

Abstract
Objectives:
In this study, some novel 2-(2-phenyl)-1H-benzo[d]imidazol-1-yl)-N’-(arylmethylene) acetohydrazide derivatives (1-12) were designed and synthesized.
Materials and Methods:
Compounds 1-12 were obtained by condensing 2-(2-phenyl)-1H-benzo[d]imidazol-1-yl)acetohydrazide (III) with the corresponding aromatic aldehyde derivatives in the presence of catalytic amounts of hydrochloric acid in ethanol.
Results:
Following the structure elucidation, epidermal growth factor receptor kinase inhibitor activity was measured. The ADP-GloTM kinase assay determines kinase activity based on the quantification of the amount of ADP produced during a kinase reaction.
Conclusion:
Almost all of the compounds’ kinase inhibitor activities were rather limited.
Keywords: Benzimidazole, acetohydrazide, epidermal growth factor receptor kinase, inhibitory
INTRODUCTION
Cancer is a lethal disease characterized by an uncontrolled proliferation of cells, invasive nature, and metastasis. In developed countries, following cardiovascular diseases, it is the second most common cause of death. According to research conducted worldwide in 2015, 8.8 million people died of cancer, which is nearly 1 in 6 of all deaths.1 With standard therapies, cancer is often not completely removed and new treatments are sought.
Tyrosine kinase belongs to the protein kinase family and it provides for protein phosphorylation.2 Phosphorylation of proteins through kinases plays an important role in signal transduction mechanisms. Changes that occur in signal transduction play an important role in cancer development and metastases because it controls functions such as cell proliferation and apoptosis.
Epidermal growth factor receptor (EGFR), the first cell surface receptor associated with cancer, is a tyrosine kinase receptor. EGFR is involved in cancer development and metastasis.3,4 Studies have shown that inhibition of EGFR can arrest or slow the development of cancer. EGFRs have been observed to be overproduced in lung, breast, prostate, bladder, ovarian, colon, and rectum cancers, and they are found to be structural in most normal epithelial cells (e.g., skin, hair follicles). Thus, inhibition of EGFR activity inhibits cell proliferation and leads to apoptosis, programmed cell death.5 EGFR, such as other tyrosine kinase receptors, may also be inhibited by both monoclonal antibodies and tyrosine kinase inhibitors. Other inhibitors of tyrosine kinase such as gefitinib, erlotinib, lapatinib are small molecules that target EGFR and are used in cancer therapy.4 It is known that compounds used as inhibitors of tyrosine kinase such as sunitinib carry benzimidazole isostere indole rings, and pazopanib and axitinib have indazole rings. Furthermore, by determining that benzimidazoles exhibit protein kinase inhibitor activity6,7,8,9, in addition to their different biologic activities, the compounds planned to be synthesized in this study are thought to be inhibitory on the EGFR receptor, which is now known to be directly related to cancer development.
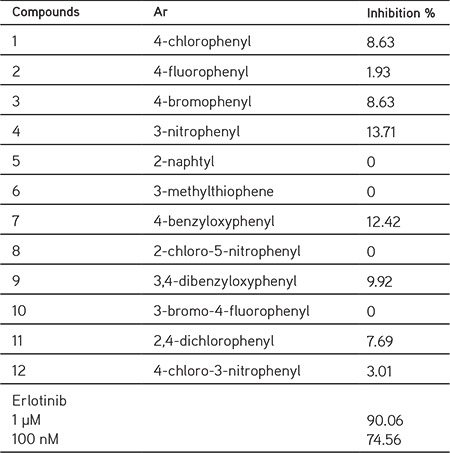
By developing novel EGFR tyrosine kinases inhibitors, we designed and synthesized new benzimidazole derivatives bearing a phenyl ring at the 2nd position and N-arylmethylideneacetohydrazide (1-12) (Table 1) at the 1st position, and evaluated their EGFR kinase inhibitory activities by comparing them with erlotinib. Following the structure elucidation, EGFR kinase inhibitor activities of the synthesized compounds were measured (Table 1).
Table 1. General structure and inhibitory effects of the synthesized compounds.
EXPERIMENTAL
General synthetic
All starting materials and chemical reagents used in the synthesis were high-grade commercial products purchased from Aldrich or Merck (Germany). Analytical thin-layer chromatography was performed using Merck precoated TLC plates, and spots were visualized with ultraviolet light. Melting points were determined using a Thermo Scientific Electrothermal IA9100 digital melting point apparatus (Bibby Scientific Limited, Staffordshire, UK) and were uncorrected. The structures of all synthesized compounds were assigned on the basis of nuclear magnetic resonance (NMR) and mass spectral analyses. 1H-NMR spectra were recorded on a Varian Mercury 400 MHz instrument (Varian Inc., Palo Alto, CA, USA) using a tetramethylsilane internal standard and DMSO-d6; coupling constants (J) are reported in Hertz. All chemical shifts are reported as δ (ppm) values. ES-MS were obtained using a Waters ZQ Micromass LC-MS spectrometer (Waters Corporation, Milford, MA, USA) with the positive electrospray ionization method. All instrumental analyses were performed at the Central Instrumentation Laboratory of the Pharmacy Faculty of Ankara University, Ankara, Turkey.
General procedure for the preparation of 2-(2-phenyl)-1H-benzimidazol-1-yl)-N’-(arylmethylidene) acetohydrazide derivatives (1-12)
Aromatic aldehyde derivative (0.02 mol) was added to a solution of the acyl hydrazide III (0.02 mol) in absolute EtOH (5 mL) containing a catalytic amount of 37% hydrochloric acid. The mixture was refluxed for 60 min, poured into cold water, and neutralized with 10% aqueous sodium bicarbonate solution. Crude product was filtered off and crystallized from dimethylformamide (DMF) water.
N’-[(4-Chlorophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (1)
M.P: 267°C; 1H NMR δ 5.06 and 5.52 (2s, 2H, CH2), 7.25-7.28 (m, 2H, Ar-H), 7.48-7.57 (m, 6H, Ar-H), 7.70-7.75 (m, 5H, Ar-H), 8.03 and 8.24 (2s, 1H, CH=N), 11.88 (brs, 1H, NH); ESI-MS (m/z): 389.4 (100%) (M+H), 391.5 (40%) (M+2+H).
N’-[(4-Fluorophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (2)
M.P: 214-217°C; 1H NMR δ 5.05 and 5.51 (2s, 2H, CH2), 7.24-7.29 (m, 4H, Ar-H), 7.52-7.57 (m, 4H, Ar-H), 7.70-7.80 (m, 5H, Ar-H), 8.04 and 8.24 (2s, 1H, -CH=N), 11.79, 11.93 (2s, 1H, NH); ESI-MS (m/z): 373.36 (M+H).
N’-[(4-Bromophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (3)
M.P: 287°C; 1H NMR δ 5.06 and 5.52 (2s, 2H, CH2), 7.25-7.30 (m, 2H, Ar-H), 7.52-7.80 (m, 11H, Ar-H), 8.02 and 8.22 (2s, 1H, CH=N), 11.86 (brs, 1H, NH); ESI-MS (m/z): 433.38 (100%) (M+H), 435.34 (95%) (M+2+H).
N’-[(3-Nitrophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (4)
M.P: 280°C; 1H NMR δ 5.09 and 5.57 (2s, 2H, CH2), 7.25-7.29 (m, 2H, Ar-H), 7.51-7.59 (m, 4H, Ar-H), 7.69-7.80 (m, 4H, Ar-H), 8.17, 8.52 (m, 3H, Ar-H), 8.53 and 8.55 (2s, 1H, -CH=N), 12.04, 12.20 (2s, 1H, NH); ESI-MS (m/z): 400.5 (M+H).
N’-[(2-Napthalene-2-yl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (5)
M.P: 285°C; 1H NMR δ 5.00 and 5.56 (2s, 2H, CH2), 7.24-7.28 (m, 2H, Ar-H), 7.49-7.58 (m, 6H, Ar-H), 7.69-7.92 (m, 3H, Ar-H), 7.94-7.98 (m, 4H, Ar-H), 8.11-8.38 (m, 2H, Ar-H and CH=N), 11.86, 11.99 (2s, 1H, NH); ESI-MS (m/z): 405.44 (M+H).
N’-[(3-Methylthiophene-2-yl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (6)
M.P: 267-269°C; 1H NMR δ 2.31 (2s, 3H, CH3), 5.01 and 5.38 (2s, 2H, CH2), 6.96 (dd, 1H, J=4.8 Hz, J=1.6 Hz, Ar-H), 7.25-7.29 (m, 2H, Ar-H), 7.52-7.58 (m, 5H, Ar-H), 7.69-7.72 (m, 2H, Ar-H), 7.78-7.81 (m, 1H, Ar-H), 8.27 and 8.48 (2s, 1H, CH=N), 11.63, 11.82 (2s, 1H, NH); ESI-MS (m/z): 375.48 (M+H).
N’-[(4-Benzyloxyphenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (7)
M.P: 240-241°C; 1H NMR δ 5.03 and 5.49 (2s, 2H, CH2), 5.15 (s, 2H, OCH2), 7.05-7.10 (m, 2H, Ar-H), 7.25-7.57 (m, 11H, Ar-H), 7.64-7.81 (m, 5H, Ar-H), 7.98 and 8.18 (2s, 1H, CH=N), 11.67, 11.81 (2s, 1H, NH); ESI-MS (m/z): 461.51 (M+H).
N’-[(2-Chloro-5-nitrophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (8)
M.P: 299-300°C; 1H NMR δ 5.08 and 5.57 (2s, 2H, CH2), 7.22-7.30 (m, 2H, Ar-H), 7.47-7.59 (m, 4H, Ar-H), 7.67-7.92 (m, 4H, Ar-H), 8.19-8.23 (m, 1H, Ar-H), 8.42 (s, 1H, Ar-H), 8.61 and 8.67 (d, d, J=2.8 Hz, CH=N), 12.18 (br s, 1H, NH); ESI-MS (m/z): 434.37 (100%) (M+H), 436.39 (39%) (M+2+H).
N’-[(3,4-Dibenzyloxyphenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (9)
M.P: 178-180°C; 1H NMR δ 5.03 and 5.19 (2s, 2H, CH2), 5.14 (s, 2H, OCH2), 5.17 (s, 2H, OCH2), 7.07-7.45 (m, 15H, Ar-H), 7.53-7.58 (m, 4H, Ar-H), 7.71-8.18 (m, 4H, Ar-H and CH=N), 11.71 (br s, 1H, NH), ESI-MS (m/z): 567.48 (M+H).
N’-[(3-Bromo-4-fluorophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (10)
M.P: 258-260°C; 1H NMR δ 5.06 and 5.54 (2s, 2H, CH2), 7.25-7.28 (m, 2H, Ar-H), 7.41-7.57 (m, 5H, Ar-H), 7.70-7.79 (m, 4H, Ar-H), 8.01-8.21 (m, 2H, Ar-H and CH=N), 11.90 (br s, 1H, NH); ESI-MS (m/z): 451.29 (100%) (M+H), 453.26 (98%) (M+2+H).
N’-[(2,4-Dichlorophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (11)
M.P: 147-148°C; 1H NMR δ 5.10 and 5.58 (2s, 2H, CH2), 7.31-7.34 (m, 2H, Ar-H), 7.47-7.50 (m, 1H, Ar-H), 7.56-7.66 (m, 4H, Ar-H), 7.73-7.81 (m, 4H Ar-H), 7.93-8.03 (m, 1H, Ar-H), 8.38 and 8.58 (2s, 1H, CH=N), 12.04, 12.29 (2s, 1H, NH); ESI-MS (m/z): 423.3 (100%) (M+H), 425.30 (65%) (M+2+H).
N’-[(4-Chloro-3-nitrophenyl)methylidene]-2-(2-phenyl-1H-benzimidazol-1-yl)acetohydrazide (12)
M.P: 269-270°C; 1H NMR δ 5.09 and 5.57 (2s, 2H, CH2), 7.25-7.29 (m, 2H, Ar-H), 7.52-7.57 (m, 4H, Ar-H), 7.70-7.85 (m, 4H, Ar-H), 8.03 (dd, Jo=8 Hz, Jm=2 Hz, Ar-H), 8.09 (s, 1H, Ar-H), 8.30, 8.38 and 8.43 (s, d, d, J=1.6 Hz, -CH=N), 12.05, 12.18 (2 br s, 1H, NH); ESI-MS (m/z): 434.4 (100%) (M+H), 436.5 (33.5%)(M+2+H).
EGFR kinase assay
It is important to evaluate the possible interaction of solubilized EGFR kinase activity with the amount of enzyme and with the reaction of the solvents (5% DMSO) of the substances for which inhibitor activity is to be assessed. For this purpose, kinase titration was first performed with EGFR kinase at a range of 0.2-200 ng. As the amount of enzyme was increased, kinase reaction was increased (more ATP was formed, more ADP was formed; results not shown).
After these preliminary experiments and system validation, the inhibitory activities of the synthesized substances on EGFR kinase were evaluated. Erlotinib, a substance known to inhibit EGFR kinase in these experiments was also used as a positive control in all experiments.
Screening of inhibitor activity
Kinase reactions were performed using 50 µM ATP, 5 ng EGFR kinase ± 1 µM inhibitor in each reaction medium. Erlotinib was tested in two different concentrations: 100 nM and 1 µM to confirm dose-dependency of the inhibitory activity. Five percent DMSO (solvent) was used as a control in all experiments. These reactions constituted the maximum enzyme activity (100% activity).
Standard curve for ATP-ADP transformation
In the first step, the standard curves for the ATP-ADP transformation to be used in determining the amount of ADP formed in the kinase reaction were established. While these curves were being generated, the ATP and ADP stock solutions were mixed at specified ratios and the resulting luminescence was read. The purpose of these curves was to determine the luminescence signal interval obtained by the amount of the designated EGFR kinase. ATP/ADP conversion curves were generated using 1 µM, 5 µM, 50 Mm, and 500 µM ATP-ADP concentrations. These experiments are important for determining luciferase activity and signal range obtained in the kinase assay.
RESULTS AND DISCUSSION
Chemistry
The benzimidazole acetohydrazide derivatives were synthesized according to the following synthetic scheme: 2-Phenyl-1H-benzo[d]imidazole (I) was prepared via the reaction of o-phenylenediamine, benzaldehyde and sodium metabisulfite in DMF.10 The reaction of I with ethyl chloroacetate yielded ethyl 2-(2-phenyl)-1H-benzo[d]imidazol-1-yl) acetate (II).11 Treatment of the ester (II) with hydrazine hydrate gave the desired hydrazide compound, 2-(2-phenyl)-1H-benzo[d]imidazol-1-yl)acetohydrazide (III).12 Compounds 1-12 were found by condensing acyl hydrazide III with the corresponding aromatic aldehydes in the presence of sulfuric acid13 (Scheme 1). The structure of the newly synthesized compounds was confirmed and supported by spectroscopic data. The mass spectrum of each of compound appeared as M+H peak, consistent with the molecular formula of the assigned structure. N-acylhydrazones may be present as four isomers depending on the geometric isomerism relative to the imino group (E, Z isomers) and conformers related to the amide linkage (syn/anti amide conformers).14 In 1H-NMR spectra measured in polar solvent dimethyl sulfoxide-d6, two signals representing the imino hydrogen (CH=N) were observed at 7.98-8.61 and 8.18-8.67 ppm, the methylene protons (CH2CO) were also typically observed as two singlet signals at 5.01-5.10 and 5.19-5.58 ppm, respectively.
Scheme 1.
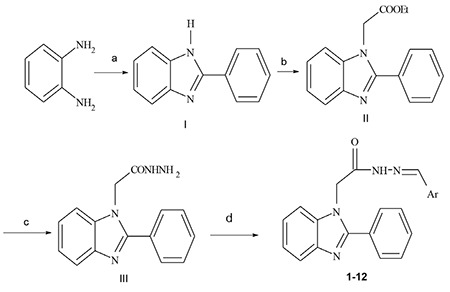
Synthetic route to compounds 1-12
(a) Na2S2O3 adduct of benzaldehyde/DMF; (b) Ethyl chloroacetate/KOH; (c) Hydrazine/ EtOH; (d) Corresponding aromatic aldehyde/EtOH/HClcat
Biological activities
The ADP-GloTM kinase assay15 determines kinase activity based on the quantification of the amount of ADP produced during a kinase reaction. EGFR is a tyrosine kinase receptor and was the first receptor to be proposed for cancer therapy. Currently, there are two approaches that target EGFRs in cancer treatment: monoclonal antibodies and small-molecule inhibitors of EGFR tyrosine kinase enzymatic activity. Erlotinib selectively inhibits tyrosine kinase activity of EGFRs.
In the present study, we measured the activity of EGFR kinase and evaluated the inhibitory efficiencies of newly synthesized compounds by comparing them with erlotinib.
The assays were performed in three steps (Figure 1). A kinase reaction with 5 ng EGFR kinase and 50 µM ATP was the first step. The amounts of both EGFR and ATP were determined in the preliminary optimization assays (results not shown). The total volume of the kinase reaction was 25 µL. The amount of the EGFR and PolyE4Y1 substrate was 1 µg, based on the recommendations of the literature. In the second step, an equal volume of ADP-GloTM reagent was added to terminate the kinase reaction and deplete the remaining ATP. On the third and final step, a kinase detection reagent was added to convert the ADP produced during the kinase reaction to ATP and to determine the amount of newly synthesized ATP through a luciferase/luciferin reaction.
Figure 1.
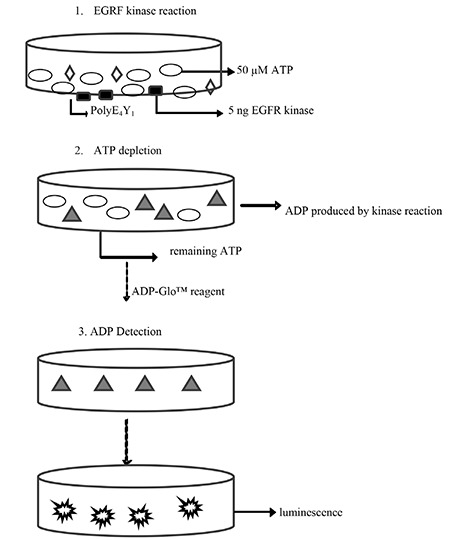
Schematic representation of the protocol
Inhibitory efficiency was determined by comparing the enzyme activities in the presence of the inhibitors with the maximum enzyme activity. Maximum kinase activity (100% kinase) was determined as the luminescence acquired in the kinase reaction where EGFR kinase, PolyE4Y1, ATP and diluting agent, and 5% DMSO were present in the reaction medium.
CONCLUSION
In the reactions where inhibitory efficiencies were determined, the reaction medium contained either erlotinib or one of the newly synthesized compounds in a single concentration of 1 µM. At this concentration, erlotinib inhibited EGFR kinase activity by nearly 90%. Inhibitory efficiencies of the new compounds are shown in Table 1. The results indicated that almost all of the compounds’ kinase inhibitor activities were somewhat limited. Compounds that have 3-nitrophenyl (4) and 4-benzyloxyphenyl (7) substituents as aryl groups were found to have slight inhibitory potencies in the kinase assay at 13.71% and 12.42%, respectively.
Footnotes
Conflict of Interest: No conflict of interest was declared by the authors.
References
- 1. [Internet] http://www.who.int/cancer .
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Bianco R, Gelardi T, Damiano V, Ciardiello F, Tortora G. Rational bases for the development of EGFR inhibitors for cancer treatment. Int J Biochem Cell Biol. 2007;39:1416–1431. doi: 10.1016/j.biocel.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 5.Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res. 2014;4:411–435. [PMC free article] [PubMed] [Google Scholar]
- 6.Hasegawa M, Nishigaki N, Washio Y, Kano K, Harris PA, Sato H, Mori I, West RI, Shibahara M, Toyoda H, Wang L, Nolte RT, Veal JM, Cheung M. Discovery of novel benzimidazoles as potent inhibitors of TIE-2 and VEGFR-2 tyrosine kinase receptors. J Med Chem. 2007;50:4453–4470. doi: 10.1021/jm0611051. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Tan C, Gao C, Zhang C, Luan X, Chen X, Liu H, Chen Y, Jiang Y. Discovery of benzimidazole derivatives as novel multi-target EGFR, VEGFR-2 and PDGFR kinase inhibitors. Bio Med Chem. 2011;19:4529–4535. doi: 10.1016/j.bmc.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Yadav S, Sinha D, Singh SK, Singh VK. Novel benzimidazole analogs as inhibitors of EGFR tyrosine kinase. Chem Biol Drug Des. 2012;80:625–630. doi: 10.1111/j.1747-0285.2012.01407.x. [DOI] [PubMed] [Google Scholar]
- 9.Abdel-Ghaffar NF. Synthesis, reactions, structure-activity relationship of 2-benzimidazole analogs as anticancer agents and study their molecular docking. Der Pharma Chem. 2013:243–257. [Google Scholar]
- 10.Ridley HF, Spickett RGW, Timmis GM. A new synthesis of benzimidazoles and aza analogs. J Heterocyclic Chem. 1965;2:453–456. [Google Scholar]
- 11.Heaney H, Ley SV. N-Alkylation of indole and pyroles in dimethyl sulphoxide. J Chem Soc Perkin I. 1973:499–500. [Google Scholar]
- 12.Smith PAS. In: Adams R, Bachmann WE, Fieser LF, Johnson JR, Snyder HR. (Eds). Organic Reactions. John Wiley & Sons, Inc. London; Chapman & Hall, Limited. 1949;3:337–389. [Google Scholar]
- 13.Carvalho SA, Feitosa LO, Soares M, Costa TE, Henriques MG, Salomão K, de Castro SL, Kaiser M, Brun R, Wardell JL, Wardell SM, Trossini GH, Andricopulo AD, da Silva EF, Fraga CA. Design and synthesis of new (E)-cinnamic N-acylhydrazones as potent antitrypanosomal agents. Eur J Med Chem. 2012;54:512–521. doi: 10.1016/j.ejmech.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 14.Patorski P, Wyrzykiewicz E, Bartkowiak G. Synthesis and Conformational Assignment of N-(E)-Stilbenyloxymethylenecarbonyl-Substituted Hydrazones of Acetone and o-(m- and p-) Chloro- (nitro-) benzaldehydes by Means of 1H and 13C NMR Spectroscopy. J Spectrosc. 2013;2013:1–12. [Google Scholar]
- 15.ADP-Glo™ Kinase Assay. Technical Manual. Promega. [Internet]