported in 15/18 subjects; one subject reported several severe AEs related to a probable viral infection 8 days post-treatment. AEs were reported most frequently in the respiratory and gastrointestinal systems (supplementary Table S3); 25/71 AEs were considered drug related. There were no changes in SNOT-22 and NERS from baseline (supplementary Tables S4 and S5).
4. Discussion
We have shown that repeated administration of the antisense oligonucleotide eluforsen generated an encouraging signal of improved CFTR biological activity in subjects with CF who are homozygous for the F508del-CFTR mutation. This is the first demonstration of improved CFTR function using an antisense oligonucleotide therapeutic approach in CF subjects. These data further strengthen available preclinical data on eluforsen, where improved CFTR function was also demonstrated as measured by in vitro functional assays using primary cultures from F508del-CFTR homozygous donors and NPD in F508del-CFTR CF mice [24,26]. Together, these data support eluforsen development for patients with CF homozygous for the F508del-CFTR mutation.
We have shown that eluforsen administered three times weekly for 4 weeks to the nasal mucosa in the F508del-CFTR homozygous cohort improved total chloride transport from day 15 onwards. This was supported by various sensitivity analyses of the total chloride transport response and by improved (i.e. decreased) basal PD observed on day 26. The latter finding is consistent with the hypothesis that increased CFTR activity would reduce ENaC activity due to the known regulatory relationship between CFTR and ENaC activity [6–10]. Although encouraging, improvement was only observed at day 26, and not all sodium transport parameters improved. NPD has been used as an outcome measure in several development programmes with CFTR-directed treatments. The magnitude of the increase in total chloride transport and the corresponding improvement in sodium transport observed with eluforsen in the homozygous cohort, approaches the magnitude of the effect for ivacaftor in patients with the G551D-CFTR mutation [17,19], which later demonstrated clinical benefit in phase 3 studies [20]. Furthermore, in a phase 2 study with lumacaftor in patients with CF homozygous for the F508del-CFTR mutation, no improved total chloride transport was observed, and lumacaftor did not show clinical efficacy in this patient population [21]. In contrast, in an early single-center phase 2 study of ataluren in patients with CF due to nonsense mutations [27], improved total chloride transport (without corresponding improved sodium transport) was reported, but subsequent phase 3 studies of ataluren did not confirm these results and clinical benefit was not demonstrated [28,29]. These differences may be due to lack of standardisation in small single-centre studies, over-interpretation associated with small studies without placebo control and marginal effect sizes, or intrinsic variation of the measurement of the method.
The prolonged duration of action observed in the homozygous cohort is consistent with data from studies performed in non-human primates, which showed a prolonged half-life for eluforsen (unpublished data, ProQR Therapeutics, Leiden, the Netherlands). This long duration of action has also been described for other antisense oligonucleotides [30]. Therefore, these findings in patients are consistent with preclinical results and support proof of concept of eluforsen biological activity [24,26].
No detectable improvements in CFTR function were observed with eluforsen dosing in the F508del compound heterozygous cohort. The slight depolarisation observed for total chloride transport response over time was comparable to that observed in placebo groups in other NPD clinical trials [12,21,27]. The absence of treatment effect in this group of patients who are compound heterozygous for F508del-CFTR may represent the reduced availability of F508del-CFTR mRNA substrate for eluforsen interaction. This lack of pharmacodynamic activity in the compound heterozygous cohort strengthens the interpretation of results in the F508del-CFTR homozygous cohort as representing a specific effect.
Several aspects of the present study should be considered when interpreting the activity of eluforsen. First, NPD assessments are technically challenging and this may explain some inconsistencies in prior trial results, as was the case for studies investigating ataluren, where early uncontrolled and unblinded phase II study results in patients with nonsense-mediated CF [27] reported improved total chloride transport, while subsequent phase III studies did not confirm these NPD results or demonstrated clinical benefit [28,29]. To increase NPD assay validity for use in multicentre clinical trials, critical operational advancements have been implemented over the past several years, including rigorous standardisation of procedures (common TDN/CTN protocols), central supply of NPD reagents and materials, common equipment including electronic data capture and centralised blinded data interpretation. These operational innovations were employed for our study and resulted in quality data from N96% of tests. In studies of ivacaftor [17,19] and lumacaftor [21], these standardisation efforts have been associated with improved predictive performance of NPD. Second, the early phase nature of this study and lack of available preexisting data on eluforsen in humans supported an early proof of concept study with an open label design. To minimise bias from this choice, all centralised assessments of NPD tracings were blinded to visit and genotype, and all calculations of change were made in mixed batches of tracings. However, operator bias cannot be completely avoided with this design. Third, as indicated in the methods section, the post-hoc exclusion of subjects not meeting the NPD inclusion criterion, as a result of real time NPD review by the enrolling investigator rather than central reader assessment of all pre-dose NPD tracings, has the potential to result in bias. A consequence of the central reader approach was that only after completion of scoring by the central reader it became apparent that three subjects in the homozygous cohort and one subject in the compound heterozygous cohort did not meet the NPD inclusion criterion. Lastly, this early phase study included a small number of individuals and the point estimate around the detected effect, while reasonable in magnitude and statistically significant by the statistical analysis plan, had wide confidence intervals.
In this study, the observation of a clear directional NPD improvement in the homozygous cohort across numerous sensitivity analyses and the absence of improved NPD response in the compound heterozygous cohort indicate the capacity to differentiate a treatment response. Such findings are similar to other studies assessing CFTR function where NPD demonstrated bioactivity of novel therapeutics, including trials of CFTR gene transfer and CFTR modulators [14,16–18,27]. This partially mitigates some limitations inherent to this early phase study design and lack of a placebo group, but further investigation is required to examine the consistency, longevity and clinical benefit of the observed response.
The results of this study provide encouraging data for improved CFTR function with eluforsen treatment, based on chloride transport parameters and supportive sodium transport parameters assessed by NPD in CF subjects homozygous for the F508del-CFTR mutation. Importantly, eluforsen was well tolerated. The findings from this study are supported by an early phase dose escalation clinical study (PQ-010–001, NCT02532764) in F508del-CFTR homozygous subjects showing that inhaled eluforsen had a favourable safety profile and an early therapeutic benefit based on clinically meaningful improvements in the CF Questionnaire-Revised respiratory domain [31].
Therapeutic agents targeting RNA such as antisense oligonucleotides are a growing area of research, and with the recent success of other oligonucleotide approaches in genetic disorders, translation of these novel technologies into CF treatment may bring a new therapeutic class to the field [22,23]. The results from this early proof of concept study indicate that eluforsen may be suitable for further development and provide support for additional larger controlled studies using the inhaled pulmonary route of delivery of eluforsen to assess therapeutic benefit in patients homozygous for the F508del-CFTR mutation.
Supplementary Material
Acknowledgements
We thank the patients, caregivers and families for their participation in this study; Thomas Hofmann for his early support; Bo Liu for his role as central NPD data manager at the Center for CFTR Detection (University of Alabama, Birmingham, AL; funded by the Cystic Fibrosis Foundation); the USCystic Fibrosis Foundation TDN and the European Cystic Fibrosis Society CTN for their support, and for providing the standard operating procedure for NPD, which can be requested by contacting the TDN Coordinating Center at TDN_NRCC@seattlechildrens.org; Sonya Montgomery for her contribution to the NPD data evaluation process and safety review; Eva Stekelenburg for her instrumental role in the NPD data flow process; and Marie Boff (Cytel) for her assistance with statistical analyses. Medical writing assistance was provided by Robyn Foster (2theNth, Bollington, UK) and Paula Martín Vaquero, PhD, (Madrid, Spain), funded by ProQR Therapeutics.
Role of the funding source
This study was supported by ProQR Therapeutics (Leiden, the Netherlands), and received funding from the Cystic Fibrosis Foundation; the Netherlands Enterprise Agency (RVO) for InnovatieKrediet IK12062; National Institutes of Health awards R35HL135816, DK072482 and U54TR001005; and the European Union’s Horizon 2020 research and innovation programme under grant agreement No 633545. ProQR Therapeutics, the study sponsor, contributed to the study design, data collection, data analysis, data interpretation and writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Abbreviations:
- AE
adverse event
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- CI
confidence interval
- Cl-free+iso
chloride-free plus isoproterenol infusion
- CTN
Clinical Trials Network
- eNAC
epithelial sodium channel
- EOT
end of treatment
- NERS,
Nasal Examination Rating Scale
- NPD
Nasal Potential Difference
- PD
potential difference
- SNOT-22
Sino-Nasal Outcome Test
- TDN
Therapeutic Development Network
Footnotes
2 International Nonproprietary Name (INN), eluforsen, is under review.
Conflict of interest statement
WdH and NT are full-time employees of ProQR Therapeutics. NP-L and NH were full-time employees of ProQR Therapeutics during design development, execution and reporting of this study. JB and FB are statistical consultants paid by ProQR Therapeutics. JPC, DPN, JAN, KDB and SMR report grants related to the submitted work. IS-G, DPN, KDB, JSE, and SMR report grants outside the submitted work. IS-G, KDB, JSE and MAM report activity on advisory boards during the conduct of the study and outside the submitted work.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jcf.2018.10.015.
References
- [1].Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245:1066–73. 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- [2].Mall MA, Hartl D. CFTR: cystic fibrosis and beyond. Eur Respir J 2014;44:1042–54. 10.1183/09031936.00228013. [DOI] [PubMed] [Google Scholar]
- [3].UK Cystic Fibrosis Registry. Annual Data Report. https://www.cysticfibrosis.org.uk/~/media/documents/the-work-we-do/uk-cf-registry/full-registry-report-2015.ashx?la=en; 2015, Accessed date: 8 March 2017.
- [4].Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry 2015 Annual Data Report. Bethesda, Cystic Fibrosis Foundation; https://www.cff.org/Our-Research/CF-Patient-Registry/2015-Patient-Registry-Annual-Data-Report.pdf; 2016. [accessed 8 March 2017]. [Google Scholar]
- [5].Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev 1999;79(1 Suppl):S23–45. 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- [6].Vankeerberghen A, Cuppens H, Cassiman JJ. The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions. J Cyst Fibros 2002;1:13–29. 10.1016/S1569-1993(01)00003-0. [DOI] [PubMed] [Google Scholar]
- [7].Mehta A CFTR: more than just a chloride channel. Pediatr Pulmonol 2005;39:292–8. 10.1002/ppul.20147. [DOI] [PubMed] [Google Scholar]
- [8].Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na + conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest 1998;102:15–21. 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science 1995;269:847–50. 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- [10].Huang P, Gilmore E, Kultgen P, Barnes P, Milgram S, Stutts MJ. Local regulation of cystic fibrosis transmembrane regulator and epithelial sodium channel in airway epithelium. Proc Am Thorac Soc 2004;1:33–7. 10.1513/pats.2306012. [DOI] [PubMed] [Google Scholar]
- [11].Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med 1981;305:1489–95. 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- [12].Rowe SM, Accurso F, Clancy JP. Detection of cystic fibrosis transmembrane conductance regulator activity in early-phase clinical trials. Proc Am Thorac Soc 2007;4: 387–98. 10.1513/pats.200703-043BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rowe SM, Clancy JP, Wilschanski M. Nasal Potential Difference measurements to assess CFTR ion channel activity. Methods Mol Biol 2011;741:69–86. 10.1007/978-1-61779-117-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].De Boeck K, Kent L, Davies J, Derichs N, Amaral M, Rowe SM, et al. CFTR biomarkers: time for promotion to surrogate end-point. Eur Respir J 2013;41:203–16. 10.1183/09031936.00057512. [DOI] [PubMed] [Google Scholar]
- [15].Alton EW, Currie D, Logan-Sinclair R, Warner JO, Hodson ME, Geddes DM. Nasal Potential Difference: a clinical diagnostic test for cystic fibrosis. Eur Respir J 1990;3: 922–6. [PubMed] [Google Scholar]
- [16].Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–72. 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med 2010; 363:1991–2003. 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Alton EW, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med 2015;3:684–91. 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rowe SM, Liu B, Hill A, Hathorne H, Cohen M, Beamer JR, et al. Optimizing Nasal Potential Difference analysis for CFTR modulator development: assessment of ivacaftor in CF subjects with the G551D-CFTR mutation. PLoS One 2013;8:e66955 10.1371/journal.pone.0066955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].European Medicines Agency assessment report: Kalydeco. http://www.ema.europa.eu/ema/; 2012, Accessed date: 8 March 2017.
- [21].Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012;67: 12–8. 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kole R, Leppert BJ. Targeting mRNA splicing as a potential treatment for Duchenne muscular dystrophy. Discov Med 2012;14:59–69. [PubMed] [Google Scholar]
- [23].Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 2016;388:3017–26. 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- [24].Beumer W, Swildens J, Henig N, Anthonijsz H, Biasutto P, Leal T, et al. QR-010, an RNA therapy, restores CFTR function using in vitro and in vivo models of ΔF508 CFTR. J Cyst Fibros 2015;14(Suppl 1):S1. Presented at the European Cystic Fibrosis Conference, 10–13 June 2015, Brussels, Belgium; 2015. 10.1016/S1569-1993(15)30002-3. [DOI] [Google Scholar]
- [25].Liu B, Hathorne H, Hill A, et al. Normal values and receiver operating characteristics of NPD as a diagnostic measure. Presented at the North American Cystic Fibrosis Conference, 21–23 October, 2010, Baltimore, MD (USA); 2010. [Google Scholar]
- [26].Beumer W, Beka M, Panin M, et al. QR-010 restores CFTR-mediated chloride transport in F508del CF mice assessed by transepithelial Nasal Potential Difference (NPD). Pediatr Pulmonol 2014;49(Suppl 38):227. Presented at the 28th Annual North American Cystic Fibrosis Conference, 9–11 October 2014, Atlanta, Georgia (United States); 2014. [Google Scholar]
- [27].Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet 2008;372:719–27. 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- [28].Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am J Respir Crit Care Med 2010;182:1262–72. 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- [29].Therapeutics PTC. PTC therapeutics announces results from pivotal phase 3 clinical trial of ataluren in patients living with nonsense mutation cystic fibrosis. PR News-wire https://www.prnewswire.com/news-releases/ptc-therapeutics-announces-results-from-pivotal-phase-3-clinical-trial-of-ataluren-in-patients-living-with-nonsense-mutation-cystic-fibrosis-300416860.html; 2017, Accessed date: 8 March 2017.
- [30].Spinraza™. Full Prescribing Information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209531s002lbl.pdf; ; November 2017, Accessed date: 28 June 2018.
- [31].Elborn JS, Bouisset F, Boff M, Checchio T, Perquin J, Lamontagne N, et al. , on behalf of the PQ-010–001 Study Investigators. A phase 1b, dose-escalation study of QR-010, a novel antisense oligonucleotide administered in subjects with cystic fibrosis homozygous for the F508del CFTR mutation. Presented at the North American Cystic Fibrosis Conference, November 2–4, 2017, Indianapolis, IN (USA); 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.