

Abstract
Severe acute respiratory syndrome (SARS)‐like coronavirus sequences were identified in two separate complementary DNA (cDNA) pools. The first pool was from a Carassius auratus (crusian carp) cell line and the second was from Ctenopharyngodon idella (grass carp) head kidney tissue. BLAST analysis suggests that these sequences belong to SARS‐like coronaviruses, and that they are not evolutionarily conserved in other species. Investigation of the submitting laboratories revealed that two laboratories from the Institute of Hydrobiology at the Chinese Academy of Sciences in Wuhan, China performed the research and submitted the cDNA libraries to GenBank. This institution is very close in proximity to the Wuhan South China Seafood Wholesale Market where SARS‐CoV‐2 first amplified in the human population. It is possible that these sequences are an artifact of the bioinformatics pipeline that was used. It is also possible that SARS‐like coronaviruses are a common environmental pathogen in the region that may be in aquatic habitats.
Keywords: epidemiology, SARS coronavirus, virus classification, water‐borne epidemics
Highlights
SARS‐like coronavirus sequences were found in two carp cDNA pools from separate laboratories in Wuhan, China.
1. INTRODUCTION
Severe acute respiratory syndrome (SARS)‐CoV‐2 emerged in the Chinese city of Wuhan in December 2019, and causes a respiratory illness called COVID‐19, which can spread from person to person. As of 2 March 2020, there have been 89 868 cases and 3069 deaths, and the virus has spread to six continents. There are no specific antivirals or vaccines for this disease.
SARS‐CoV‐2 is a member of the Coronaviridae family and includes a number of viruses that cause the common cold (eg, 229E, OC43, NL63, and HKU1). 1 SARS‐CoV‐2 is also related to SARS‐CoV and Middle East respiratory syndrome coronavirus (MERS‐CoV), which cause severe disease that is associated with high mortality rates. 2 , 3 Coronaviruses originate in bats but are zoonotic and can circulate in a number of mammals and birds. 1 SARS‐CoV emerged into the human population because of the exotic food trade in China, which allowed for a transmission event between a civet cat and a human. 4 MERS‐CoV has emerged into the human population because of the use of camels as livestock, which has allowed for multiple transmission events between camels and humans. 5
The intermediate animal host of SARS‐CoV‐2 is still unknown. 2 , 3 In this study, I mined the NCBI expressed sequence tag (est) database to discover sequences related to SARS‐CoV‐2. SARS‐like virus sequences that were highly homologous to SARS‐CoV‐2 were identified in two separate complementary DNA (cDNA) pools. The first pool was from a Carassius auratus (crusian carp) cell line and the second was from Ctenopharyngodon idella (grass carp) head kidney tissue. 6 , 7 The sequence from C. auratus cDNA was 152 amino acids long and the sequence from C. idealla cDNA was 88 amino acids long. BLAST analysis suggests that these sequences belong to SARS‐like coronaviruses, and that they are not evolutionarily conserved in other species.
Investigation of the submitting laboratories revealed that two laboratories from the Institute of Hydrobiology at the Chinese Academy of Sciences in Wuhan, China performed the research. This institution is less than 10 miles from the Wuhan South China Seafood Wholesale Market where SARS‐CoV‐2 first amplified in the human population. It is important to highlight the limitation of this study, which relies solely on a bioinformatics pipeline, which may be flawed. However, these data raise the possibility that SARS‐like coronaviruses are a common environmental pathogen in the region. Further research is required to test cell lines and the aquatic environment in and around the epicenter of the epidemic for SARS‐like coronaviruses.
2. MATERIALS AND METHODS
2.1. RNA extraction, library construction, and sequencing
RNA extraction, library construction, and sequencing was performed previously by Zhong et al 6 and Adams et al 7 and uploaded to GenBank on the expressed sequence tag (est) database.
2.2. Genetic, bioinformatics, and phylogenetic analyses
Translated nucleotide BLAST (tblastn) database searches were performed by searching for SARS‐CoV‐2 protein sequences (Figure S1) in the NCBI expressed sequence tag (est) database. Two cDNA clones were identified and accession numbers are GE213092 and JK851329.1. Standard Protein Blast was performed using the identified protein sequences against the NCBI nonredundant database and Clustal Omega was used to align protein and nucleic acid sequences. Phylogeny.fr “One Click” Mode was used to align, curate, generate the phylogeny, and for tree rendering.
3. RESULTS
3.1. Bioinformatic detection of coronavirus sequences
To identify novel coronavirus sequences, translated nucleotide BLAST (tblastn) database searches were performed by searching for SARS‐CoV‐2 sequences in the NCBI expressed sequence tag (est) database. SARS‐CoV‐2 protein sequences (amino acids 266‐13 468 and 13 468‐21 555) used to search the database (Figure S1). Tblastn analysis using this sequence identified two cDNA clones that were highly homologous to SARS‐like coronaviruses. These clones were from two separate cDNA pools. The first cDNA pool was made from Carassius auratus (crucian carp) blastulae embryonic cell line and contained a sequence of 152 amino acids that covered 2% of the SARS‐CoV‐2 genome and was 93.42% identical. Standard Protein BLAST analysis found that this sequence represents a portion of a coronavirus RNA‐dependent RNA polymerase (RdRp) and was homologous to SARS‐like coronaviruses (Table 1). The second cDNA pool was made from Ctenopharyngodon idella (grass carp) head kidney and contained a sequence of 88 amino acids that covered 1% of the SARS‐CoV‐2 genome and was 93.18% identical. Standard Protein BLAST found that this sequence represents a portion of a coronavirus helicase protein and was also homologous to SARS‐like coronaviruses (Table 2). Protein and nucleic acid alignments of each cDNA clone were performed to compare with the most related coronavirus sequences (Figures 1 and 2). Phylogenetic analysis showed that the C. auratus SARS‐like coronavirus sequence clusters with other SARS‐like coronaviruses (Figure 3).
Table 1.
Standard protein BLAST results using the Carassius auratus coronavirus sequence against NCBI's nonredundant database
| Description | Query coverage | Percent identity |
|---|---|---|
| RNA‐dependent RNA polymerase [Bat coronavirus (BtCoV/A1018/2005)] | 98% | 97.32% |
| RNA‐dependent RNA polymerase [Bat SARS‐like coronavirus] | 96% | 100.0% |
| RNA‐dependent RNA polymerase [Bat SARS‐like coronavirus] | 96% | 100.0% |
| RNA‐dependent RNA polymerase [Bat coronavirus] | 98% | 97.32% |
| RNA‐dependent RNA polymerase [Bat SARS‐like coronavirus] | 96% | 99.32% |
Abbreviation: SARS, severe acute respiratory syndrome.
Table 2.
Standard protein BLAST results using the Ctenopharyngodon idella sequence against NCBI's nonredundant database
| Description | Query coverage | Percent identity |
|---|---|---|
| Replicase 1B [SARS coronavirus TW‐HP1] | 100% | 93.18% |
| Helicase [severe acute respiratory syndrome coronavirus 2] | 100% | 94.32% |
| nsp13‐pp1ab [severe acute respiratory syndrome‐related coronavirus] | 100% | 93.18% |
| ORF1 [Bat CoV 279/2005] | 100% | 93.18% |
| Nonstructural polyprotein 1ab [Bat SARS‐like coronavirus] | 100% | 94.32% |
Abbreviation: SARS, severe acute respiratory syndrome.
Figure 1.

Clustal Omega protein alignment of (A) Carassius auratus SARS‐like coronavirus sequence and BtCoV/A1018/2005 and (B) Ctenopharyngodon idella SARS‐like coronavirus sequence and SARS coronavirus TW‐HP1. SARS, severe acute respiratory syndrome
Figure 2.

Clustal Omega nucleotide alignment of (A) C. auratus SARS‐like coronavirus sequence and Bat SARS‐CoV Rs672/2006 and (B) C. idella SARS‐like coronavirus sequence and SARS coronavirus BJ202. SARS, severe acute respiratory syndrome
Figure 3.
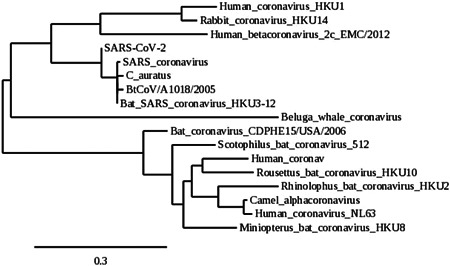
Phylogenetic position of C. auratus SARS‐like coronavirus sequence. The evolutionary history of the C. auratus SARS‐like coronavirus sequence and a number of coronaviruses from different clades was inferred by generating homologous amino acid sequences using MUSCLE multiple sequence alignment and Gblocks 0.91b software, followed by PhyML and TreeDyn software using the Phylogeny.fr “One Click” Mode. SARS, severe acute respiratory syndrome
3.2. Locating the submitting laboratories
We then identified the submitting laboratories through the NCBI database and found that both cDNA libraries were submitted by laboratories at the Institute of Hydrobiology, Chinese Academy of Sciences, in Wuhan, China. This institution is situated on a waterway and is less than 10 miles from the Wuhan South China Seafood Wholesale Market where the current SARS‐CoV‐2 epidemic first amplified in the human population (Figure 4). The cDNA libraries were submitted by the two laboratories at the Institute of Hydrobiology on 08 March 2011 and 04 February 2014, respectively, although related manuscripts were published many years prior.
Figure 4.
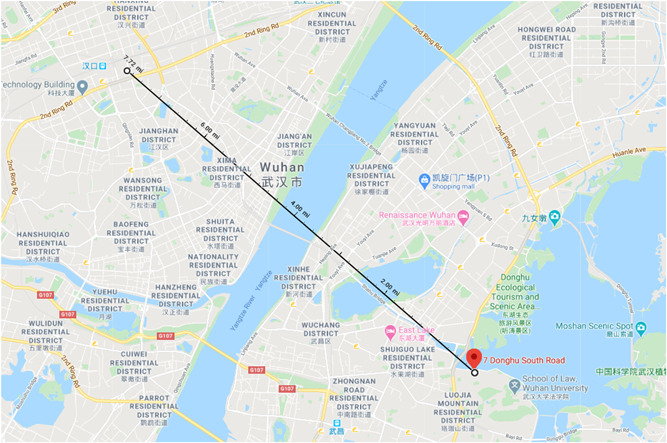
Google Map of both the Institute of Hydrobiology at the Chinese Academy of Sciences and the Wuhan South China Seafood Wholesale Market. The Institute of Hydrobiology is pinned and a distance measurement to the Wuhan South China Seafood Wholesale Market is shown
4. DISCUSSION
These data reveal two SARS‐like coronavirus sequences in separate cDNA pools of carp cells and tissue that originated from two separate laboratories at the Institute of Hydrobiology, Chinese Academy of Sciences, in Wuhan, China. Both cDNA samples were submitted many years ago. It is unclear how these sequences relate to the ongoing SARS‐CoV‐2 epidemic, and it is possible that these sequences are artifacts and do not represent coronavirus contamination of the cDNA samples.
cDNA samples can often harbor contaminating nucleic acid from the surrounding environment and can be a source to search for environmental DNA. In the case of the C. auratus, cDNA was produced from a cell line that was established in 1985. It is possible that this cell line contains an adventitious agent that is related to SARS‐CoV‐2. C. idella cDNA was produced from head kidney tissue from a live fish. This suggests that some carp species may be permissive to coronavirus infection. It is unclear how the identified sequences relate to the ongoing SARS‐CoV‐2 epidemic, although the identification of these sequences in the cells and tissues of animals in the proximity of the outbreak epicenter suggests that environmental surveillance should focus on adventitious agents in cell culture and the nearby aquatic environment.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supplemental Figure 1. SARS‐CoV‐2 protein sequence that was used to search the NCBI expressed sequence tag (est) database using tblastn
Conway MJ. Identification of coronavirus sequences in carp cDNA from Wuhan, China. J Med Virol. 2020;92:1629–1633. 10.1002/jmv.25751
REFERENCES
- 1. Corman VM, Muth D, Niemeyer D, Drosten C. Hosts and sources of endemic human coronaviruses. Adv Virus Res. 2018;100:163‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2 [published online ahead of print February 27, 2020]. J Med Virol. 2020:1‐10. 10.1002/jmv.25731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu Z, Xiao X, Wei X, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐CoV‐2 [published online ahead of print February 26, 2020]. J Med Virol. 2020:1‐7. 10.1002/jmv.25726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai MM. SARS virus: the beginning of the unraveling of a new coronavirus. J Biomed Sci. 2003;10:664‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coleman CM, Frieman MB. Emergence of the Middle East respiratory syndrome coronavirus. PLoS Pathog. 2013;9:e1003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhong XP, Wang D, Zhang YB, Gui JF. Identification and characterization of hypoxia‐induced genes in Carassius auratus blastulae embryonic cells using suppression subtractive hybridization. Comp Biochem Physiol B Biochem Mol Biol. 2009;152:161‐70. [DOI] [PubMed] [Google Scholar]
- 7. Adams MD, Kelley JM, Gocayne JD, et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991;252:1651‐6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. SARS‐CoV‐2 protein sequence that was used to search the NCBI expressed sequence tag (est) database using tblastn