

Abstract
Cancer is a provocative issue across the globe and treatment of uncontrolled cell growth follows a deep investigation in the field of drug discovery. Therefore, there is a crucial requirement for discovering an ingenious medicinally active agent that can amend idle drug targets. Increasing pragmatic evidence implies that histone deacetylases (HDACs) are trapped during cancer progression, which increases deacetylation and triggers changes in malignancy. They provide a ground-breaking scaffold and an attainable key for investigating chemical entity pertinent to HDAC biology as a therapeutic target in the drug discovery context. Due to gene expression, an impending requirement to prudently transfer cytotoxicity to cancerous cells, HDAC inhibitors may be developed as anticancer agents. The present review focuses on the basics of HDAC enzymes, their inhibitors, and therapeutic outcomes.
Keywords: Histone deacetylase inhibitors, apoptosis, multitherapeutic approach, cancer
INTRODUCTION
In recent years, immense progress has been made in the management of cancer, due to which the life expectancy of cancer patients has been improved remarkably. Cancer is represented by inappropriate cell proliferation or transformation.1In cancerous cells, genes undergo various modification processes either by mutation or epigenetics. A number of potential approaches have been proposed for the treatment of cancer, but histone deacetylase inhibitors (HDACIs) are the emerging ones.2 Various reports in the literature revealed that certain histone deacetylase (HDAC) family members are aberrantly expressed in several tumors and have a nonredundant function in controlling the hallmarks of cancerous cells. They are classified into two types, i.e., Zn-dependent (class I and class II) and nicotinamide adenine dinucleotide (NAD)-dependent (class III) enzymes. Currently, researchers around the globe are paying more attention to the modification of the Zn-dependent portion of the histone family. At present, there are a total of 11 HDAC family members identified on the basis of their similarity chain (Figure 1).3,4
Figure 1.
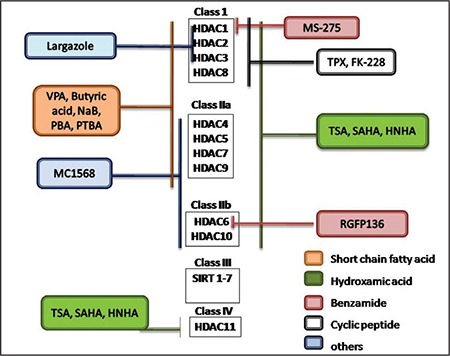
Schematic representation of different histone deacetylase and inhibitors
HDACs are enzymes that catalyze the deacetylation of lysine remnants located at the N-terminal of several protein substrates, such as nucleosomal histones. Histone acetylation has an important role in gene expression. Histone acetyl transferases and HDACs are the two types of enzymes that are primarily amenable for the catalysis of particular lysine residues of histones.5 Enzymes inhibitors are well known to stimulate cell cycle arrest, p53 sovereign, initiation of cyclin dependent kinase inhibitor, i.e., p21, tumor discriminating apoptosis, and segregation of normal and malignant cells. HDACIs have attracted significant interest recently for the treatment of cancer as well as of other human disorders.6
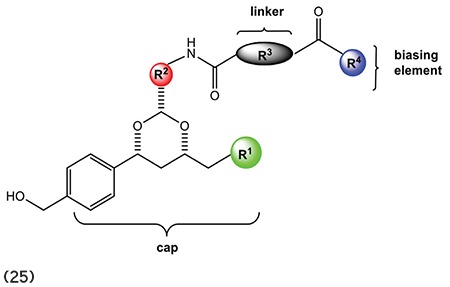
A number of HDAC inhibitors have been reported to date that cause tumor cell growth arrest at doses that are apparently nontoxic and appear to be very selective.1 HDACIs consists of three defined structural parts of an ideal pharmacophore, i.e., (a) recognition cap group (b) hydrophobic linker, and (c) the zinc-binding group (Figure 2).7,8 Earlier, HDACIs highlighted the alteration of the surface recognition site (capping group) and the zinc ion binding group.5 Some selective HDACIs help in identifying the specific position of the HDAC protein responsible for cancer. This prospective identification by HDACIs plays an important role to improve the therapeutic profile of new generation HDACIs. In addition to changing the metal binding site, the hydrophobic site is also varied, concentrating on modifying the linker site by varying unsaturation and adding a ring (e.g., aryl, cyclohexyl) inside the series,9 but still selective and potent HDACIs are yet to be investigated.
Figure 2.
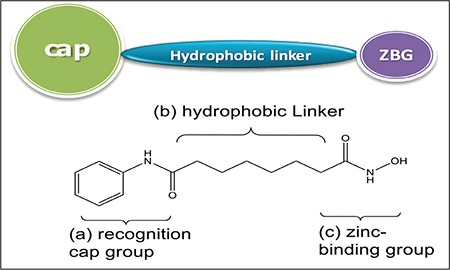
Pharmacophore requirements for histone deacetylase inhibitors
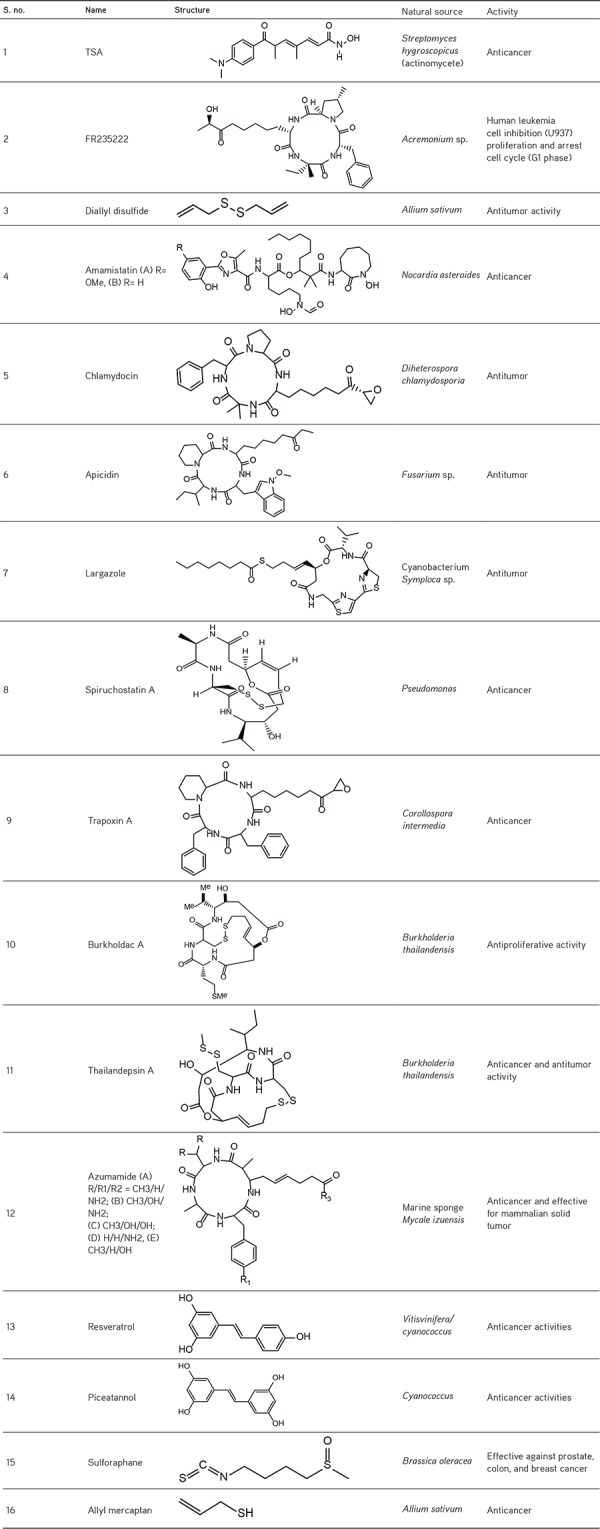
On the basis of chemical structures and enzymatic activities, HDACIs are (Figure 3)10 chemically classified as hydroxamates (vorinostat, panobinostat, givinostat, quisinostat, abexinostat, belinostat, tefinostat, resminostat, pracinostat), benzamides (entinostat, mocetinostat, chidamide), aliphatic acids (valproic acid), and cyclic peptides (romidepsin).11 These HDACIs possess specific structural components that trigger diverse functions like interruption in the cell cycle, angiogenesis, and immunomodulation by acting on histone and non-histone proteins.9 A large number of HDACIs originate from natural sources and show substantial effects against cancer cells. Some examples of natural HDACIs are given in Table 1.12,13,14
Figure 3.
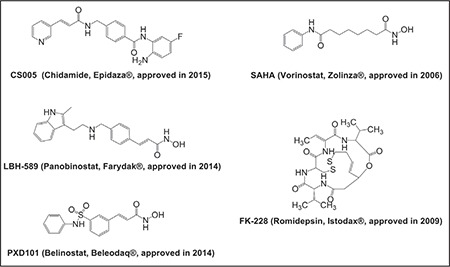
Some of the approved histone deacetylase inhibitors
Table 1. Naturally occurring HDACIs.
Food and Drug Administration approved and clinical trial drugs
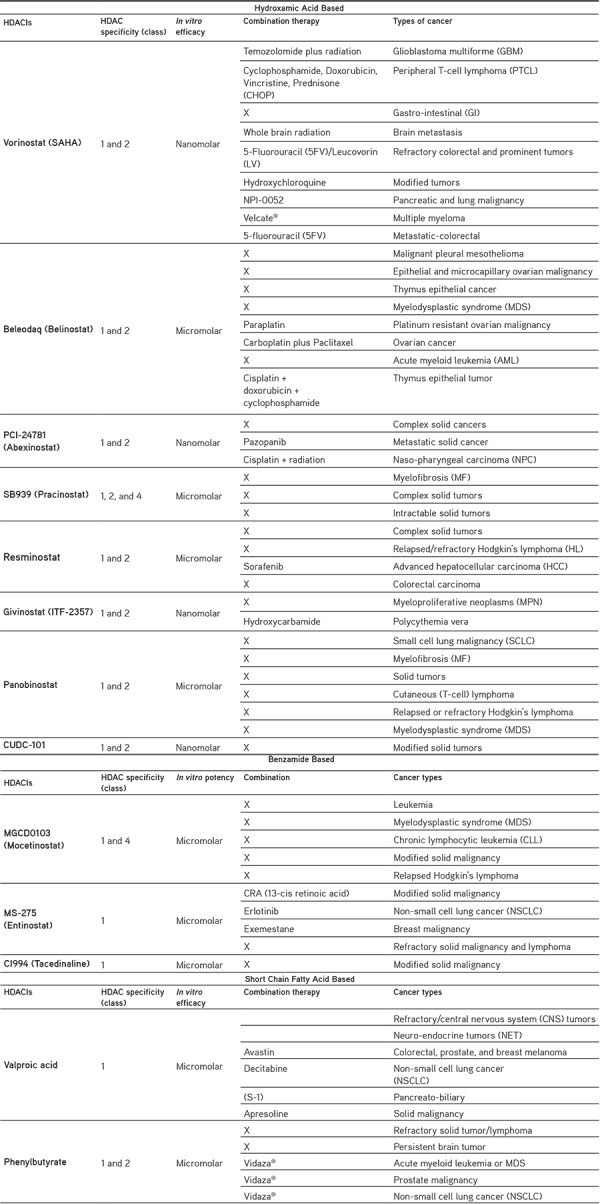
Vorinostat, romidepsin, belinostat, and romidepsin are HDACIs that are approved by the Food and Drug Administration (FDA) for the treatment of cutaneous T-cell lymphoma (CTCL). More than 80 HDACIs drugs are under clinical trial at present and 11 of them are particular for solid and hematological tumors. Single and combination drugs for the treatment of other types of cancer are shown in Table 2.3
Table 2. Various HDACIs in clinical trials.
Hydroxamic acids
A number of HDACIs have been identified and some are under clinical trial with a hydroxamic acid scaffold for the treatment of various types of cancer. The hydroxamic acid-based drug molecule consists of three defined structural parts of an ideal pharmacophore, i.e., (a) recognition cap group, (b) hydrophobic linker, and (c) zinc-binding group. HDACIs act by binding to the cap bearing amino group, a linker with 4-6 carbon unit and zinc binding group for the inhibition of enzyme.15
Trichostatin A is the first hydroxamate-based HDACI that was isolated from Streptomyces hygroscopicus to inhibit HDACs. Only the R-isomer of Trichostatin A was found to be active against HDACs.16
Vorinostat (N-hydroxy-N’-phenyl-octanediamide), marketed under the name Zolinza® by Merck, was the one of the first HDACIs permitted for the treatment of CTCL by the FDA, in 2006.17 Vorinostat hinders all classes of HDAC proteins (I, II, and IV), except class III HDAC, which is NAD+ dependent.18,19
Panobinostat (LB589) is a new drug developed by Novartis for the treatment of various cancers20 and was approved by the FDA in 2015 for the treatment of multiple myeloma.21,22,23
Givinostat (ITF2357) has been reported as a hydroxamic acid-based HDACI that revealed positive effects in patients with Hodgkin’s lymphoma, multiple myeloma, and severe lymphocytic leukemia. The European Union has designated givinostat as an orphan drug for the treatment of systemic juvenile idiopathic arthritis and polycythemia vera.24
Abexinostat (PCI-24781) has been reported as a potent hydroxamate-based HDACI having a wide spectrum of anticancer activity. It is used alone or together with proteasome inhibitors in the treatment of neuroblastoma cell lines.25 Abexinostat is used with the usual chemotherapy agents, or is used for different types of carcinomas, e.g., tissue soft-tissue sarcoma (sarcoma models of human).26
Belinostat (Beleodaq or PXD101) is a novel hydroxamate-type HDAC inhibitor that exhibits in vitro cytotoxicity at low micromolar concentrations and it is active for the treatment of ovarian cancer, CTCL, thymoma or thymic carcinoma, and myelodysplastic syndrome. This drug showed remarked effects in single or combined therapy.27
CUDC 101 is multitarget inhibitor of enzymes and receptors like HDAC, tyrosine kinases, epidermal growth factor receptor, and human epidermal growth factor receptor-2 and it possesses potent anti-proliferative and pro-apoptotic activities.28
Pracinostat (SB939) is another clinical trial (phase II) compound with HDAC inhibitory activity. Studies postulated that the activity or acceptability of compound 8 is in transitional/high risk myelofibrosis affected patients.29 The drug has also been tested for modified solid tumors30 but yielded no promising results. The drug also showed greater effectiveness in children with refractory solid tumors.31
Resminostat prevents cell growth and robustly induces apoptosis in multiple myeloma cell lines in small µm concentration.32 This drug shows a significant effect when dispensed in combination with other drugs (melphalan, bortezomib).33 In phase II clinical trials, it showed positive effects in Hodgkin’s lymphoma and was also evaluated for higher colorectal malignancy.34
Quisinostat (JNJ-26481585) is an experimental drug discovered by Johnson and Johnson through clinical studies. The data suggest that it is a “pan” inhibitor and it was found to be effective for the treatment of CTCL and leukemia myeloid in nanomolar concentration.35
Tefinostat or CHR-2845 (cyclopentyl 2-((4-(N1hydroxyoctanediamido) cyclohexyl) methylamino)-2-phenylacetate) comes under the hydroxamic acid category used as a particular substrate for hCE-1 (intracellular carboxyl-esterase), whose expression is limited to cells of the family of monocytes/macrophages. It is a monocyte or macrophage focused HDACI that is cleaved to an active acid and has significant effects towards myeloid leukemia. The phase I clinical trial of the drug showed remarkable effects on hematological malignancies and lymphoid tumors.36
CHR-3996 is a next generation HDACI based on hydroxamic acid and showed greater potency towards class I HDAC with latent anti-neoplastic effect and also showed potential effect for modified malignancies in clinical trials.37,38
Benzamide derivatives
This is another class of HDACI having 20 amino anilide moiety which targets specifically class I HDACs. They bind to zinc-chelating moiety for the interaction with the catalytic Zn2+ in HDACs’ active site.39
Entinostat (MS-275) was found to potentially inhibit various cancer cells like NSCLC, breast malignancy, lympho-blastic cancer, colon and renal cancer, and meta-static tumors and also has a notable effect in different phases of clinical trials and with selectivity towards class 1.40
Mocetinostat (MGCD0103) is an isotype of HDACI and showed in vitro activity against HDAC1 selectively and some activity against the various isoforms of HDAC (2, 3, and 11).41 The drug showed greater potency in hematological leukemia,42 lymphoma cancer,43 and solid malignancy.44
Chidamide (Epidaza) is an HDAC inhibitor developed and approved in China (in 2015) that showed potential effects in the treatment of pancreatic cancer.45
Short chain fatty acids
The chemical class of short chain fatty acids has been also introduced as HDAC inhibitors. Their mode of action is based on the presence of a COOH group covering the acetate release channel with a Zn binding site and they vie for the acetate group freed from the deacetylation reaction. The best examples of short chain fatty acids are valproic acid and sodium butyrate, which are under clinical trial.46
Valproic acid is used as anti-convulsant and mood-stabilizing agent. Recently it was introduced as a pan-HDACI in the third phase of clinical trials for the treatment of cancer, i.e., cervical or ovarian. It shows significant therapeutic effects either alone or in combination therapy.47,48
AN-9 is used for chronic NSCLC and lymphocytic and lymphoma malignancies.49
Cyclic peptides
Romidepsin belongs to the class of depsipeptides, and has recently been tested in phase-II clinical trials as well as critical trials in cutaneous and peripheral T-cell lymphomas. An unprejudiced response was seen in 10 of 28 evaluable CTCL affected patients, from an overall response rate of 36%, comprising 3 and 7 complete and partial responses, respectively. Myelotoxicity, nausea, vomiting, and cardiac dysrhythmias are some of the serious side effects. Hematologic and solid malignancies seen in cancer affected patients may be treated with depsipeptides, which are also under clinical trial in single or combination therapy.50
Toxicity in clinical trials
Antitumor drugs seem to have more serious toxicity than any other class of drugs. In some cases, thrombocytopenia, neutropenia, anemia, fatigue, and diarrhea are the certain side effects of inhibitors (grades III and IV). By the discontinuation of the (HDAC) drug, some volunteers suffering from thrombocytopenia along with nausea, vomiting, anorexia, constipation, and dehydration were also seen.
Inhibitors of HDAC also have some adverse effects like any class of anticancer agents. The inhibitors (grades III and IV) cause certain side effects like thrombocytopenia, neutropenia, anemia, fatigue, and diarrhea.51,52 In some cases, HDAC causes thrombocytopenia but it can be easily resolved by discontinuation of the drug.40 Some other side effects were also seen, like nausea, vomiting, anorexia, constipation, and dehydration. Deaths of volunteers in clinical trials involving HDACIs have been reported. For example, during trials of mocetinostat in patients with critical Hodgkin’s lymphoma four died, of which two were treatment related deaths.53 Similarly, some other deaths were recorded during clinical trials involving vorinostat and givinostat.52,54 Hence, before starting clinical trials some amendments are necessary to reduce the toxicity of HDACIs and curtail the cytotoxicity effects in patients.
Approaches towards the development of HDACIs
Most HDACIs have been recognized but not considered to be competitive inhibitors. The enzymes are inhibited by insertion of the same catalytic site as the usual enzyme substrates called competitive inhibitors. A competitive HDACI normally contributing to the ordinary function of the common model of pharmacophore depends upon the crystal structures of enzyme inhibitor (HDAC-like protein comes from Aquifex aeolicus) complex.
Noncompetitive inhibitors selectively disrupting the HDACs’ interaction with precise DNA binding proteins and some other regulatory proteins (like 14-3-3 protein) might be potent selective outlines (Figure 4).55 Alteration of identified HDACIs is important to recognize the chemical entity that affects the potency of inhibitors and is an important initiative for further investigating a novel chemical molecule.
Figure 4.
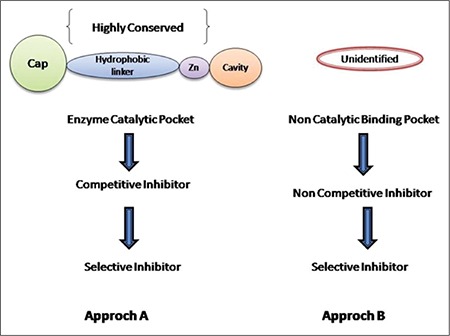
Different approaches for selective histone deacetylase inhibition
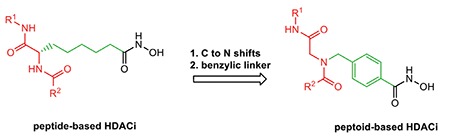
Some of the new HDACIs with peptoid-based cap groups were synthesized and found to be more selective against HDAC6 isoform than towards other HDAC isoforms (Figure 5).56 The compounds obtained from this hypothesis were found to be more active, showing extraordinary chemo-sensitizing effects and remarkable activity against Cal27 and CisR.56 Selective inhibition of HDAC6 is a promising target nowadays for a wide range of diseases such as neurodegenerative disorders (Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease), cancers, and hematological malignancies.
Figure 5.
Peptoid-based novel chemical entity effective against HDAC6 isoform
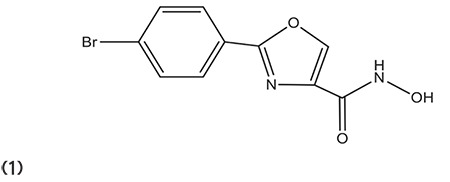
During the identification of some selective HDAC6 inhibitors, a biarylhydroxamate structure was explored without any branching. The heterocycles (thiazole, oxazole, and oxadiazole) attached to the hydroxamate showed a huge impact on HDAC6 potency and selectivity. Compound 1 containing oxazole moiety was identified by Senger et al.57 as a potent and selective inhibitor in vitro and in cell culture. (Graphic 1)
Graphic 1.
Zhang et al.58 outlined the synthesis, characterization, and biological evaluation of suberoylanilide hydroxamic acid (SAHA)-based derivatives with greater binding towards HDAC8 than the SAHA. Compound 2 shows pronounced activity while inhibiting the cancerous cell lines of human glioma, i.e., MGR2, U251, and U373. (Graphic 2)
Graphic 2.
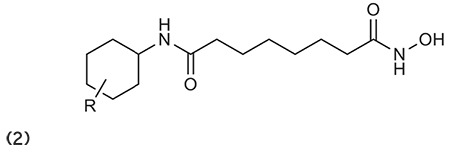
Bicyclic heterocyclic compounds are well known and widely used in medicinal chemistry, always attracting remarkable attention in the pharmaceutical industry due to their wide therapeutic value. A series of novel acrylamide derivatives based on the lead compound of MS-275 has been synthesized by Li et al.59 The synthesized compounds were quantized for antiproliferative activities against cancerous cell lines (HCT-116, MCF-7, and A549). Furthermore, compound 3 manifested an adequate pharmacokinetic profile with 76% bioavailability in rats, and can probably be regarded as a novel compound for drug discovery. (Graphic 3)
Graphic 3.
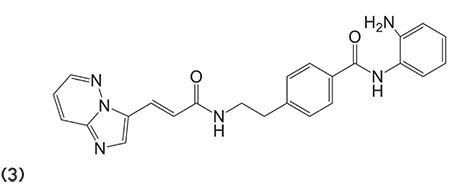
Chavan and Mahajan60 outlined and synthesized a number of derivatives having semi- or thio-carbazone moiety containing hydroxamic acid with average to high G score. Numerous compounds exhibited potent anti-proliferative effects for the MCF7, HCT15, and Jurkat cancer cell lines. Compound 4 showed potential activity against colon cancer. (Graphic 4)
Graphic 4.
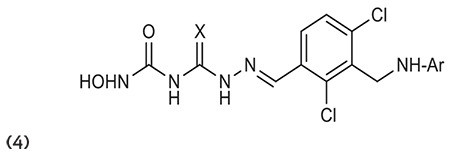
Zhang et al.61 described colchicine bearing hydroxamate moiety with HDAC inhibitory activity that possesses good effect against HDACs and tubulin. Compounds 5a-b show modest inhibition of HDAC activity and significant action on cytotoxicity. (Graphic 5)
Graphic 5.
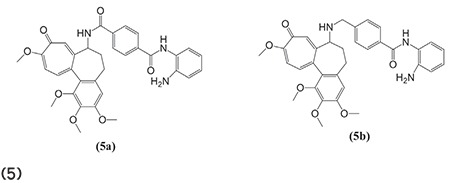
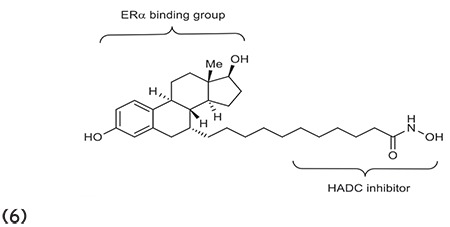
Mendoza-Sanchez et al.62 outlined the fusion of antiestrogens with known HDACIs to obtain more effective antiproliferative compounds for the treatment of breast cancer. The fused compound 6 had antiestrogenic and HDACI activity. The benzamide bifunctional molecule was found to be active for class I deacetylases (HDAC3) and class II deacetylases (HDAC6) and was potent in nM concentration in breast cancer models. (Graphic 6)
Graphic 6.
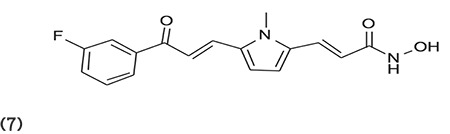
Fleming et al.63 reported the advanced synthesis and structural modification of MC1568 (7), which was found to be selective for class IIa HDACI. (Graphic 7)
Graphic 7.
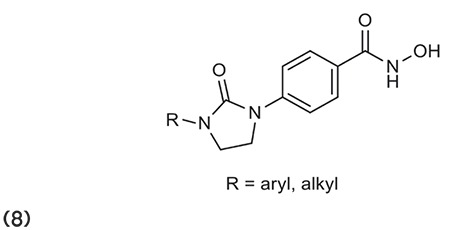
Cheng et al.64 reported the synthesis of phenyl-imidazolidin-2-one derivatives as selective HDACIs. Compound 8 of the series possesses remarkable antitumor activity against cancer cell lines (HCT-116, PC3, and HL-60) in comparison to SAHA. It also showed a major antitumor effect in the xenograft model of HCT 116 mice. (Graphic 8)
Graphic 8.
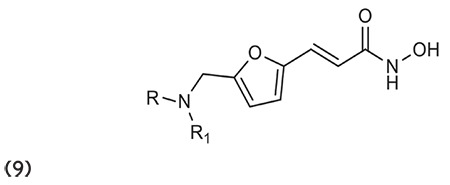
Feng et al.65 described the influence of the insertion of a branched hydrophobic group, e.g., N-hydroxyfurylacrylamide, at the cap side of HDACI and was reported to determine the activity in terms of inhibition against tumor cells. All the synthesized compounds were reported to have high selectivity towards HDAC1 and the compound like 9 showed magnificent selectivity next to HDAC6. (Graphic 9)
Graphic 9.
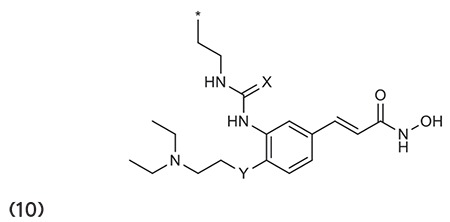
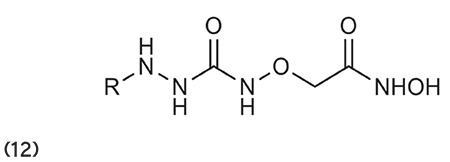
Ning et al.66 stated that the substitution of urea/thiourea on disubstituted cinnamic-based hydroxamates (10) has a remarkable HDAC inhibitory effect and antiproliferative activity against tumor cell lines. (Graphic 10)
Graphic 10.
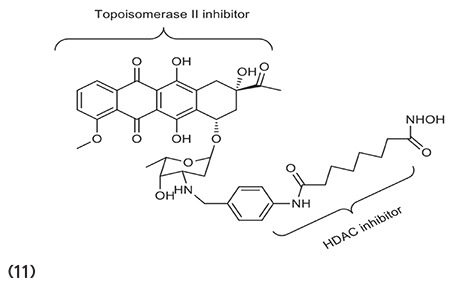
Guerrant et al.67 reported a bifunctional approach to produce chemoactive agents in a single structural design that has 2-fold activity against HDAC and topoisomerase II. Results revealed that compound 11 inhibits both these enzymes with strong inhibitory capacity against different cancerous cell lines. (Graphic 11)
Graphic 11.
Marek et al.68 reported a novel series by incorporating an alkoxy-amide linkage in hydroxamic acid-based compounds. Compound 17 exhibited the same effects contrasted to SAHA in a pan-HDAC cell-based assay and improved cytotoxic outcome against various cancer cell lines (A-2780, Cal-27, Kyse-510, and MDA-MB-231). Compound 12 exerted significant activity against HDAC enzyme and inhibited HDAC4 and 5 in nM concentrations. (Graphic 12)
Graphic 12.
Hou et al.69 described a potent chiral compound (NK-HDAC-51) that exhibited more potent activity than vorinostat in both enzyme- and cell-based assays due to its better physicochemical properties, e.g., Log-D, solubility, micromole stability of liver (t1/2), stability of plasma (t1/2), and apparent permeability. (Graphic 13)
Graphic 13.
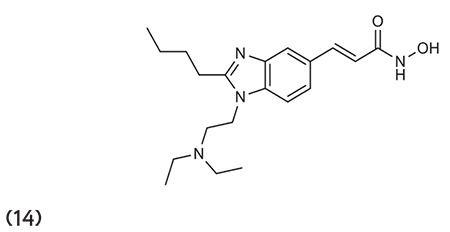
Wang et al.70 outlined the synthesis of 3-(1,2-disubstituted-1H-benzimidazol-5-yl)-N-hydroxy-acryl-amides HDACIs. In vivo studies against various tumor models (HCT-116, PC3, A-2780, MV411, and Ramos) showed that compound 14 is highly effective and has very good pharmacokinetics, safety, and pharmaceutical properties. (Graphic 14)
Graphic 14.
Chun et al.71 synthesized a series of compounds like 15 for anticancer activity and antiproliferative effects against MCF7, MDA-MB 231, MCF 7/Dox, MCF 7/Tam, SK-OV 3, LNCaP, and PC3 human cancer cell lines by the synthesis of suberoylanilide hydroxamate derivatives.71 (Graphic 15)
Graphic 15.
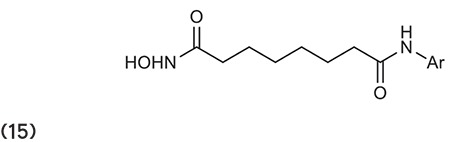
Zhang et al.8 reported a new series of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives for the inhibition of HDACs. Compounds like 16 show potent activity and have better inhibitory activity than vorinostat. (Graphic 16)
Graphic 16.
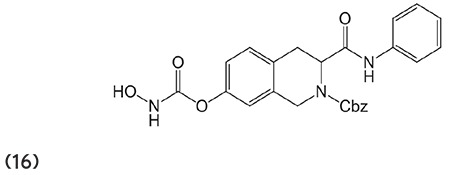
Koncic et al.72 carefully examined a number of hydroxamic acid derivatives of NSAIDs (17) and appraised their antioxidant, radical scavenging activity with regard to butylated hydroxyanisole. (Graphic 17)
Graphic 17.
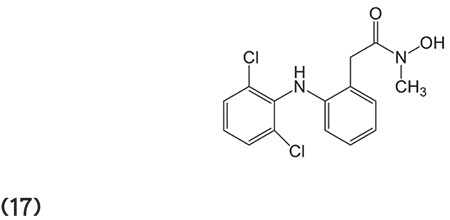
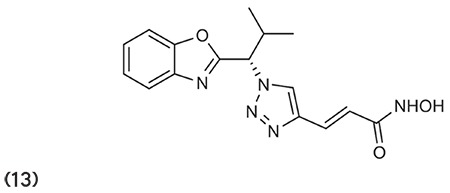
Kozikowski et al.73 outlined a novel series of hydroxamate-based HDACIs synthesized by cycloaddition method. Compounds like 18 have greater potency against HDAC6 with an IC50 value of 2 picomolar. Some compounds were found to be capable of preventing cell growth in pancreatic cancer approximately 10 times more effectively than vorinostat. (Graphic 18)
Graphic 18.
Kim et al.74 reported that novel d-lactam-based HDACIs that have various substituted benzyl, bi-aromatic cap groups were prepared through metathesis reaction. Compound 19 showed inhibitory activity against five different human cancer cell lines (PC3, AC-HN, NUGC3, HCT15, and MBA-MB-231). (Graphic 19)
Graphic 19.
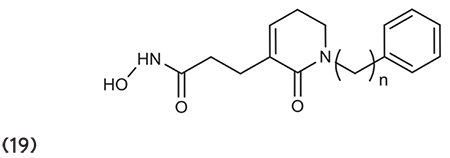
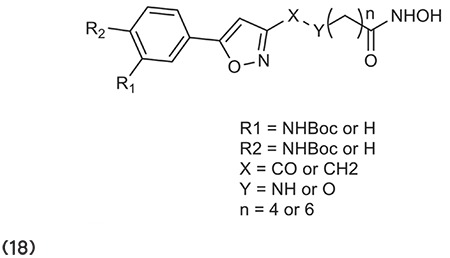
Kahnberg et al.75 described various derivatives of 2-aminosuberic acid. Compound 31 has the ability to kill a range of tumor cells including MM96L melanoma cells, out of whole compounds. Compound 20 exhibits hyperacetylation of histones in both normal and cancerous cells, induces p-21 expression, and discriminates the survival of cancer cells to a nonproliferating phenotype. (Graphic 20)
Graphic 20.
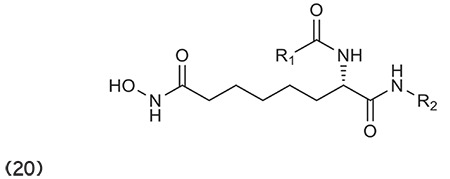
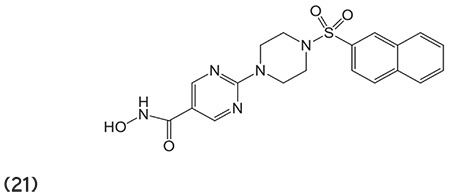
Angibaud et al.76 described a series of novel pyrimidyl-5-hydroxamic acids for HDAC inhibition. Moreover, amino-2-pyrimidinylcan is used as a linker to provide enzymatic potency to HDACIs. (Graphic 21)
Graphic 21.
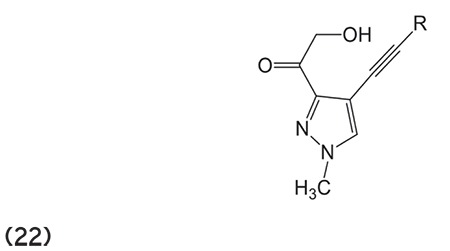
Mshvidobadze et al.77 developed a variety of pyrazolohydroxamic acid molecules that showed greater efficiency against HDAC enzyme. (Graphic 22)
Graphic 22.
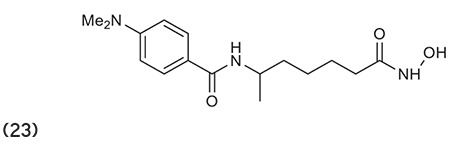
Van Ommeslaeghe et al.78 reported potent amide type HDACIs and molecular modeling confirms the flexibility of the linker chain of compound 23, which is important for the orientation of the dimethyl-amino-benzoyl group in the enzyme. (Graphic 23)
Graphic 23.
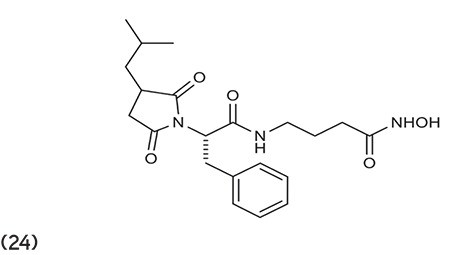
Curtin et al.79 outlined the synthesis and evaluation of a series of succinimide hydroxamic acids, which were prepared and evaluated for HDAC inhibition and antiproliferation. Compound 24 was found to be more potent. (Graphic 24)
Graphic 24.
Sternson et al.80 synthesized a series of potent compounds like 25 having 1,3-dioxane moiety that showed HDAC inhibitory activity. (Graphic 25)
Graphic 25.
CONCLUSIONS
Currently the management of cancer has been improved significantly, and although there are various medications for the treatment of cancer they still seem to be ineffective. Thus it is a major challenge for researchers to develop safe, effective agents with an improved therapeutic index. HDACIs, a new category of anticancer agents, exert innumerable biological effects, i.e., stimulation of cell differentiation, cell demise, cell-cycle arrest, and bringing on of autophagic cell death. Development of specific HDACIs with an enhanced therapeutic index leads to successful target accomplishment that proceeds to increased efficacy. Additionally, recent clinical studies postulate that the inhibitor of HDAC enzyme responds to both hematological and solid tumor malignancies. A low therapeutic range is one of the major drawbacks of existing HDACIs. Inhibitors of HDAC enzyme are used either in monotherapy or in combination therapy with different targeted agents. Combination therapy is more viably successful than monotherapy because it uses chemotherapeutic and biotherapeutic agents having lower toxicity and better clinical outcomes in tumors.
The present review highlights the structure–activity relationship of various HDACIs synthesized across the globe, which will be helpful for designing new potential agents. Special attention was paid to the existing synthesized medicinal compounds over the past few years and their therapeutic application, which will be helpful for future advancement. Apart from cancer, HDACIs are presently used in different remedial areas such as neurodegenerative disorders, cardiovascular disease, liver fibrosis, retinal degenerative disease, regulation of immune response, anti-inflammatory, conjunctivitis, and asthma. We have also tried to summarize the current developments in the structural scaffold of HDACIs such as surface recognition site, linker region, and metal binding moiety. The recent summation by various research groups has been incorporated to understand the advancement of potential inhibitors.
Acknowledgments
The authors are thankful to the Vice-chancellor, Banasthali Vidyapith, for providing the necessary research facilities. The financial assistance provided by DST-CURIE, New Delhi, is duly acknowledged.
Footnotes
Conflict of interest: No conflict of interest was declared by the authors.
References
- 1.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;15:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 2.Wang C, Henkes LM, Doughty LB, He M, Wang D, Meyer-Almes FJ, Cheng YQ. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J Nat Prod. 2011;74:2031–2038. doi: 10.1021/np200324x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen PJ, Huang C, Meng XM, Li J. Epigenetic modifications by histone deacetylases: Biological implications and therapeutic potential in liver fibrosis. Biochimie. 2015;116:61–69. doi: 10.1016/j.biochi.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 5.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2015;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mai A, Massa S, Rotili D, Simeoni S, Ragno R, Botta G, Nebbioso A, Miceli M, Altucci L, Brosch G. Synthesis and biological properties of novel, uracil-containing histone deacetylase inhibitors. J Med Chem. 2006;49:6046–6056. [Google Scholar]
- 7.Paris M, Porcelloni M, Binaschi M, Fattori D. Histone deacetylase inhibitors: from bench to clinic. J Med Chem. 2008;51:1505–1529. doi: 10.1021/jm7011408. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Feng J, Liu C, Zhang L, Jiao J, Fang H, Su L, Zhang X, Zhang J, Li M, Wang B, Xu W. Design, synthesis and preliminary activity assay of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid derivatives as novel Histone deacetylases (HDACs) inhibitors. Bioorg Med Chem. 2010;18:1761–1772. doi: 10.1016/j.bmc.2010.01.060. [DOI] [PubMed] [Google Scholar]
- 9.Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439–1460. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Li X, Hou J, Huang Y, Xu W. Design, synthesis, and antitumor evaluation of histone deacetylase inhibitors with L-phenylglycine scaffold. Drug Des Devel Ther. 2015;9:5553–5567. doi: 10.2147/DDDT.S94037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehnert JM, Kelly WK. Histone deacetylase inhibitors: biology and mechanism of action. Cancer J. 2007;13:23–29. doi: 10.1097/PPO.0b013e31803c72ba. [DOI] [PubMed] [Google Scholar]
- 12.Salvador LA, Luesch H. Discovery and mechanism of natural products as modulators of histone acetylation. Curr Drug Targets. 2012;13:1029–1047. doi: 10.2174/138945012802008973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim B, Hong J. An overview of naturally occurring histone deacetylase inhibitors. Curr Top Med Chem. 2015;14:2759–2582. doi: 10.2174/1568026615666141208105614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Losson H, Schnekenburger M, Dicato M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules. 2016;21:1608. doi: 10.3390/molecules21111608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 16.Blumenschein GR Jr, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL, Chiao JH, Chen C, Frankel SR. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–87. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 17.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 18.Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 19.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 20.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 21.Muraoka M, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Shitara N, Chong JM, Iwama T, Miyaki M. p300 gene alterations in colorectal and gastric carcinomas. Oncogene. 1996;12:1565–1569. [PubMed] [Google Scholar]
- 22. [Internet] http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm435296.html .
- 23.Tan J, Cang S, Ma Y, Petrillo RL, Liu D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J Hematol Oncol. 2010;3:5. doi: 10.1186/1756-8722-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He R, Chen Y, Chen Y, Ougolkov AV, Zhang JS, Savoy DN, Billadeau DD, Kozikowski AP. Synthesis and biological evaluation of triazol-4-ylphenyl-bearing histone deacetylase inhibitors as anticancer agents. J Med Chem. 2010;53:1347–1356. doi: 10.1021/jm901667k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sholler GS, Currier EA, Dutta A, Slavik MA, Illenye SA, Mendonca MC, Dragon J, Roberts SS, Bond JP. PCI-24781 (abexinostat), a novel histone deacetylase inhibitor, induces reactive oxygen species-dependent apoptosis and is synergistic with bortezomib in neuroblastoma. J Cancer Ther Res. 2013;2:2–21. doi: 10.7243/2049-7962-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez G, Liu J, Ren W, Wei W, Wang S, Lahat G, Zhu QS, Bornmann WG, McConkey DJ, Pollock RE, Lev DC. Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res. 2009;15:3472–3483. doi: 10.1158/1078-0432.CCR-08-2714. [DOI] [PubMed] [Google Scholar]
- 27.Plumb JA, Finn PW, Williams RJ, Bandara MJ, Romero MR, Watkins CJ, La Thangue NB, Brown R. Mol Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Cancer Ther. 2003;2:721–728. [PubMed] [Google Scholar]
- 28.Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu H, Wang DG, Yin L, Samson M, Forrester J, Zifcak B, Xu GX, DellaRocca S, Zhai HX, Cai X, Munger WE, Keegan M, Pepicelli CV, Qian C. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70:3647–3656. doi: 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- 29.Quintás-Cardama A, Kantarjian H, Estrov Z, Borthakur G, Cortes J, Verstovsek S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk Res. 2012;36:1124–1127. doi: 10.1016/j.leukres.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Razak AR, Hotte SJ, Siu LL, Chen EX, Hirte HW, Powers J, Walsh W, Stayner LA, Laughlin A, Novotny-Diermayr V, Zhu J, Eisenhauer EA. Phase I clinical, pharmacokinetic and pharmacodynamic study of SB939, an oral histone deacetylase (HDAC) inhibitor, in patients with advanced solid tumours. Br J Cancer. 2011;104:756–762. doi: 10.1038/bjc.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zorzi AP, Bernstein M, Samson Y, Wall DA, Desai S, Nicksy D, Wainman N, Eisenhauer E, Baruchel S. A phase I study of histone deacetylase inhibitor, pracinostat (SB939), in pediatric patients with refractory solid tumors: IND203 a trial of the NCIC IND program/C17 pediatric phase I consortium. Pediatr Blood Cancer. 2013;60:1868–1874. doi: 10.1002/pbc.24694. [DOI] [PubMed] [Google Scholar]
- 32.Mandl-Weber S, Meinel FG, Jankowsky R, Oduncu F, Schmidmaier R, Baumann P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br J Haematol. 2010;149:518–528. doi: 10.1111/j.1365-2141.2010.08124.x. [DOI] [PubMed] [Google Scholar]
- 33.Walewski J, Paszkiewicz-Kozik E, Borsaru G, Moicean A, Warszewska A, Strobel A, Biggi A, Hauns B, Mais A, Henning SW. In Proceedings of the (52nd). ASH Annual Meeting and Exposition. Orlando; FL, USA. 2010:4–7. [Google Scholar]
- 34.Bitzer M, Ganten TM, Woerns MA, Siveke JT, Dollinger MM, Scheulen ME, Wege H, Giannini EG, Cillo U. Trevisani F. J Clin Oncol. 2013;31:15083–15088. [Google Scholar]
- 35.Arts J, King P, Mariën A, Floren W, Beliën A, Janssen L, Pilatte I, Roux B, Decrane L, Gilissen R, Hickson I, Vreys V, Cox E, Bol K, Talloen W, Goris I, Andries L, Du Jardin M, Janicot M, Page M, van Emelen K, Angibaud P. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15:6841–6851. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- 36.Zabkiewicz J, Gilmour M, Hills R, Vyas P, Bone E, Davidson A, Burnett A. Knapper S. Oncotarget. 2016;7:16650–16662. doi: 10.18632/oncotarget.7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moffat D, Patel S, Day F, Belfield A, Donald A, Rowlands M, Wibawa J, Brotherton D, Stimson L, Clark V, Owen J, Bawden L, Box G, Bone E, Mortenson P, Hardcastle A, van Meurs S, Eccles S, Raynaud F, Aherne W. Discovery of 2-(6-{[(6-fluoroquinolin-2-yl)methyl]amino}bicyclo[3.1.0] hex-3-yl)-N-hydroxypyrimidine-5-carboxamide (CHR-3996), a class I selective orally active histone deacetylase inhibitor. J Med Chem. 2010;53:8663–8678. doi: 10.1021/jm101177s. [DOI] [PubMed] [Google Scholar]
- 38.Banerji U, van Doorn L, Papadatos-Pastos D, Kristeleit R, Debnam P, Tall M, Stewart A, Raynaud F, Garrett MD, Toal M, Hooftman L, De Bono JS, Verweij J, Eskens FA. A phase I pharmacokinetic and pharmacodynamic study of CHR-3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin Cancer Res. 2012;18:2687–2694. doi: 10.1158/1078-0432.CCR-11-3165. [DOI] [PubMed] [Google Scholar]
- 39.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu J, Yan TP, Lu AH, Rahil J, Wang J, Lefebvre S, Li Z, Vaisburg AF, Besterman JM. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 40.Pili R, Salumbides B, Zhao M, Altiok S, Qian D, Zwiebel J, Carducci MA, Rudek MA. Phase I study of the histone deacetylase inhibitor entinostat in combination with 13-cis retinoic acid in patients with solid tumours. Br J Cancer. 2012;106:77–84. doi: 10.1038/bjc.2011.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fournel M, Bonfils C, Hou Y, Yan PT, Trachy-Bourget MC, Kalita A, Liu J, Lu AH, Zhou NZ, Robert MF, Gillespie J, Wang JJ, Ste-Croix H, Rahil J, Lefebvre S, Moradei O, Delorme D, Macleod AR, Besterman JM, Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 42.Blum KA, Advani A, Fernandez L, Van Der Jagt R, Brandwein J, Kambhampati S, Kassis J, Davis M, Bonfils C, Dubay M, Dumouchel J, Drouin M, Lucas DM, Martell RE, Byrd JC. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br J Haematol. 2008;147:507–514. doi: 10.1111/j.1365-2141.2009.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.V El-Khoury V, Moussay E, Janji B, Palissot V, Aouali N, Brons NH, Van Moer K, Pierson S, Van Dyck E, Berchem G. The histone deacetylase inhibitor MGCD0103 induces apoptosis in B-cell chronic lymphocytic leukemia cells through a mitochondria-mediated caspase activation cascade. Mol Cancer Ther. 2010;9:1349–1360. doi: 10.1158/1535-7163.MCT-09-1000. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guha M. HDAC inhibitors still need a home run, despite recent approval. Nat Rev Drug Discov. 2015;14:225–226. doi: 10.1038/nrd4583. [DOI] [PubMed] [Google Scholar]
- 46.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133(7 Suppl):2485–2493. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 47.Bouzar AB, Boxus M, Defoiche J, Berchem G, Macallan D, Pettengell R, Willis F, Burny A, Lagneaux L, Bron D, Chatelain B, Chatelain C, Willems L. Valproate synergizes with purine nucleoside analogues to induce apoptosis of B-chronic lymphocytic leukaemia cells. Br J Haematol. 2009;144:41–52. doi: 10.1111/j.1365-2141.2008.07426.x. [DOI] [PubMed] [Google Scholar]
- 48.Stamatopoulos B, Meuleman N, De Bruyn C, Mineur P, Martiat P, Bron D, Lagneaux L. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia. 2009;23:2281–2289. doi: 10.1038/leu.2009.176. [DOI] [PubMed] [Google Scholar]
- 49.Mai A, Altucci L. Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Bio. 2009;41:199–213. doi: 10.1016/j.biocel.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 50.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 51.Madsen AS, Kristensen HM, Lanz G, Olsen CA. The effect of various zinc binding groups on inhibition of histone deacetylases 1-11. Chem Med Chem. 2014;9:614–626. doi: 10.1002/cmdc.201300433. [DOI] [PubMed] [Google Scholar]
- 52.Younes A, Oki Y, Bociek RG, Kuruvilla J, Fanale M, Neelapu S, Copeland A, Buglio D, Galal A, Besterman J, Li Z, Drouin M, Patterson T, Ward MR, Paulus JK, Ji Y, Medeiros LJ, Martell RE. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011;12:1222–1228. doi: 10.1016/S1470-2045(11)70265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bishton MJ, Harrison SJ, Martin BP, McLaughlin N, James C, Josefsson EC, Henley KJ, Kile BT, Prince HM, Johnstone RW. Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood. 2011;117:3658–3668. doi: 10.1182/blood-2010-11-318055. [DOI] [PubMed] [Google Scholar]
- 54.Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N, Rambaldi A. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. 2010;89:185–190. doi: 10.1007/s00277-009-0793-8. [DOI] [PubMed] [Google Scholar]
- 55.Su H, Altucci L, You Q. Competitive or noncompetitive, that’s the question: research toward histone deacetylase inhibitors. Mol Cancer Ther. 2008;7:1007–1012. doi: 10.1158/1535-7163.MCT-07-2289. [DOI] [PubMed] [Google Scholar]
- 56.Diedrich D, Hamacher A, Gertzen CG, Alves Avelar LA, Reiss GJ, Kurz T, Gohlke H, Kassack MU, Hansen FK. Rational design and diversityoriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem Commun (Camb) 2016;52:3219–3222. doi: 10.1039/c5cc10301k. [DOI] [PubMed] [Google Scholar]
- 57.Senger J, Melesina J, Marek M, Romier C, Oehme I, Witt O, Sippl W, Jung M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J Med Chem. 2016;59:1545–1555. doi: 10.1021/acs.jmedchem.5b01493. [DOI] [PubMed] [Google Scholar]
- 58.Zhang S, Huang W, Li X, Yang Z, Feng B. Synthesis, Biological Evaluation, and Computer-Aided Drug Designing of New Derivatives of Hyperactive Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitors. Chem Biol Drug Des. 2015;86:795–804. doi: 10.1111/cbdd.12554. [DOI] [PubMed] [Google Scholar]
- 59.Li Y, Wang Y, Xie N, Xu M, Qian P, Zhao Y, Li S. Design, synthesis and antiproliferative activities of novel benzamides derivatives as HDAC inhibitors. Eur J Med Chem. 2015;100:270–275. doi: 10.1016/j.ejmech.2015.05.045. [DOI] [PubMed] [Google Scholar]
- 60.Chavan V. Mahajan SS. Der Pharma Chemica. 2015;7:199–204. [Google Scholar]
- 61.Zhang X, Kong Y, Zhang J, Su M, Zhou Y, Zang Y, Li J, Chen Y, Fang Y, Zhang X, Lu W. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur J Med Chem. 2015;95:127–135. doi: 10.1016/j.ejmech.2015.03.035. [DOI] [PubMed] [Google Scholar]
- 62.Mendoza-Sanchez R, Cotnoir-White D, Kulpa J, Jutras I, Pottel J, Moitessier N, Mader S, Gleason JL. Design, synthesis and evaluation of antiestrogen and histone deacetylase inhibitor molecular hybrids. Bioorg Med Chem. 2015;23:7597–7606. doi: 10.1016/j.bmc.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 63.Fleming CL, Ashton TD, Gaur V, McGee SL, Pfeffer FM. Improved synthesis and structural reassignment of MC1568: a class IIa selective HDAC inhibitor. J Med Chem. 2014;57:1132–1135. doi: 10.1021/jm401945k. [DOI] [PubMed] [Google Scholar]
- 64.Cheng J, Qin J, Guo S, Qiu H, Zhong Y. Design, synthesis and evaluation of novel HDAC inhibitors as potential antitumor agents. Bioorg Med Chem Lett. 2014;24:4768–4772. doi: 10.1016/j.bmcl.2014.06.080. [DOI] [PubMed] [Google Scholar]
- 65.Feng T, Wang H, Su H, Lu H, Yu L, Zhang X, Sun H, You Q. Novel N-hydroxyfurylacrylamide-based histone deacetylase (HDAC) inhibitors with branched CAP group (Part 2) Bioorg Med Chem. 2013;21:5339–5354. doi: 10.1016/j.bmc.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 66.Ning C, Bi Y, He Y, Huang W, Liu L, Li Y, Zhang S, Liu X, Yu N. Design, synthesis and biological evaluation of di-substituted cinnamic hydroxamic acids bearing urea/thiourea unit as potent histone deacetylase inhibitors. Bioorg Med Chem Lett. 2013;23:6432–6435. doi: 10.1016/j.bmcl.2013.09.051. [DOI] [PubMed] [Google Scholar]
- 67.Guerrant W, Patil V, Canzoneri JC, Oyelere AK. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J Med Chem. 2012;55:1465–1477. doi: 10.1021/jm200799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, Kurz T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56:427–436. doi: 10.1021/jm301254q. [DOI] [PubMed] [Google Scholar]
- 69.Hou J, Li Z, Fang Q, Feng C, Zhang H, Guo W, Wang H, Gu G, Tian Y, Liu P, Liu R, Lin J, Shi YK, Yin Z, Shen J, Wang PG. Discovery and extensive in vitro evaluations of NK-HDAC-1: a chiral histone deacetylase inhibitor as a promising lead. J Med Chem. 2012;55:3066–3075. doi: 10.1021/jm201496g. [DOI] [PubMed] [Google Scholar]
- 70.Wang H, Yu N, Chen D, Lee KC, Lye PL, Chang JW, Deng W, Ng MC, Lu T, Khoo ML, Poulsen A, Sangthongpitag K, Wu X, Hu C, Goh KC, Wang X, Fang L, Goh KL, Khng HH, Goh SK, Yeo P, Liu X, Bonday Z, Wood JM, Dymock BW, Kantharaj E, Sun ET. Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile. Med Chem. 2011;54:4694–4720. doi: 10.1021/jm2003552. [DOI] [PubMed] [Google Scholar]
- 71.Chun P, Won HK, Jungsu K, Kang JA, Hye JL, Young P, Mee YA, Hyung SK, Hyung RM. Synthesis and Importance of Bulky Aromatic Cap of Novel SAHA Analogs for HDAC Inhibition and Anticancer Activity. Bull Korean Chem Soc. 2011;32:1891–1896. [Google Scholar]
- 72.Koncic MZ, Rajic Z, Petric N, Zorc B. Antioxidant activity of NSAID hydroxamic acids. Acta Pharm. 2009;59:235–242. doi: 10.2478/v10007-009-0017-8. [DOI] [PubMed] [Google Scholar]
- 73.Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J Med Chem. 2008;51:4370–4373. doi: 10.1021/jm8002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim HM, Hong SH, Kim MS, Lee CW, Kang JS, Lee K, Park SK, Han JW, Lee HY, Choi Y, Kwon HJ, Han G. Modification of cap group in delta-lactam- based histone deacetylase (HDAC) inhibitors. Bioorg Med Chem Lett. 2007;17:6234–6238. doi: 10.1016/j.bmcl.2007.09.034. [DOI] [PubMed] [Google Scholar]
- 75.Kahnberg P, Lucke AJ, Glenn MP, Boyle GM, Tyndall JD, Parsons PG, Fairlie DP. Design, synthesis, potency, and cytoselectivity of anticancer agents derived by parallel synthesis from alpha-aminosuberic acid. J Med Chem. 2006;49:7611–7622. doi: 10.1021/jm050214x. [DOI] [PubMed] [Google Scholar]
- 76.Angibaud P, Arts J, Van Emelen K, Poncelet V, Pilatte I, Roux B, Van Brandt S, Verdonck M, De Winter H, Ten Holte P, Marien A, Floren W, Janssens B, Van Dun J, Aerts A, Van Gompel J, Gaurrand S, Queguiner L, Argoullon JM, Van Hijfte L, Freyne E, Janicot M. Discovery of pyrimidyl-5-hydroxamic acids as new potent histone deacetylase inhibitors. Eur J Med Chem. 2005;40:597606. doi: 10.1016/j.ejmech.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 77.Mshvidobadze EV, Vasilevskya SF. Elguero J. Tetrahedron. 2004;60:11875–11878. [Google Scholar]
- 78.Van Ommeslaeghe K, Elaut G, Brecx V, Papeleu P, Iterbeke K, Geerlings P, Tourwé D, Rogiers V. Amide analogues of TSA: synthesis, binding mode analysis and HDAC inhibition. Bioorg Med Chem Lett. 2003;13:1861–1864. doi: 10.1016/s0960-894x(03)00284-1. [DOI] [PubMed] [Google Scholar]
- 79.Curtin ML, Garland RB, Heyman HR, Frey RR, Michaelides MR, Li J, Pease LJ, Glaser KB, Marcotte PA, Davidsen SK. Succinimide hydroxamic acids as potent inhibitors of histone deacetylase (HDAC) Bioorg Med Chem Lett. 2002;13:2919–2923. doi: 10.1016/s0960-894x(02)00622-4. [DOI] [PubMed] [Google Scholar]
- 80.Sternson SM, Wong JC, Grozinger CM, Schreiber SL. Synthesis of 7200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org Lett. 2001;3:4239–4242. doi: 10.1021/ol016915f. [DOI] [PubMed] [Google Scholar]