

Abstract
MicroRNAs (miRNAs), small non-coding RNAs that regulate gene expression post-transcriptionally, are involved in many complex cellular processes. Several miRNAs are differentially expressed in hematopoietic tissues and play important roles in normal differentiation, but, when aberrantly regulated, contribute to the abnormal proliferation and differentiation of leukemic cells. Recently, we reported that a small subset of miRNAs is differentially expressed in acute promyelocytic leukemia (APL) blasts and is modulated by treatment with all-trans-retinoic acid (ATRA). In particular, PML/RARα-positive blasts from APL patients display lower levels of miRNA let-7c, a member of the let-7 family, than normal promyelocytes and its expression increases after ATRA treatment. In this study, we investigated the effects of let-7c in acute myeloid leukaemia (AML) cells. We found that ectopic expression of let-7c promotes granulocytic differentiation of AML cell lines and primary blasts. Moreover, we identified PBX2, a well-known homeodomain protein whose aberrant expression enhances HoxA9-dependent leukemogenesis, as a novel let-7c target that may contribute to the AML phenotype. Together, these studies raise the possibility that perturbation of the let-7c-PBX2 pathway may have a therapeutic value in AML.
Introduction
Acute myeloid leukaemia (AML), the most common leukemia in adults, is a heterogeneous clonal disorder characterized by the accumulation of differentiation-arrested myeloid progenitor cells (blasts) in the bone marrow, ultimately leading to hematopoietic failure. Current treatments induce complete remission in most patients, but survival rate remains the lowest of all leukemias.1 AML comprises different subtypes, one of which, acute promyelocytic leukaemia (APL), is characterized by maturation arrest at the promyelocytic stage of development. Differentiation arrest is caused by expression of PML/RARα, a fusion protein with oncogenic properties, which interferes with myeloid differentiation by repressing transcription of retinoic acid (RA)-responsive genes, an effect reversed by treatment with All-Trans-Retinoic Acid (ATRA).2
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression post-transcriptionally.3 Their function has been linked to the regulation of several cellular processes, including the effects of oncogenes or tumor suppressor genes in the process of neoplastic transformation.4 A number of miRNAs are differentially expressed in hematopoietic tissues in vivo and play an important role as regulators of normal differentiation and of the aberrant proliferation and differentiation of leukemic cells.5–11 Moreover, miRNA expression signatures might discriminate AML from ALL12 and from normal bone marrow CD34+ cells.13 We have shown that a small subset of miRNAs is differentially expressed in APL blasts and modulated by ATRA treatment. Interestingly, we identified let-7c as one of the miRNAs down-regulated in APL blasts at diagnosis, compared to in vitro-differentiated normal promyelocytes.6 Let-7c belongs to the let-7 family of miRNAs that play important roles in various cellular processes, and distinct family members appear to be specifically deregulated in certain cancers.14 and references therein Low expression of let-7 family members correlates with increased transformation capacity in vitro and poor survival in vivo.14
In AMLs, expression of let-7c was down-regulated in patients with t(8;21) and inv(16) as well as in PML/RARα-positive APL blasts.6,15 Although several let-7 family targets (RAS, MYC, Lin28 and HMGA2, as well as other cell cycle progression genes) have been identified and assessed for their roles in tumorigenesis,14,16–20 putative targets of let-7c that may contribute specifically to the AML phenotype have not been experimentally validated.
Here, we investigated the functional role of let-7c in AML and show that, when ectopically expressed, it induces granulocytic differentiation. Moreover, we have identified PBX2, a gene required for HoxA9-dependent immortalization of myeloid cells, as a novel target of let-7c that may contribute to the AML phenotype and provide a target for therapeutic intervention.
Results and Discussion
To investigate the role of let-7c in AML, we assessed the consequences of its over-expression in the NB4, HL-60 and U937 myeloid cell lines. We observed that levels of the α-integrin CD11b, a membrane antigen associated with granulo-monocytic differentiation, were higher in let-7c-overexpressing cell lines and were further increased upon ATRA treatment (Figure 1a). Let-7c over-expression was also accompanied by variable increases in the levels of other membrane antigens such as the α-integrins CD11a and c and the β2-integrin CD18 (Supplementary Figure 1). Compared to mimic negative control-transfected cells, over-expression of let-7c in NB4 and HL-60 cells induced a more mature cell morphology as revealed by: i) reduced cell size and nucleus-to-cytoplasm ratio; and ii) detection of indented-shaped and convoluted nuclei (Figure 1b and Supplementary Figure 2). In HL-60 and U937 cell lines, over-expression of let-7c had no effect on cell proliferation, as revealed by DNA content analysis and cell counts; instead, let-7c over-expression led to a slight inhibition of NB4 cell proliferation (Supplementary Figure 3 and data not shown).
Figure 1. Over-expression of miRNA let-7c affects differentiation in AML cell lines and primary blasts.
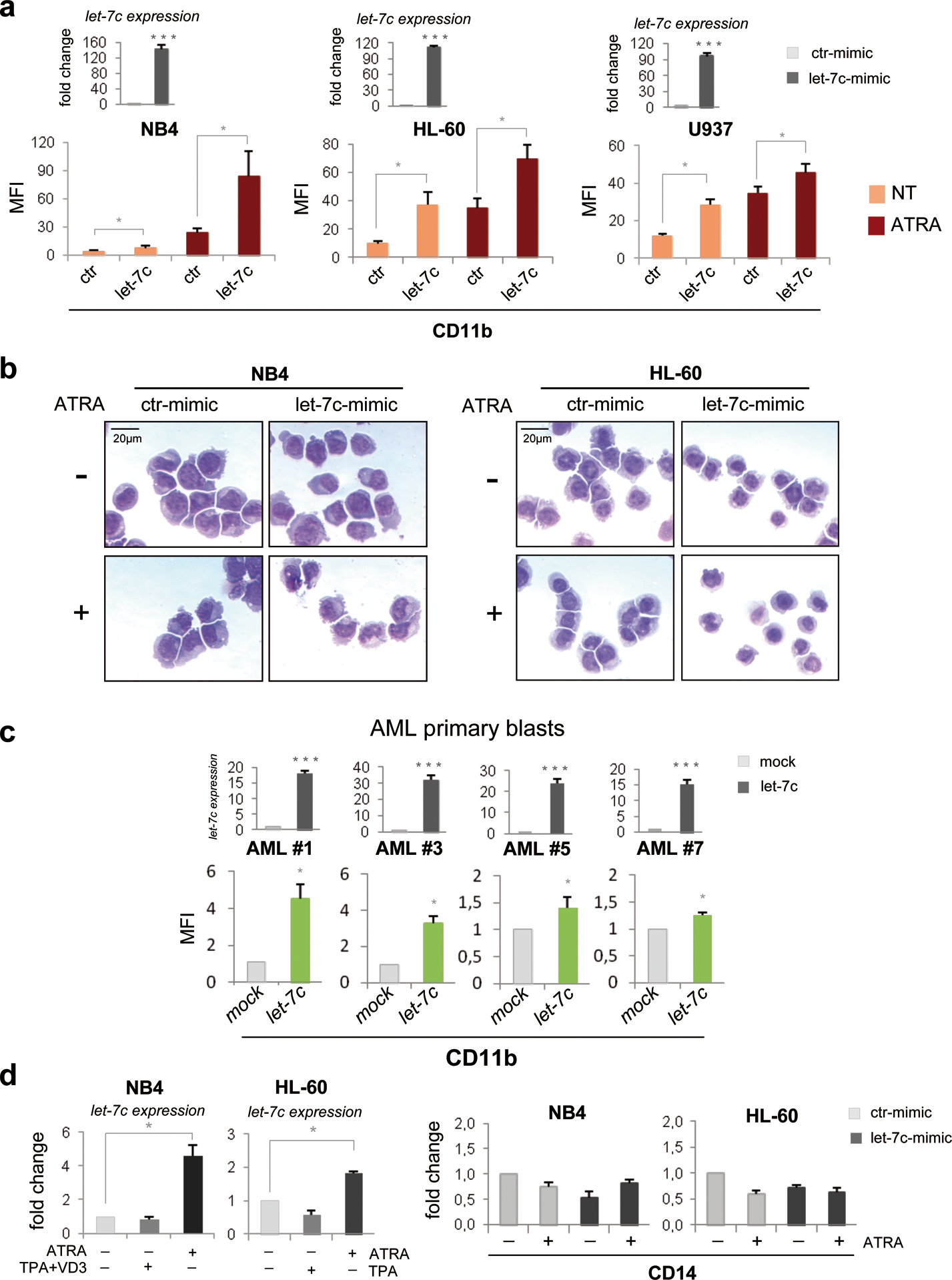
(a) Myeloid cell lines were electroporated (Nucleofector 4D system, Lonza, buffer SF) either with a miRIDIAN hsa-let-7c mimic (let-7c-mimic; 600 nM, Dharmacon) or miRIDIAN mimic negative control #2 (ctr-mimic; 600 nM, Dharmacon). In standardization experiments, transfection efficiency ranged from 41% to 65%, based on the uptake of the fluorescent transfection indicator siGLO (Dharmacon RNATechnologies, Lafayette, CO; data not shown). 24 h after transfection, cells were treated with ATRA (1μM) for 48 hours and analyzed for let-7c and membrane antigen CD11b expression. Total RNA was isolated using Trizol Reagent (Invitrogen).
(upper panel) Quantification of ectopic let-7c expression in the indicated myeloid cell lines was assessed by reverse transcriptase reaction using stem-loop primers followed by quantitative real-time polymerase chain reaction (qRT-PCR) performed in accordance with manufacturer’s instructions (Applied Biosystems Inc., Foster City, CA, USA) on an Applied Biosystem 7500 Real Time PCR System SDS v1,2. Samples were normalized for RNU19 small RNA expression. Real-time PCR was performed in triplicate, including no-template controls. Values are reported as fold changes of let-7c-mimic compared to ctr-mimic. Error bars indicate SEM (n = 3). ***P value <0.001 (Student’s t-test).
(lower panel) CD11b expression was assessed by FACSCalibur flow cytometer. Acquisition and analysis was performed using Cell Quest Pro Software (Becton Dickinson). Briefly, untreated or ATRA-treated NB4, HL-60 and U937 cells were harvested, washed twice in PBS + 5% FBS and incubated 30 min on ice in the dark with a 1:10 fluorochrome-labeled anti-CD11b PE-conjugated antibody or isotype control (BD Biosciences Pharmingen). Values were expressed as Mean Fluorescence Intensity (MFI). Error bars indicate SEM (n= 3). *P value <0.05 (Student’s t-test).
(b) May Grünwald-Giemsa-stained cytospins of untreated or ATRA-treated (1μM, 48 hours) myeloid cell lines overexpressing let-7c-mimic or ctr-mimic oligonucleotides, analyzed at 400× magnification under a microscope (Eclipse 1000; Nikon, Tokyo, Japan) equipped with a digital camera. A representative field is shown. The scale bar represents 20 μm.
(c) (upper panel) Quantification of let-7c in lentivirally transduced primary AML blasts. Samples were normalized for levels of RNU19 small RNA. Real-time PCR was performed in triplicate, including no-template controls. Values are reported as fold changes compared to empty vector-transduced samples. Error bars indicate SEM (n = 3). ***P value <0.001 (Student’s t-test). (lower panel) CD11b expression measured by FACS analysis in primary AML blasts after 72h of infection with a lentiviral vector expressing let-7c (let-7c) or with the empty lentiviral vector (mock). GFP-positive cells were analyzed by fluorescence activated cell sorting as described.7 Values are expressed as MFI fold changes compared to mock infected cells. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test). Methods: Leukemia blasts were obtained from bone marrow collected at diagnosis from four AML patients with normal karyotype. Informed consent was obtained from the patients and the study was approved by the review board and ethic committee of “SC Ematologia” Azienda Ospedaliera Universitaria, Policlinico Tor Vergata in accordance with the Declaration of Helsinki. Blasts isolation and molecular characterization of major fusion proteins were performed as described.34 RNA extraction, RT-PCR reactions, mature miRNA quantification and CD11b expression was performed as described in the legend of Figure 1a. For the lentivirus let-7c construction, a pU1-let-7c plasmid was generated by cloning a fragment of the pri-let-7c (from −68 to +146 bp relative to the 5’ end of pre-let-7c sequence) into the pSP65-U1 cassette plasmid. The U1-let-7c expression cassette was then sub-cloned into the EcoRV site of the lentiviral vector pRRLSIN-cPPT-PGK-GFP-WPRE. The 3th generation packaging kit was purchased from Invitrogen and used according to the manufacturer’s instructions. Infective particles were produced and utilized as previously described.7
(d) (left panel) Mature let-7c expression in the indicated myeloid cell lines treated with differentiation-inducing agents was assayed as described in the legend of Figure 1a. Samples were normalized for expression of RNU19 small RNA. For induction of granulocytic differentiation, ATRA (Sigma) was added at a final concentration of 1 μM for 48 hours. For induction of monocytic differentiation, HL-60 cells were treated with TPA alone (50 nM, 48 hours), whereas NB4 cells were treated with TPA and Vitamin D3 (200 nM each) as described.21,22 Values are reported as fold changes of let-7c levels in treated versus untreated cells. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test). (right panel) Expression of monocytic-specific membrane antigen CD14 in let-7c-mimic- or ctr-mimic-transfected NB4 and HL-60 cells, untreated or treated with ATRA (1μM; 48 hours). cDNAs were amplified using the TaqMan Gene Expression assay (Hs 02621496_s1 Applied Biosystems). Samples were normalized to levels of β-Actin mRNA transcripts. Values are reported as fold changes compared to untreated ctr-mimic transfected cells. Error bars indicate SEM (n = 3).
Consistent with these findings, we also observed increased expression of CD11b in let-7c-lentivirally transduced primary blasts from the bone marrow of four newly diagnosed AML patients (Figure 1c). Comparison of CD11b expression in three primary AML patients transfected with let-7c or a mimic negative control miRNA showed higher levels of CD11b after let-7c transfection (Supplementary Figure 4), confirming that its effects are specific. Together, these data strongly suggest that let-7c induces granulo-monocytic differentiation of AML cells and, in let-7c-expressing myeloid cell lines, this process was not accompanied by changes in cell counts and cell cycle distribution.
Under appropriate culture conditions, AML cells can be induced to differentiate along the monocytic and/or the granulocytic lineage, supporting the bipotential granulo–monocytic nature of AML blasts.21,22 To gain further insight in the let-7c-mediated AML cell differentiation, we investigated if this process was lineage-specific. As shown in Figure 1d, left panel, let-7c expression was increased upon ATRA-induced granulocytic differentiation of NB4 and HL-60 cells, but was essentially unchanged upon induction of monocytic differentiation by treatment with TPA and Vitamin D3 (NB4) or TPA alone (HL-60). In agreement with these lineage-specific changes in let-7c levels, ectopic expression of let-7c in NB4 and HL-60 cells did not induce the expression of the monocytic-specific membrane antigen CD14 (Figure 1d, right panel). Together, these findings suggest that the granulocyte-specific pro-differentiation role of let-7c in AML cells is functionally related to its expression during granulocytic differentiation.
Although numerous studies have described diverse let-7 family targets, putative let-7c-regulated targets that may contribute specifically to the AML phenotype have not been identified yet.14,16–20 Screening of potential targets by miRanda (http://www.microrna.org/microrna), TargetScan 5.2 (http://www.targetscan.org), Pictar (http://www.pictar.org) and miRDB (http://mirdb.org/miRDB/) algorithms,23,24 yielded several dozen to hundreds of putative let-7c target genes. To narrow down the analysis to a manageable number of candidates, we studied potential targets identified by at least three databases. A selected list of let-7c targets, grouped by biological function, is reported in Figure 2a, left panel. Among these, experimentally validated targets such as RAS, MYC, Lin28 and HMGA2, could be implicated in AML.10,14,25 However, our aim was to focus on novel, not experimentally validated let-7c targets, potentially involved in its pro-differentiation function in myeloid cells. To this purpose, we focused on PBX2 and PBX3 because of their involvement in leukemias.26 PBX genes belong to the Three Aminoacid Loop Extension (TALE) superfamily of homeodomain transcription factors which form heteromeric complexes with Hox and Meis proteins.27,28 PBX2 and PBX3 3’UTR contain two putative let-7c binding sites (Figure 2a, right panel). To assess whether these two mRNAs are let-7c targets, we evaluated the levels of the encoded protein in let-7c-over-expressing NB4 cells. We found that ectopic expression of let-7c caused a decrease in PBX2, but not PBX3, protein levels (Figure 2b), an effect confirmed in HL-60 and U937 myeloid cells (Figure 2c). Let-7c over-expression also induced a decrease of PBX2 mRNA expression (Figure 2d). Such decrease appears to correlate better with reduced PBX2 protein levels in U937 than in NB4 and HL-60 cells (Supplementary Figure 5), suggesting that, in these two cell lines, ectopic expression of let-7c represses PBX2 expression also by a mechanism of translational inhibition.
Figure 2. Putative let-7c targets.
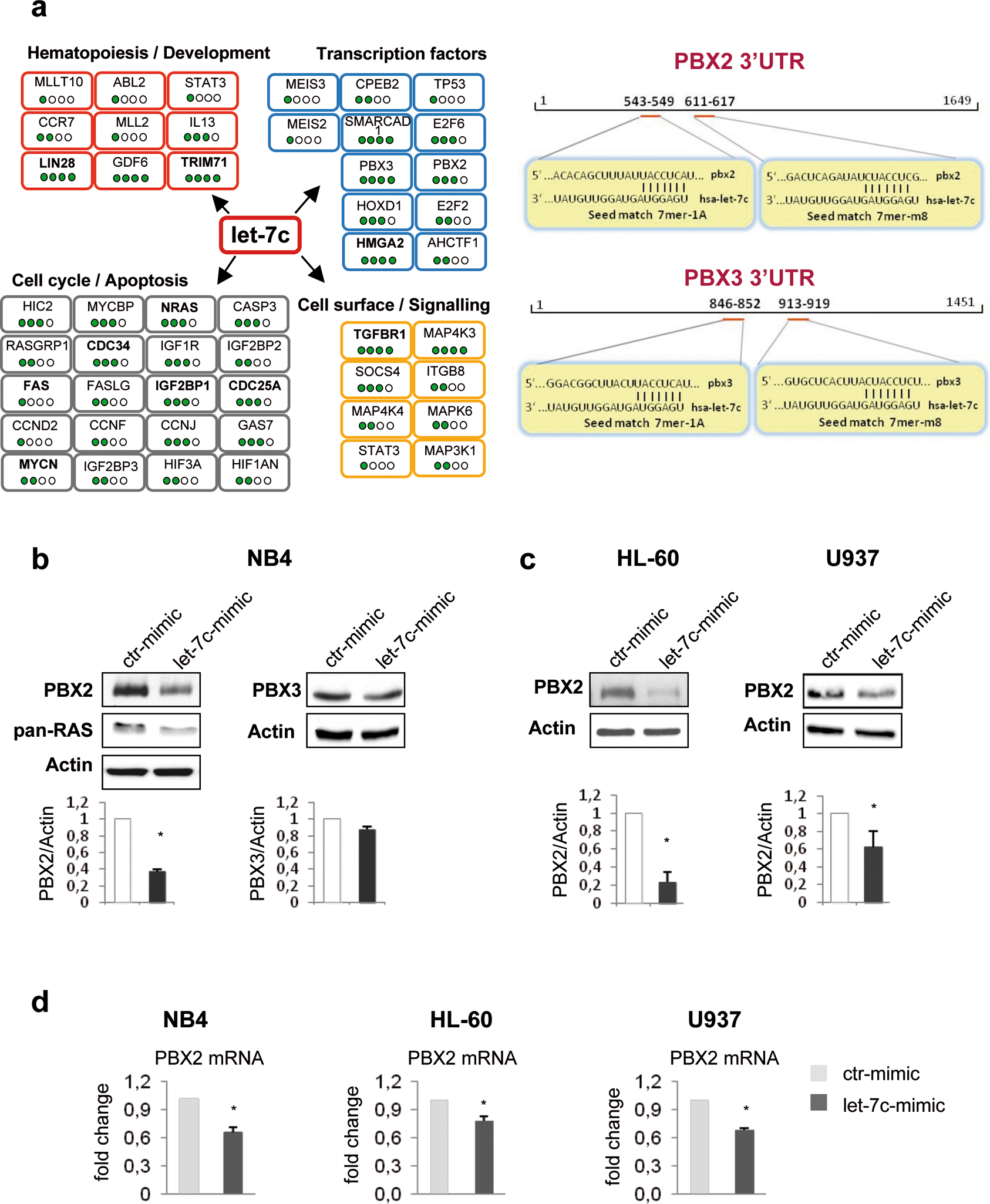
(a) (left panel) Scheme of selected candidate target genes of miRNA let-7c screened by miRanda, TargetScan 5.2, Pictar and miRDB algorithms. Filled spots represent the number of algorithms able to predict the putative target gene of the miRNA let-7c. In bold, genes already experimentally validated as let-7c targets.
(right panel) Schematic representation of putative binding sites for miRNA let-7c in the 3’UTRs of the PBX2 and PBX3 genes.
(b) Western blots of a representative experiment showing levels of endogenous PBX2 and PBX3 proteins in NB4 cells, 72h after transfection with let-7c or control RNA mimics. Immunoblotting was performed with antibodies against PBX2 and PBX3 (G-20, sc-890 and D-17, sc-891, Santa Cruz). Anti-actin (CP01, Ab-1, Calbiochem), was used as loading control whereas anti pan-RAS (OP40, Ab-3, Calbiochem), a well-known let-7 family target, was used as positive control. In the lower panel, proteins were quantified using the densitometry function of the ImageJ software (NIH, U.S.A), normalized to actin within the same sample and expressed as fold changes compared to control. Three independent experiments are presented. Numbers represent the average density (in arbitrary units) detected by the software normalized for the ctr sample. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test).
(c) Western blot assays were used to monitor the expression level of endogenous PBX2 protein in HL-60 and U937 cells, 72h after transfection with let-7c-mimic or control mimic. For each cell line, a representative experiment is shown, and the results from the densitometric analysis of three independent experiments are presented in the histograms. Mean± SEM (n= 3).*P value <0.05 (Student’s t-test).
(d) Expression of endogenous PBX2 is suppressed by let-7c. PBX2 RNA quantification in the indicate myeloid cells transfected with let-7c-mimic or control-mimic, was performed using SYBR Green-based qRT-PCR as described.6 GAPDH gene expression was used as endogenous control. Results (mean ± SEM of three independent experiments) are expressed as fold changes in let-7c mimic versus control-mimic-transfected cells. *P value <0.05 (Student’s t-test).
Next, we evaluated the effect of ATRA treatment on PBX2 protein levels in AML cells. ATRA treatment affected only slightly PBX2 expression in U937 and NB4 cells (Figure 3a, upper panel); however, it led to increased PBX2 mRNA levels (Figure 3a, lower panel). The discrepancy in PBX2 mRNA and protein levels suggests that post-transcriptional mechanisms, such as impaired translation of PBX2 mRNA, are involved in the regulation of PBX2 expression by ATRA. As expected, let-7c was markedly up-regulated in ATRA-treated NB4 and U937 cells (Figure 3b), suggesting that PBX2 post-transcriptional negative regulation is mediated, at least in part, by this miRNA. Consistent with these findings, expression of let-7c and PBX2 protein but not PBX2 mRNA was inversely correlated in 7 out of 8 AML patient samples (Figure 3c). Together, these results suggest that let-7c could dampen an increase in PBX2 expression in ATRA-treated AML cells. To investigate this possibility, we knocked-down let-7c expression in NB4 and U937 cells and assessed expression of differentiation markers and PBX2 levels in untreated and ATRA-treated cells. We found that let-7c knock-down significantly reduced CD11b up-regulation upon ATRA treatment in both cell lines (Figure 4a, middle panel). Interestingly, let-7c knock-down also led to an increase of PBX2 protein levels upon ATRA treatment (Figure 4a, lower panel).
Figure 3. Endogenous PBX2 expression during ATRA treatment.
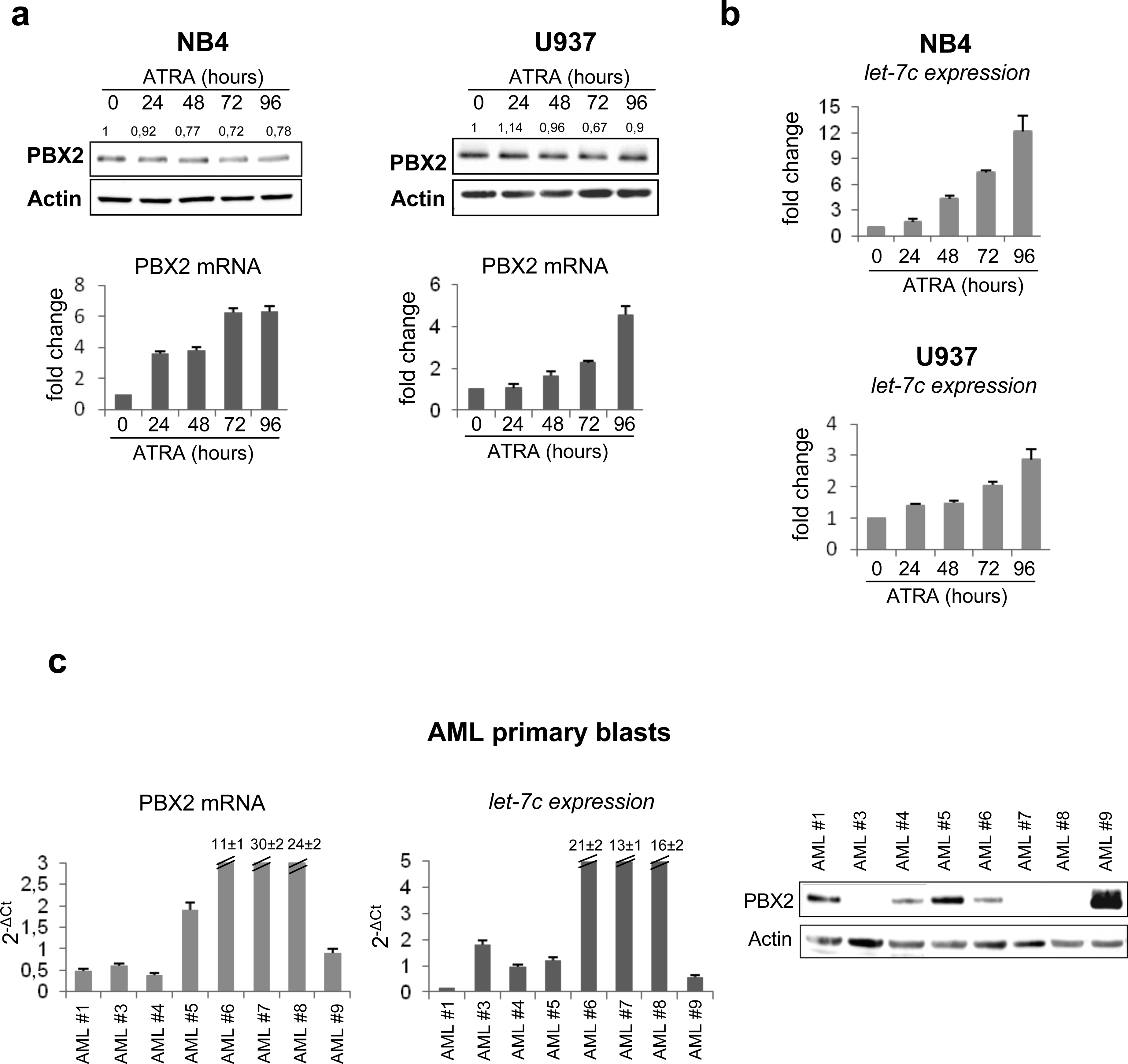
(a) (upper panel) Western blot of endogenous PBX2 protein in NB4 and U937 cells during ATRA treatment. Numbers above the lanes represent densitometric analysis of PBX2/Actin reported as fold change with respect to untreated samples. Immunoblotting and densitometry were performed as described in the legend of Figure 2b.
(lower panel) PBX2 RNA quantification in NB4 and U937 cells, during ATRA treatment, was performed using SYBR Green-based qRT-PCR as described.6 GAPDH gene expression was used as endogenous control. Results (mean ± SEM of three independent experiments) are expressed as fold changes in ATRA-treated relative to untreated cells.
(b) Let-7c expression as evaluated by stem-loop PCR in NB4 and U937 cell lines during ATRA treatment (1μM). Values are reported as fold changes compared to let-7c level in untreated cells. Error bars indicate SEM (n = 3).
(c) (left panel) RNA quantification of PBX2 by SYBR Green DNA Master mix and specific primers, available upon request, in AML primary blasts. Values are reported as 2−ΔCt. Samples were normalized to GAPDH gene expression. Error bars indicate SEM (n=3).
(middle panel) Let-7c expression evaluated by stem-loop PCR in AML primary blasts. Values are reported as 2−ΔCt. Samples were normalized to RNU19 small RNA levels. Error bars indicate SEM (n=3).
(right panel) Western Blot of endogenous PBX2 protein in AML primary blasts. RNA extraction, RT-PCR reactions, mature miRNA quantification and western blots were performed as described in the legend of Figure 1a and 2b.
Figure 4. PBX2 is a direct target of let-7c.
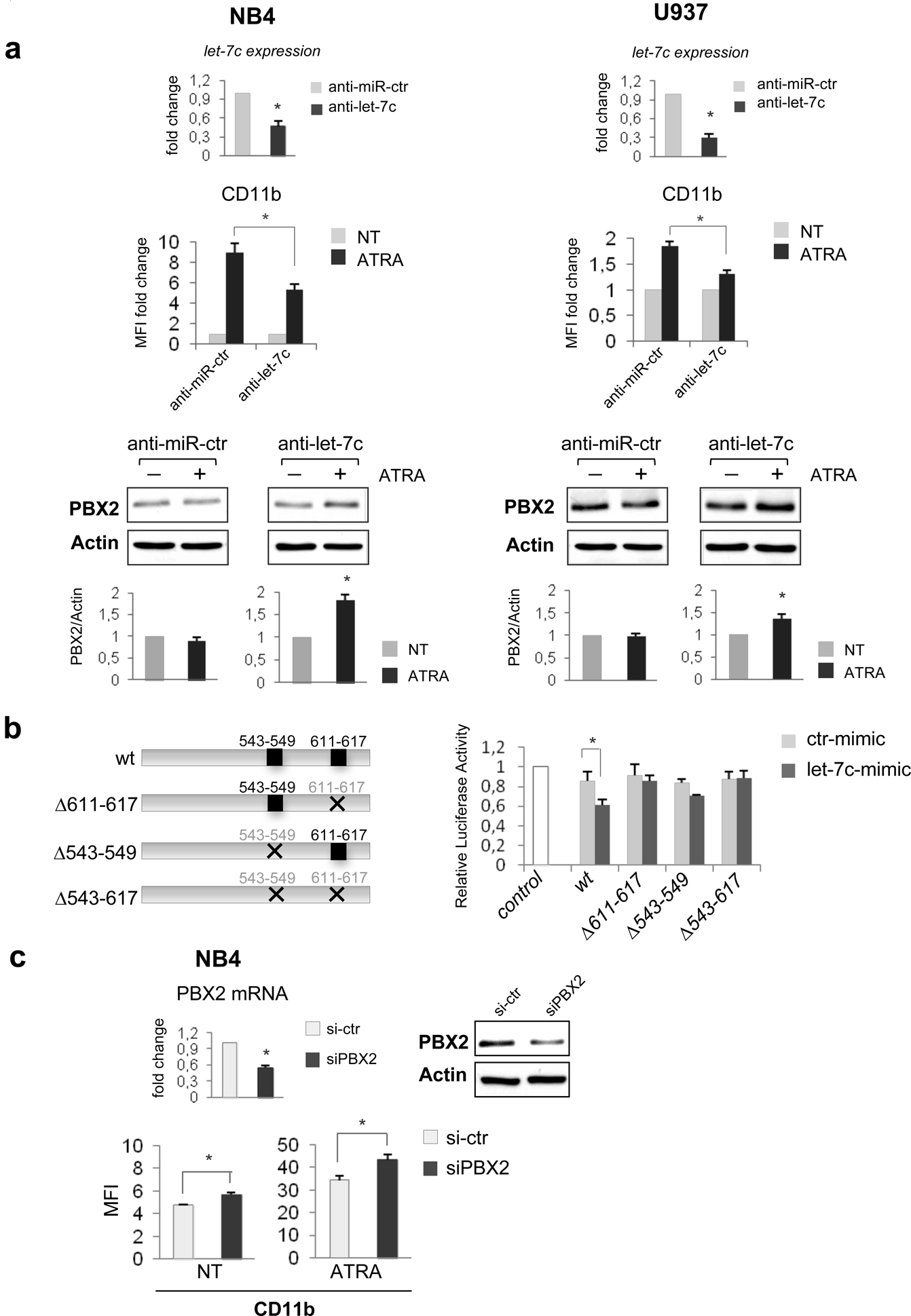
(a) (upper panel) Expression level of let-7c, evaluated by stem-loop PCR, in the indicated cell lines after transfection with anti-let-7c or negative control (anti-let-7c or anti-miR-ctr, 800 nM; LNA Exiqon).
(middle panel) CD11b expression, measured by FACS analysis, in the indicated myeloid cell lines. 24 h after transfection with anti-let-7c or anti-miR-ctr, cells were treated with ATRA (1μM) for 48 hours and analyzed for expression of the membrane antigen CD11b as described inFfigure 1a. Values are expressed as MFI fold change compared to untreated control. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test).
(lower panel) Western blots of endogenous PBX2 protein in NB4 and U937 cells, treated or untreated with ATRA (72 hours), after electroporation with anti-let-7c or anti-miR-ctr. A representative experiment for western blot, performed as described in the legend of Figure 2b, is shown. The intensity for each band was densitometrically quantified and the results of three independent experiments are presented in the histograms. Mean± SEM (n= 3).*P value <0.05 (Student’s t-test).
(b) (left panel) Schematic representation of reporter constructs containing the complete (3’UTR wt) or deletion mutants (PBX2-Δ543–549, -Δ611–617, -Δ543-Δ617) of let-7c–binding sequences in PBX2 constructs used to measure luciferase activity.
(right panel) Human 293T cells were transiently co-transfected, by Lipofectamine 2000 (Invitrogen), with 50ng of Renilla luciferase pRL-TK and 0.8 μg of firefly luciferase reporter plasmid containing wild-type or the indicated deleted PBX2 3’UTRs (see below for PBX2 3’UTRs plasmid details) and 40 pmol of either the hsa-let-7c mimic or ctr-mimic RNA oligonucleotides. The pRL-TK vector providing constitutive expression of Renilla luciferase was co-transfected as an internal control to correct differences in both transfection and harvest efficiencies. 48h post-transfection with let-7c or control RNA mimic, cells were lysed and luciferase activity quantified using the Dual Luciferase Reporter kit (Promega Inc.), as reported.35 Firefly luciferase activity of each sample was normalized by Renilla luciferase activity. Results are expressed as fold activation relative to the basal activity of pGL3 empty Control vector (control). The normalized luciferase activity, set as mean of at least three independent experiments done in duplicate, is shown. Error bars represent the mean ± SEM (n = 3). *P value of <0.05 (Student’s t-test).
The human PBX2 3’UTR (NM_002586) segment containing the target sites for let-7c were amplified by PCR from genomic DNA and cloned into pGL3 Control vector (Promega) downstream of the luciferase gene. The specific primers used are available upon request. The deletion mutants were obtained by restriction site digestions. All the plasmids were verified by sequence analysis.
(c) (upper panel) Expression of endogenous PBX2 mRNA and protein after electroporation of small interference RNAs (siRNA) targeting human PBX2 (siPBX2, 600 nmol; ON-TARGETplus SMARTpool,™ Dharmacon). The control RNA duplex (si-ctr 600 nmol; ON-TARGETplus Non-targeting pool, Dharmacon) for siRNA had no homology to any human genome sequences. For PBX2 mRNA, value is reported as fold changes compared to control-transfected cells. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test). For western blot a representative experiment, performed as described in the legend of Figure 2b, is shown.
(lower panel) CD11b expression, measured by FACS analysis, after electroporation with siPBX2 or si-ctr, in the NB4 cell line, treated or untreated with ATRA (48h). Values are expressed as MFI. Error bars indicate SEM (n = 3). *P value <0.05 (Student’s t-test).
To confirm that PBX2 is a direct target of let-7c, a human PBX2 3’UTR fragment with or without let-7c–binding site sequences (PBX2-Δ543–549, Δ611–617, -Δ543–617; Figure 4b, left panel) was cloned downstream of the firefly luciferase reporter gene. Of interest, the relative luciferase activity of the reporter with wild-type 3’UTR was suppressed upon co-transfection with mimic let-7c, whereas that of the deletion mutant reporters was not, suggesting that let-7c suppresses gene expression through interaction with its binding sequence in the 3’UTR of PBX2 (Figure 4b, right panel). Finally, to evaluate whether PBX2 is a target involved in the pro-differentiation function of let-7c in AML, we assessed the effects of PBX2 knock-down on cell differentiation. As shown in Figure 4c, PBX2 down-regulation induced an increase in CD11b expression, suggesting that it contributes to the pro-differentiating effect of let-7c. However, PBX2 knock-down did not induce CD11b expression as effectively as forced expression of let-7c (compare Figure 4c with Figure 1a), suggesting that this miRNA could induce AML differentiation, in part, independently of PBX2. Indeed, analysis of additional let-7 targets characterized in other cell contexts and with a potential role in normal and malignant hematopoiesis,10,14,16–20,25 revealed that levels of RAS and Lin28 were clearly down-regulated in let-7c over-expressing NB4 cells (Supplementary Figure 6). By contrast, expression of MYC was slightly decreased, and levels of HMGA2 did not change at all (Supplementary Figure 6). These data, together with those recently published by other groups,10,29,30 suggest that Lin28 and RAS are let-7c target genes involved in its pro-differentiation function in myeloid cells. In particular, knockdown of Lin28A in bone marrow cells led to hematopoietic lineage-skewing, with an increase in myeloid cells and a decrease in B-cell numbers29 and let-7-mediated megakaryocytic cell differentiation was blocked by Lin28 overexpression.30 Regarding RAS, activating mutations of N-RAS are common in AML31 and K-Ras cooperates with PML-RARα in inducing an APL-like disease with high penetrance and short latency.31 Moreover, the down-modulation of RAS protein by ATRA in NB4 cells is, in part, mediated by let-7 family members.10
In summary, we have shown that let-7c promotes granulocytic differentiation in AML, and have identified and experimentally validated PBX2 as a novel let-7c target in myeloid cells.
Since PBX2 regulates normal and abnormal biological processes through complex formation with Meis1 and HoxA9,27,28 it would be of interest to assess whether subsets of HoxA9-regulated genes are also modulated upon perturbation of let-7c expression. Moreover, since Meis1-HoxA9 expression is required for MLL-dependent leukemogenesis,33 it would be interesting to test whether let-7c overexpression can also suppress proliferation of leukemic cells expressing MLL-chimeric proteins.
Supplementary Material
Acknowledgments
We thank Dr. Alessandro Fatica from “Sapienza” University in Rome for a kind gift of lentiviral vector pRRLSIN-cPPT-PGK-GFP-WPRE and Dr. Gianluca Bossi for kindly providing information on lentiviral utilization; Alessandra Boe for technical assistance for cell sorting and FACS analysis. We also acknowledge Dr. Silvia Bacchetti for helpful comments on the manuscript.
Supported by grants: Associazione Italiana Leucemie (AIL) and Ministry of Health, Ricerca oncologica-Project of integrated program to MGR.
B. Calabretta was supported, in part, by NCI grant CA95111
Footnotes
A. Pelosi and S. Careccia are PhD students University of Rome “Sapienza”. A. Pelosi is a recipient of a fellowship from AIRC. S. Careccia is a recipient of the fellowship AIL “Alberto Landi”.
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the ONCOGENE website
REFERENCES
- 1.Estey E, Döhner H. Acute myeloid leukemia. Lancet 2006; 368: 1894–1897. [DOI] [PubMed] [Google Scholar]
- 2.de Thè H, Chen Z. Acute promyelocytic leukemia: novel insights into the mechanisms of cure. Nat Rev 2010; 10: 775–783. [DOI] [PubMed] [Google Scholar]
- 3.Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J Pathol 2011; 223: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857–866. [DOI] [PubMed] [Google Scholar]
- 5.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303: 83–86. [DOI] [PubMed] [Google Scholar]
- 6.Careccia S, Mainardi S, Pelosi A, Gurtner A, Diverio D, Riccioni R et al. A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 2009; 28: 4034–4040. [DOI] [PubMed] [Google Scholar]
- 7.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-a and C/EBPα regulates human granulopoiesis. Cell 2005;123: 819–831. [DOI] [PubMed] [Google Scholar]
- 8.Felli N, Fontana L, Pelosi E, Botta L, Bonci D, Facchiano F et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A 2005; 102: 18081–18086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontana L, Pelosi E, Greco P, Recanicchi S, Testa U, Liuzzi F et al. MicroRNAs 17–5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 2007; 9: 775–787. [DOI] [PubMed] [Google Scholar]
- 10.Garzon R, Pichiorri F, Palumbo T, Visentini M, Ageilan R, Cimmino A et al. MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 2007; 26: 4148–4157. [DOI] [PubMed] [Google Scholar]
- 11.Havelange V, Garzon R. MicroRNAs: emerging key regulators of hematopoiesis. Am J Hematol 2010; 85: 935–942. [DOI] [PubMed] [Google Scholar]
- 12.Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB et al. MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl Acad Sci U S A 2007; 104: 19971–19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isken F, Steffen B, Merk S, Dugas M, Markus B, Tidow N et al. Identification of acute myeloid leukaemia associated microRNA expression patterns. Br J Haematol 2008; 140: 153–161. [DOI] [PubMed] [Google Scholar]
- 14.Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 2010; 17: F19–36. [DOI] [PubMed] [Google Scholar]
- 15.Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Lowenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 2008; 111: 5078–5085. [DOI] [PubMed] [Google Scholar]
- 16.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A et al. RAS is regulated by the let-7 microRNA family. Cell 2005; 120: 635–647. [DOI] [PubMed] [Google Scholar]
- 17.Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007; 315: 1576–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C et al. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008; 216: 440–450. [DOI] [PubMed] [Google Scholar]
- 19.Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 2007; 67: 7713–7722. [DOI] [PubMed] [Google Scholar]
- 20.Viswanathan SR, Daley GQ. Lin28: A microRNA regulator with a macro role. Cell 2010; 140: 445–449. [DOI] [PubMed] [Google Scholar]
- 21.Bhatia M, Kirkland JB, Meckling-Gill KA. Overexpression of poly(ADP-ribose) polymerase promotes cell cycle arrest and inhibits neutrophilic differentiation of NB4 acute promyelocytic leukemia cells. Cell Growth Differ 1996; 7: 91–100. [PubMed] [Google Scholar]
- 22.Brown G, Drayson MT, Durham J, Toellner KM, Hughes PJ, Choudhry MA et al. HL60 cells halted in G1 or S phase differentiate normally. Exp Cell Res 2002; 281: 28–38. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics 2008; 24:325–332. [DOI] [PubMed] [Google Scholar]
- 24.Wang X miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA 2008; 14: 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 2009. 41: 843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laurent A, Bihan R, Omilli F, Deschamps S, Pellerin I. PBX proteins: much more than Hox cofactors. Int J Dev Biol 2008; 52: 9–20. [DOI] [PubMed] [Google Scholar]
- 27.Schnabel CA, Jacobs Y, Cleary ML. HoxA9-mediated immortalization of myeloid progenitors requires functional interactions with TALE cofactors Pbx and Meis. Oncogene 2000; 19: 608–616. [DOI] [PubMed] [Google Scholar]
- 28.Shen WF, Rozenfeld S, Kwong A, Köm ves LG, Lawrence HJ, Largman C. HOXA9 forms triple complexes with PBX2 and MEIS1 in myeloid cells. Mol Cell Biol 1999; 19: 3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhuri AA, So AY, Mehta A, Minisandram A, Sinha N, Jonsson VD, et al. Onco-mir-miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc Natl Acad Sci U S A 2012; 109: 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Zhang J, Gao L, McClellan S, Finan MA, Butler TW, et al. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ 2012; 19: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowen DT, Frew ME, Hills M, Gale RE, Wheatley K, Groves MJ, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 2005; 106: 2113–2119. [DOI] [PubMed] [Google Scholar]
- 32.Chan IT, Kutok JL, Williams IR, Cohen S, Moore S, Shigematsu H, et al. Oncogenic K-ras cooperates with PML-RAR alpha to induce an acute promyelocytic luekemia-like disease. Blood 2006; 108: 1708–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong P, Iwasaki M, Somervaille TC, So CW, Cleary ML. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev 2007; 21: 2762–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rizzo MG, Giombini E, Diverio D, Vignetti M, Sacchi A, Testa U et al. Analysis of p73 expression pattern in acute myeloid leukemias: lack of DeltaN-p73 expression is a frequent feature of acute promyelocytic leukemia. Leukemia 2004; 18: 1804–1809. [DOI] [PubMed] [Google Scholar]
- 35.Mainardi S, Pelosi A, Palescandolo E, Riccioni R, Fontemaggi G, Diverio D et al. deltaN-p73 is a transcriptional target of the PML/RARalpha oncogene in myeloid differentiation. Cell Death Differ 2007; 14: 1968–1971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.