

Abstract
The city of Wuhan, Hubei province, China, was the origin of a severe pneumonia outbreak in December 2019, attributed to a novel coronavirus (severe acute respiratory syndrome coronavirus 2 [SARS‐CoV‐2]), causing a total of 2761 deaths and 81109 cases (25 February 2020). SARS‐CoV‐2 belongs to genus Betacoronavirus, subgenus Sarbecovirus. The polyprotein 1ab (pp1ab) remains unstudied thoroughly since it is similar to other sarbecoviruses. In this short communication, we performed phylogenetic‐structural sequence analysis of pp1ab protein of SARS‐CoV‐2. The analysis showed that the viral pp1ab has not changed in most isolates throughout the outbreak time, but interestingly a deletion of 8 aa in the virulence factor nonstructural protein 1 was found in a virus isolated from a Japanese patient that did not display critical symptoms. While comparing pp1ab protein with other betacoronaviruses, we found a 42 amino acid signature that is only present in SARS‐CoV‐2 (AS‐SCoV2). Members from clade 2 of sarbecoviruses have traces of this signature. The AS‐SCoV2 located in the acidic‐domain of papain‐like protein of SARS‐CoV‐2 and bat‐SL‐CoV‐RatG13 guided us to suggest that the novel 2019 coronavirus probably emerged by genetic drift from bat‐SL‐CoV‐RaTG13. The implication of this amino acid signature in papain‐like protein structure arrangement and function is something worth to be explored.
Keywords: coronavirus, pp1ab protein, SARS, SARS‐CoV‐2, virus, Wuhan
Highlights
The amino acid signature in pp1ab of the novel coronavirus SARS‐CoV‐2 helps to elucidate its evolutive origin.
SARS‐CoV‐2 probably rose directly from viruses infecting bats rather than pangolins.
The amino acid signature in SARS‐CoV‐2 could be a target for a specific diagnosis of COVID‐19.
1. INTRODUCTION
A recent outbreak of severe pneumonia was traced in the city Wuhan, Hubei province, China, causing 2761 deaths and at least 81109 cases (25 February 2020). The causative agent of the disease is a member of the Coronaviridae family, designed as severe acute respiratory syndrome coronavirus 2 isolate Wuhan‐Hu‐1 (SARS‐CoV‐2‐WH‐HU1: MN908947.3). 1 Reports indicated that SARS‐CoV‐2 is closely related to three Chinese bat SARS‐like coronaviruses (Bat‐SL‐CoVs) forming a monophyletic cluster, denominated clade 2, within subgenus Sarbecovirus. 1 , 2 , 3 , 4 , 5 , 6
The polyprotein 1ab (pp1ab) is the largest protein of coronaviruses that through proteolytic cleavage is divided into 16 mature nonstructural proteins (nsps). The nsps are involved in replication and transcription of the viral genome and are responsible for the cleavage of the polyprotein, thus making them attractive antiviral drug targets. 7
Due to the lack of remarkable differences between pp1ab of SARS‐CoV‐2 with those from other sarbecoviruses, 3 , 5 pp1ab of SARS‐CoV‐2 has not been thoroughly analyzed. Despite the high similarity between pp1ab proteins, it could be possible to identify distinguishable regions representing molecular signatures for the specific detection of virus strains or to track its evolutive history. In this short communication, we expound a comparative sequence analysis of pp1ab protein of SARS‐CoV‐2.
2. MATERIALS AND METHODS
The analysis was performed using the phylogenetic‐structural sequence analysis; sequence comparisons were made in a phylogenetic order. 8 Thus, pp1ab from SARS‐CoV‐2 isolates are compared first, then the polyprotein of SARS‐CoV‐2 is contrasted against those from clade 2 of sarbecoviruses. Finally, the protein is set against those from clade 1 and 3. Protein alignments were performed using the alignment tool MAFFT v7 (default parameters). Pairwise comparisons were performed with the Sequence Demarcation Tool‐V1.2 (SDT; default parameters). Simplot analyses were conducted with SimPlot v3.5.1. using a sliding window of 200 moving in steps of 30. Pp1ab of SARS‐CoV‐2 was the reference sequence (MN908947.3). The phylogenetic relationship of SARS‐CoV‐2 was carried out using algorithms included in MEGA v10.0.4. The alignment of 44 full genomes of members of the genus Betacoronaviruses was performed using the alignment tool MAFFT v7. The evolutionary relationships were inferred with the Neighbor‐Joining method. The phylogeny test was carried out by the bootstrap method (5000 replicates).
3. RESULTS AND DISCUSSION
According to the phylogenetic‐structural sequence analysis, first, we compared pp1ab proteins with 144 isolates of SARS‐CoV‐2 from patients around the world (Table S1). The analysis displayed that most pp1ab proteins have not changed; only six amino acid changes were detected (Table S2). We consider an amino acid change if two or more sequences have the same mutation. One of these mutations (L3606F), placed in the position 37 of nsp6 protein (L37F), is shared by ten sequences from viruses isolated in China, USA, France, Hong Kong, Italy, and Singapore (Table S2). Coronavirus nsp6 is a transmembrane protein that is associated with nsp3 and nsp4 proteins to form the organelle‐like replicative structures (double‐membrane vesicles). 9 Prediction of transmembrane helices (TMHs) segments in nsp6 protein showed that L37F does not alter the secondary structure of the adjacent transmembrane domains (Figure S1). In fact, the mutation L37F is predicted to be outside of the membrane as part of an unstructured coil segment (32SLFFFL/FYEN) that connects the first (12‐31 residues) and the second (41‐60 residues) TMHs (Figure S1). Strikingly, the position 37 of nsp6 protein is a Val residue that is conserved in all analyzed sarbecoviruses (Data S1), except in SARS‐CoV‐2 (Leu). So the mutation of the aliphatic Leu residue for the aromatic Phe residue in this conserved position probably has functional implications; although Leu and Phe are both hydrophobic residues, the Phe residue could also perform cation‐π interactions that could affect the protein‐protein interactions in the L37F mutant. The structural impact of this mutation can not be determined since experimental data in the Protein Data Bank are not available for homology modeling of nsp6 using a single or multiple templates (eg, SWISS‐MODEL server, Phyre2, etc.).
Interestingly, a virus isolated from a Japanese male (GISAID: EPI_ISL_407084), with no critical pneumonia (patient status described in GISAID database), has eight deleted amino acids at position 32 to 39 aa of pp1ab (nsp1) (Table S2). Since nsp1 is a virulence factor that inhibits host gene expression, 10 the implications of this deletion are worth to be addressed. However, since the deletion is present in just one isolated virus, this finding needs to be confirmed by other genome sequences.
For the second part of the analysis, we compared pp1ab of SARS‐CoV‐2 to those of the clade 2 of sarbecoviruses (bat‐SL‐CoV‐ZC45: MG772933; bat‐SL‐CoV‐ZXC21S: MG772934; bat‐SL‐CoV‐RaTG13: MN996532). The pairwise comparison performed with SDT indicated that the most related protein was the one belonging to bat‐SL‐CoV‐RaTG13 (98.6%). Also, proteins from bat‐SL‐CoV‐ZC45 and bat‐SL‐CoV‐ZXC21S showed a pairwise identity above 95%. To determine if the identity is preserved throughout its length, we compared the pp1ab sequences from these viruses via similarity plot analysis. The analysis showed that pp1ab of bat‐SL‐CoV‐ZC45 and bat‐SL‐CoV‐ZXC21S displayed three dissimilarity regions that surround residues 1000 (87.7%), 4670 (91.4%), and 6590 (91.6%) (Figure S2). The pairwise identity between SARS‐CoV‐2 and bat‐SL‐CoV‐RaTG13 is conserved throughout its length.
Scrutiny on the alignment allowed us to note that regions of residues 4631 and 6565 of bat‐SL‐CoV‐ZC45 and bat‐SL‐CoV‐ZXC21S were more similar to those from viruses of clade 3 rather than SARS‐CoV‐2 or bat‐SL‐CoV‐RaTG13. These findings explain why the ORF1b of bat‐SL‐CoV‐ZC45 and bat‐SL‐CoV‐ZXC21S are clustered with those of sarbecoviruses from Clade 3, 1 suggesting a recombination history between these viruses with clade 3 members.
When we analyzed the position 1000 of the pp1ab alignment, we identified rich glutamine (~22%) amino acid signature in pp1ab of SARS‐CoV‐2 (AS‐SCoV2). This signature is 42 aa in length (DSQQTVGQQDGSEDNQTTTIQTIVEVQPQLEMELTPVVQTIE), placed between the amino acids 983 and 1024 of pp1ab, which correspond to the N‐terminal of papain‐like protease (165 aa to 206 aa). Specifically, AS‐SCoV2 is located in the flexible acidic‐domain (AC domain) rich in glutamic acid (Figure 1). 11 AC domain is flanked by the ubiquitin‐like and the ADP‐ribose‐1″‐phosphatase domains. 10 The functional implication of the AC domain extension conferred by AS‐SCoV2 needs to be studied.
Figure 1.

Alignment of pp1ab proteins from sarbecoviruses. Pp1ab proteins of sarbecoviruses (green: clade 1; blue clade 2: blue; clade 3: red) were aligned using the multiple sequence alignment program MAFFT v7 and manually edited for maximizing coincidences. The figure shows the AC domain of pp1ab. Conserved residues are yellow highlighted. AS‐SCoV2 conserved residues are blue highlighted. N‐terminal region of the papain‐like protein is represented above the alignment. AS‐SCoV2, SARS‐CoV‐2; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
The AS‐SCoV2 is partially conserved in the pp1a protein of bat‐SL‐CoV‐ZC45 (identity: 40.5%; indels: 6) and virtually missing in bat‐SL‐CoV‐ZXC21S (identity: 16%; indels: 27), while AS‐SCoV2 is very conserved in pp1ab of bat‐SL‐CoV‐RaTG13 (identity: 76%; Indels: 0) (Figure 1). These results suggest that SARS‐CoV‐2 is closely related to bat‐SL‐CoV‐RaTG13 rather than bat‐SL‐CoV‐ZC45 or bat‐SL‐CoV‐ZXC21S. The alignment of the pp1ab from members of the subgenus Sarbecovirus showed that the AS‐SCoV2 is exclusive of the novel coronavirus and slightly preserved in bat‐SL‐CoV from clade 2 (Figure 1). We found the same results when we compared the pp1ab of members of the genus Betacoronavirus (Table S3; Data S1). A search in the nucleotide and protein GenBank databases displayed that AS‐SCoV2 is codified only in genomes of SARS‐CoV‐2. Also, no proteins other than pp1ab of SARS‐CoV‐2 and bat‐SL‐CoV‐RaTG13, partially, have the signature. Because we determined that the presence of AS‐SCoV2 in human coronaviruses is restricted only to SARS‐CoV‐2, we suggest that their respective 126 nucleotide sequence can be used to design alternative specific PCR diagnosis for SARS‐CoV‐2.
The genome sequences of two sarbecoviruses isolated from the pangolin (Manis javanica) were recently released in GISAID database, one of them was collected in Guangzhou, China in 2019 (Pangolin‐BetaCoV‐Guandong‐2019; EPI_ISL_410721) and the other one was collected in Guangxi, China in 2017 (Pangolin‐BetaCoV‐Guangxi‐2017; EPI_ISL_410538). Phylogenetic analysis based on genome sequence showed that both viruses collected from pangolins are clustered in the clade 2 (Figure 2). The pairwise comparison of pp1ab protein showed that the protein of Pangolin‐BetaCoV‐Guandong‐2019 is 94% identical to those of SARS‐CoV‐2 while the pp1ab of Pangolin‐BetaCoV‐Guangxi‐2017 has an identity of 92%. Since both pangolin viruses are grouped in clade 2 and their pp1ab proteins are identical to those of SARS‐CoV‐2, we analyzed the alignment of its pp1ab protein to determine if the AS‐SCoV2 is present (Figure 1). The homologous region of AS‐SCoV2 pp1ab of Pangolin‐BetaCoV‐Guandong‐2019 has 34 aa that showed an identity of 35%, whereas the homologous region of AS‐SCoV2 in Pangolin‐BetaCoV‐Guangxi‐2017 have 36 aa which are completely different from those of sarbecoviruses (Figure 1). These findings suggest that the pp1ab of SARS‐CoV‐2 is more closely related to pp1ab of Bat‐SL‐CoV‐RaTG13 than to pp1ab of coronaviruses isolated from Chinese pangolins. Since the region that encodes the pp1ab protein represents about 71% of SARS‐CoV‐2 genome, we suggest that it is less likely that the novel human coronavirus has been arising directly from the viruses isolated from pangolins.
Figure 2.
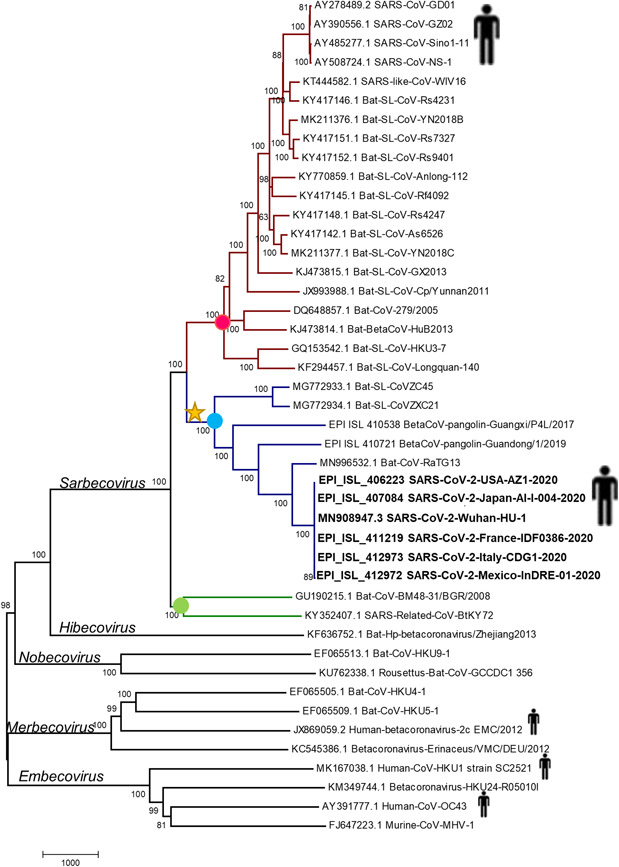
Phylogenetic analysis of SARS‐CoV‐2. The Phylogenetic relationship was inferred with the Neighbor‐Joining method based on the genome alignment. Bootstrap values (5000 iterations) are indicated for each node. Dots represent the clade 1 (green), clade 2 (blue), and clade 3 (red). Star indicates the probable acquisition of ancestral AS‐SCoV2. The evolutionary distances were computed using the number of differences method. The scale bar, placed below the tree, indicates the number of base differences per sequence. AS‐SCoV2, SARS‐CoV‐2; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
First reports focused on the genetic characterization of SARS‐CoV‐2 suggested that this virus has a recombinant origin. 2 Our results indicate that most probably, a recombination event did not happen in the first half of the viral genome (ORF1ab). Under this idea, an alternative explanation for SARS‐CoV‐2 origin is that bat‐SL‐CoV‐RaTG13, collected 6 years ago, is the progenitor of SARS‐CoV‐2, which has evolved since the collection date by genetic drift before infecting humans. Three observations support the hypothesis: 1. The high pairwise identity of pp1ab from SARS‐CoV‐2 and bat‐SL‐CoV‐RaTG13 is preserved throughout its length (Figure S2). 2. The exclusive AS‐SCoV2 of the novel coronavirus is conserved in bat‐SL‐CoV‐RaTG13 (Figure 1). 3. The high pairwise identity (96.3%) shared by SARS‐CoV‐2 and bat‐SL‐CoV‐RaTG13 is preserved in its whole genome, only a slight dissimilarity region is displayed in the ORF of spike protein. 6
Although the origin of SARS‐CoV‐2 does not appear to be caused by recent genetic recombination, at least acquisition of genetic material must have given place to AS‐SCoV2. Since members of subgenus Sarbecovirus from clade 1 and clade 3 have at the minimum 25 missing aa in its homologous region of AS‐SCoV2 and members of clade 2 have sequence traces of AS‐SCoV2, except by BetaCoV‐pangolin‐Guangxi‐2017 (Figures 1), we suggest the viral ancestor of clade 2 members probably gained the genetic material (Figure 2). In this scenario, the ancestral AS‐SCoV2 have been changed over the time by genetic drift evolving to the actual AS‐SCoV2. This probably explains why we could not infer the origin of these signature based on the available sequences from databases, although there is a possibility that the organism that donated this segment has not been discovered yet. Regarding the pp1ab of Pangolin‐BetaCoV‐Guangxi‐2017, we suggest that a recombination event in the genomic region of the that encodes the first 3500 amino acids of pp1ab probably happened since the AS‐SCoV2 is no present in this virus and the Simplot analysis showed several dissimilarity peaks in this region when the pp1ab of Pangolin‐BetaCoV‐Guangxi‐2017 is compared with either of clade 2 members of sarbecoviruses (Figure S3).
The AS‐SCoV2 located in the acidic‐domain of papain‐like protein from SARS‐CoV‐2 and bat‐SL‐CoV‐RaTG13 guided us to suggest that the novel human‐pathogenic coronavirus probably emerged by genetic drift from bat‐SL‐CoV‐RaTG13. The implication of AS‐SCoV2 in papain‐like protein structure arrangement and function is something worth to be explored.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENT
This research was funded by CONACYT of Mexico (grant number: APN‐2015‐01‐741) to Y.C.C. D.A.G.R was supported by a fellowship from CONACYT.
Cárdenas‐Conejo Y, Liñan‐Rico A, García‐Rodríguez DA, Centeno‐Leija S, Serrano‐Posada H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human‐pathogenic coronavirus (SARS‐CoV‐2). J Med Virol. 2020;92:688–692. 10.1002/jmv.25758
REFERENCES
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ji W, Wang W, Zhao X, Zai J, Li X. Homologous recombination within the spike glycoprotein of the newly identified coronavirus may boost cross‐species transmission from snake to human. J Med Virol. 2020;92:433‐440. 10.1002/jmv.25682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chan JF‐W, Kok K‐H, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221‐236. 10.1080/22221751.2020.1719902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou P, Yang X‐L, Wang X‐G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Báez‐Santos YM St., John SE, Mesecar AD. The SARS‐coronavirus papain‐like protease: structure, function, and inhibition by designed antiviral compounds. Antiviral Res. Elsevier B.V. 115, 2020:21‐38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Argüello‐Astorga G, Herrera‐Estrella L. Evolution of light‐regulated plant promoters. Annu Rev Plant Physiol Plant Mol Biol. 1998;9:525‐555. 10.1146/annurev.arplant.49.1.525 [DOI] [PubMed] [Google Scholar]
- 9. Hagemeijer MC, Rottier PJM, Haan CAM. Biogenesis and dynamics of the coronavirus replicative structures. Viruses. 2012;4:3245‐3269. 10.3390/v4113245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Züst R, Cervantes‐Barragán L, Kuri T, et al. Coronavirus non‐structural protein 1 Is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007;3:e109. 10.1371/journal.ppat.0030109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Serrano P, Johnson MA, Almeida MS, et al. Nuclear magnetic resonance structure of the N‐terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J Virol. 2007;81:12049‐12060. 10.1128/JVI.00969-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information