

Abstract
The recently emerged 2019 Novel Coronavirus (SARS‐CoV‐2) and associated COVID‐19 disease cause serious or even fatal respiratory tract infection and yet no approved therapeutics or effective treatment is currently available to effectively combat the outbreak. This urgent situation is pressing the world to respond with the development of novel vaccine or a small molecule therapeutics for SARS‐CoV‐2. Along these efforts, the structure of SARS‐CoV‐2 main protease (Mpro) has been rapidly resolved and made publicly available to facilitate global efforts to develop novel drug candidates. Recently, our group has developed a novel deep learning platform – Deep Docking (DD) which provides fast prediction of docking scores of Glide (or any other docking program) and, hence, enables structure‐based virtual screening of billions of purchasable molecules in a short time. In the current study we applied DD to all 1.3 billion compounds from ZINC15 library to identify top 1,000 potential ligands for SARS‐CoV‐2 Mpro protein. The compounds are made publicly available for further characterization and development by scientific community.
Keywords: SARS-CoV-2, COVID-19, deep learning, virtual screening, protease inhibitors
1. Introduction
Coronaviruses (CoVs) are enveloped viruses containing a single positive‐stranded RNA, and causing a wide array of respiratory, gastrointestinal, and neurological diseases in human hosts.1, 2 It has been established that strains of CoVs were at the source of the 2002 severe acute respiratory syndrome (SARS) and 2012 middle east respiratory syndrome (MERS) epidemics.3 In late December 2019, a novel CoV of SARS‐CoV‐2 was identified to be the cause of atypical pneumonia outbreak in Wuhan, China, named COVID‐19.4 The rapidly increasing number of infected patients worldwide prompted the World Health Organization to declare a state of global health emergency to coordinate scientific and medical efforts to rapidly develop a cure for patients.5 While drug repurposing may be a short‐term and non‐specific solution to treat COVID‐19 patients,6 development of more targeted inhibitors is highly desirable.
Previous research efforts to develop anti‐viral agents against members of Coronaviridae family demonstrated that the Angiotensin‐converting enzyme II (ACE2) entry receptor, the RNA‐dependent RNA polymerase (RdRp) and the main protease (Mpro) proteins may represent suitable drug targets.7 Although initially promising, inhibitors targeting ACE2 (hence aiming to block critical coronavirus‐host interactions) did not advance clinically due to significant side effects.8 Likewise, RdRp inhibitors appeared to be not very specific and demonstrated overall lower potency, that also translated into common side effects in patients.1, 9 Nevertheless, rapid drug repurposing efforts have identified Remdesivir, a RdRp inhibitor, as a promising antiviral drug against COVID‐19.10, 11 Clinical trials are currently ongoing to determine the full efficacy spectrum of the compound in patients (clinicaltrials.gov, NCT0428070512). Concurrently, CoV infected patients administered with protease inhibitors, lopinavir/ritonavir, have shown improved outcome,1, 13 demonstrating the potential of the main protease (Mpro) as the most promising drug target in CoVs14, 15 Hence, a recently published X‐ray crystal structure of the SARS‐CoV‐2 Mpro provides an excellent ground for structure‐based drug discovery efforts.16
Earlier efforts to target SARS‐CoV resulted in identification of several covalent Mpro inhibitors targeting the catalytic dyad of the protein defined by His41 and Cys14517 residues. However, covalent inhibitors are often marked by adverse drug responses, off‐target side effects, toxicity and lower potency.18, 19, 20, 21, 22 Therefore, noncovalent protease inhibitors may have advantages for the treatment of this kind of infections. Still, the majority of approved drugs administered as anti‐SARS were designed for other viral strains (Table S1 in supplementary material). Notably, no CoV‐protease specific inhibitor has yet successfully completed a clinical development program to date.19, 23
The impact of current COVID‐19 outbreak and the likelihood of future CoV epidemics strongly advocate for rapid development of new treatments and fast intervention protocols. Few research groups have already suggested potential repurposing strategies for clinically approved drugs24, 25, 26 or proposed de novo agents27 as therapeutic solutions for SARS‐CoV‐2. However, previously reported docking (virtual screening) campaigns with Mpro targets were able to process only few millions or even thousands compounds.6, 28, 29, 30 The main reason for that is that conventional docking is too computationally expensive and slow, while the libraries of available chemicals are growing exponentially.31 To address this general challenge, we have recently developed a novel deep learning‐based approach for accelerated screening of large chemical libraries, consisting of billions of entities. This Deep Docking (DD) platform utilizes quantitative structure‐activity relationship (QSAR) models trained on docking scores of database subsets to approximate in an iterative manner the docking outcome of the remaining entries. Importantly, DD does not provide any novel scoring function for docking, thus its accuracy relies completely on the docking program that is used. The development of deep learning scoring functions has been already attempted, but results have shown various degrees of success which could be due to a lack of appropriate datasets.32, 33 Likely, as the very nature of docking is approximate, the improvements are likely to come from better approximation of physical‐chemical processes, including solvation, enthalpic and entropic factors, rather than from a better training base and procedures.34, 35 Thus, our method represents not just feasible, but also practical options for utilizing deep learning in virtual screening. Herein we have used DD for large‐scale virtual screening against the SARS‐CoV‐2 Mpro active site.
2. Materials and Methods
To assess the performance of fast Glide SP protocol36 to virtually screen against the Mpro target, we collected 81 known SARS Mpro small molecule inhibitors that are reported by Pillaiyar et al.,37 and Turlington et al..38 Then, we generated 50 molecular decoys for each active molecules using the methodology implemented in the Database of Useful Decoys: Enhanced (DUD−E).39 All compounds were prepared for docking with the OpenEye package. Most probable tautomer and ionization states at pH 7.4 were calculated with OpenEye QUACPAC package40 and starting 3D conformations were generated using Omega pose routine.41 The structure of SARS Mpro bound to a noncovalent inhibitor (PDB 4MDS, 1.6 Å resolution) was obtained from the Protein Data Bank (PDB),42 and prepared using Protein Preparation Wizard.43 Docking was performed using Glide SP module.36 Receiver operating curve areas under the curve (ROC AUC) were then calculated.
We used DD to virtually screen all ZINC15 (1.36 billion compounds)44 against the SARS‐CoV‐2 Mpro. The model was initialized by randomly sampling 3 million molecules and dividing them evenly into training, validation and test set. The structure PDB 6LU7 (resolution 2.16 Å)45 of the SARS‐CoV‐2 Mpro bound to the N3 covalent inhibitor was obtained from the PDB, and prepared as before. Molecule preparation and docking were performed similarly as before, and computed scores were used for DNN initialization. We then ran 4 iterations, adding each time 1 million of docked molecules sampled from previous predictions to the training set and setting the recall of top scoring compounds to 0.75. At the end of the 4th iteration, the top 3 million molecules predicted to have favorable scores were then docked to the protease site. The set of protease inhibitors (7,800 compounds) from the BindingDB repository was also docked to the same site.46 Our computational setup consisted of 13 Intel(R) Xeon(R) Gold 6130 CPUs @ 2.10GHz (a total of 390 cores) for docking, and 40 Nvidia Tesla V100 GPUs with 32GB memory for deep learning.
3. Results and Discussion
Although drug repurposing and high‐throughput screening have identified potential hit compounds with strong antiviral activity against COVID‐19,47 no noncovalent inhibitors for SARS‐CoV‐2 Mpro have been reported to date. Glide protocols were recently deployed to identify potential hit compounds as protease inhibitors, notably against FP‐2 and FP‐3 (P. falciparum cysteine protease),48 nsP2 (Chikunguya virus protease),49 and more recently against SARS‐CoV‐2 MPro.47 Therefore, Glide was shown to be adequate and effective in docking ligands with high fidelity compared to other available academic and commercial docking software.50, 51 Nonetheless, we performed our own benchmarking study to evaluate the viability of using Glide SP to screen the SARS‐CoV‐2 Mpro.
We first evaluated the feasibility of virtual screening using a closely related protein, the SARS Mpro (96 % of sequence identity,) for which different series of noncovalent inhibitors with low micromolar to nanomolar acitivity have been discovered.37
Our benchmarking study revealed good ability of Glide SP to dock known inhibitors. First, the co‐crystallized ligand (SID 24808289 from Turlington et al.38) was accurately redocked to its binding site (root mean square deviation (r.m.s.d.) of 0.86 Å between Glide and x‐ray pose, Figure 1a). Second, ROC AUC value for Glide SP used to dock 81 Mpro inhibitors and ∼4,000 decoys was 0.72, similarly to the more computationally expensive Glide XP protocol (Figure 1b), and 0.74 when active molecules were diluted in 1 million random compounds extracted from ZINC15 (Figure S1 in supplementary material). Thus, in light of recent studies advocating for extending virtual screening to large chemical libraries when docking works well at smaller scales,31 we decided to use Glide SP as DD docking program to screen ZINC15 against SARS‐CoV‐2 Mpro.
Figure 1.
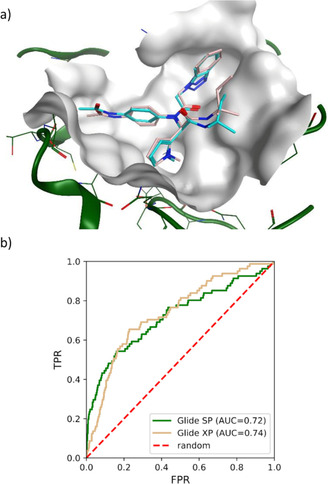
Evaluation of Glide SP docking protocol on SARS Mpro inhibitors. a) Redocking of ligand 7 to the SARS Mpro active site (PDB 4MDS) resulted in 0.86 Å of r.m.s.d (root mean square deviation) between computational (pink) and x‐ray (cyan) poses. b) ROC curves and AUC obtained by docking 81 inhibitors and ∼4,000 decoys to the Mpro active site with Glide SP and XP protocols.
DD relies on a deep neural network trained with docking scores of small random samples of molecules extracted from a large database to predict the scores of remaining molecules and, therefore, discard low scoring molecules without investing time and resources to dock them. The combination of an iterative process to improve model training and the use of simple 2D QSAR descriptors such as Morgan fingerprints makes DD particularly suited for fast virtual screening of emerging giga‐sized chemical libraries using standard computational resources. We have recently showed the wide range of applicability of DD by using the method to dock all ZINC15 compounds to 12 targets representing major protein families of therapeutic interest.52
The use of DD platform enabled us to dock 1.3 billion compounds from ZINC15 database44 into SARS‐CoV‐2 Mpro active site using standard Glide SP protocols in a week. In our benchmark study on SARS Mpro, AUC ROC for Glide SP improved from 0.72 to 0.78, when a ligand efficiency (LE) cutoff of −0.20 kcal/mol was introduced prior to ranking molecules by their docking scores (Figure S2 in supplementary material). Thus, the top 1,000 hits selected from the DD run were picked following the same strategy.
The SARS Mpro cleaves the replicase polyproteins, pp1a and pp1b, at 11 specific positions, using core sequences in the polyprotein substrate to determine cleavage sites.53 The positions of the residues on the polyproteins are named depending on their relative position to the cleavage site. Position P1 corresponds to the residue just before the cleavage site, followed by P2, P3, P4, P5, and up until the N‐terminal of the cleavage site. Position P1′ corresponds to the residue immediately following the cleavage site, followed by P2′, P3′, P4′, P5′ and up until the C‐terminal of the cleavage site.54 The protease recognizes specific residues at each position of the polyproteins to determine a cleavage site and initiate the replication‐transcription complex necessary for viral replication.55
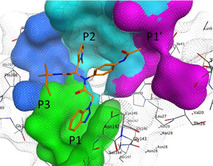
Based on the consensus recognition sequence of the polyproteins, a substrate‐analogue inhibitor, CMK, was designed to mimic positions P1 to P6 of the substrate in the SARS Mpro substrate‐binding sites. The compound is characterised by its chloromethyl ketone warhead and its core sequence of Val(P6)‐Asn(P5)‐Ser(P4)‐Thr(P3)‐Leu(P2)‐Gln(P1) to occupy the same volume as the residues of the recognition sequence.56 An X‐ray crystallography structure of the SARS Mpro with the CMK inhibitor revealed the mode of inhibitor binding to the substrate‐binding sites of the main protease, providing a crucial structural basis for rational drug design and guiding drug discovery efforts against the SARS Mpro (PDB 1UK457). Therefore, the pocket of the Mpro can be partitioned into different sections, depending on the volume occupied by polyprotein residues at each positions.
Using insight gained from the crystal structure, Turlington et al. first developed a moderate noncovalent inhibitor, SID 24808289, with an IC50 of 6.2 μM,38 and demonstrated that additional positions could be explored for efficient inhibition of SARS Mpro. The binding pose of the compound is shown in Figure 2, and the compound occupies the same volume as positions P1 to P4, and P1′. The compound still maintained key interactions with catalytic dyad of Gln189 and Met49 through hydrophobic contacts (P2), and through hydrogen bonds to Cys145, His163, and Glu166 (P1).
Figure 2.

P1, P2, P3 and P1′ groups in the substrate‐binding subsites of SARS MPro (PDB 4MDS). The compound, SID 24808289, is represented as orange sticks. The surface of the protease binding site is partitioned into different sections depending on the pocket occupancy of the compound.
The number 1 series of compounds identified from our virtual screening is presented in Table 1. They are predicted to have consistent binding pose, similar to the noncovalent compound SID 24808289, as shown in Figure 3a. The predicted interaction between ZINC000541677852 and SARS‐CoV‐2 Mpro is shown in Figure 3b. This series of compounds occupied the same volume as the P1, P2 and P3 groups with the common favored hydrophobic interactions of the phenyl ring (P2), and two hydrogen bonds to Cys145 and Leu141 respectively (P1).
Table 1.
Top hit series identified from our DD.
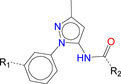
|
Compound |
R1 |
R2 |
Glide score (kcal/mol) |
|---|---|---|---|
|
ZINC000541677852 |
CF3 −−−− |
|
‐11.32 |
|
ZINC000636416501 |
Cl−−−− |
|
‐10.85 |
|
ZINC000543523838 |
Br−−−− |
|
‐10.75 |
|
ZINC000544491494 |
Br−−−− |
|
‐10.65 |
|
ZINC000544491491 |
Br−−−− |
|
‐10.50 |
|
ZINC000541676760 |
CF3 −−−− |
|
‐10.48 |
|
ZINC000543523837 |
Br−−−− |
|
‐10.43 |
|
ZINC000152979101 |
Br−−−− |
|
‐10.33 |
|
ZINC000152975931 |
CF3 −−−− |
|
‐10.03 |
|
ZINC001627499877 |
Br−−−− |
|
‐9.32 |
|
ZINC001362111980 |
Cl−−−− |
|
‐9.13 |

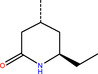
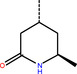
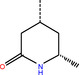
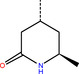
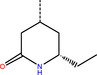
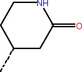
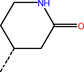
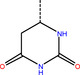
Figure 3.
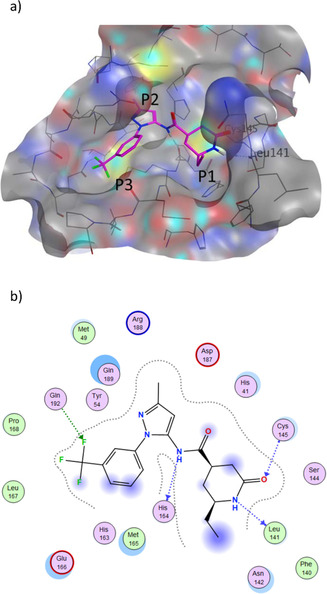
a) Predicted binding pose of our top hit compound (shown in magenta sticks) in the SARS‐CoV‐2 pocket with the annotated pocket occupancy b) Representation of the interactions of ZINC000541677852 in the SARS‐CoV‐2 pocket. Blue dotted lines represent hydrogen bonds with protein backbone atoms, and green dotted lines represent hydrogen bonds with sidechain atoms.
We have also analyzed the origin of top 1,000 ZINC hits (selected by LE), and observed that 99 % of them are not present in the ZINC15 in‐stock library (∼11 millions of molecules), commonly used in routine docking campaigns, demonstrating that the DD methodology can access complete and diverse chemical space beyond classical docking. The Glide SP scores of the top 1,000 candidates we selected were significantly better than top 1,000 molecules from a 1 million random sample of ZINC15 entries, and even better than top candidates from BindingDB protease inhibitor library, which were docked to the same site (Figure 4).
Figure 4.
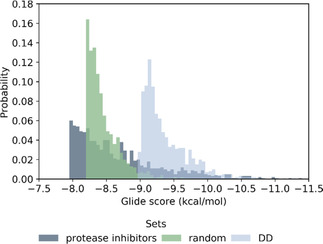
Score probability of top 1,000 ranked compounds extracted from docking of a set of protease inhibitors (7,800 compounds), a random sample of ZINC15 (1 million molecules) and top 1 million molecules from DD.
We also evaluated the chemical diversity of the newly identified set of inhibitors compared to the protease library. Calculation of Murcko frameworks58 for hits from such library and DD hits revealed a similar number of frameworks present in the two sets (603 and 587 scaffolds, respectively). Encouragingly, we observed just two common frameworks, clearly indicating that screening 1.36 billion enables identification of new chemical classes that can potentially inhibit SARS‐COV‐2 Mpro. Thus, DD allowed us to rapidly narrow down ZINC15 to a smaller dataset enriched with high scoring compounds, which consists of novel molecules with highly favourable docking scores as well as significantly different structures than known protease inhibitors.
Our DD screening identified 585 new scaffolds for SARS‐CoV‐2 that are not shared with known protease inhibitors, although they can establish all the critical interactions with the protease active site, thus providing a completely new set of chemicals for testing and optimization. Collectively, our results strongly support the use of docking the largest available compound library for identifying novel potent scaffolds or chemicals, as concluded by Lyu et al..31
4. Conclusions
The use of DD methodology in conjunction with Glide allowed rapid estimation of docking scores for 1.3 billion chemical structures into an active site of novel SARS‐CoV‐2 Mpro. The candidate inhibitors in the top‐1,000 hit list are chemically diverse, exhibit superior docking scores compared to known protease inhibitors, and can be readily sourced from established vendors. The structures of the identified compounds are made publicly available and should facilitate international efforts in rapid development of suitable drug candidates against COVID‐19.
5. Data Dissemination
List of the top 1,000 identified compounds, as well as docking results in SDF format are publicly available at https://drive.google.com/drive/folders/1xgA8ScPRqIunxEAXFrUEkavS7y3tLIMN?usp=sharing.
Conflict of Interest
None declared.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Acknowledgements
This work was funded by the CIHR Canadian 2019 Novel Coronavirus (2019‐nCoV) Rapid Research grant # DC0190GP.
A.-T. Ton, F. Gentile, M. Hsing, F. Ban, A. Cherkasov, Mol. Inf. 2020, 39, 2000028.
References
- 1. Zumla A., Chan J. F. W., Azhar E. I., Hui D. S. C., Yuen K.-Y., Nat. Rev. Drug Discovery 2016, 15, 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Wit E., van Doremalen N., Falzarano D., Munster V. J., Nat. Rev. Microbiol. 2016, 14, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., Qin C., Viruses 2019, 11, DOI 10.3390/v11010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hui D. S., Azhar E. I., Madani T. A., Ntoumi F., Kock R., Dar O., Ippolito G., Mchugh T. D., Memish Z. A., Drosten C., Zumla A., Petersen E., Int. J. Infect. Dis. 2020, 91, 264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.“Coronavirus latest: Chinese cases spike after changes to diagnosis method,” can be found under http://www.nature.com/articles/d41586-020-00154-w, 2020.
- 6.M. Wang, R. Cao, L. Zhang, X. Yang, J. Liu, M. Xu, Z. Shi, Z. Hu, W. Zhong, G. Xiao, Cell Res 2020, DOI 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed]
- 7.G. Li, E. De Clercq, Nat Rev Drug Discov 2020, d41573-020-00016-0.
- 8. Han D. P., Penn-Nicholson A., Cho M. W., Virology 2006, 350, 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cameron C. E., Castro C., Curr. Opin. Infect. Dis. 2001, 14, 757–764. [DOI] [PubMed] [Google Scholar]
- 10. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell Res 2020, 30, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.C. J. Gordon, E. P. Tchesnokov, J. Y. Feng, D. P. Porter, M. Gotte, J. Biol. Chem. 2020, jbc.AC120.013056. [DOI] [PMC free article] [PubMed]
- 12.“Adaptive COVID-19 Treatment Trial,” can be found under https://clinicaltrials.gov/ct2/show/NCT04280705, n.d..
- 13. Chu C. M., Thorax 2004, 59, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu I.-L., Mahindroo N., Liang P.-H., Peng Y.-H., Kuo C.-J., Tsai K.-C., Hsieh H.-P., Chao Y.-S., Wu S.-Y., J. Med. Chem. 2006, 49, 5154–5161. [DOI] [PubMed] [Google Scholar]
- 15. Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., Brown E. D., Chem. Biol. 2004, 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao, PDB 2020, DOI 10.2210/pdb6lu7/pdb.
- 17. Paasche A., Zipper A., Schäfer S., Ziebuhr J., Schirmeister T., Engels B., Biochemistry 2014, 53, 5930–5946. [DOI] [PubMed] [Google Scholar]
- 18. Lee H., Mittal A., Patel K., Gatuz J. L., Truong L., Torres J., Mulhearn D. C., Johnson M. E., Bioorg. Med. Chem. 2014, 22, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh A. K., Xi K., Johnson M. E., Baker S. C., Mesecar A. D., in Annual Reports in Medicinal Chemistry, Elsevier, 2006, pp. 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tuley A., Fast W., Biochemistry 2018, 57, 3326–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Turk B., Nat. Rev. Drug Discovery 2006, 5, 785–799. [DOI] [PubMed] [Google Scholar]
- 22. Ghosh A. K., Gong G., Grum-Tokars V., Mulhearn D. C., Baker S. C., Coughlin M., Prabhakar B. S., Sleeman K., Johnson M. E., Mesecar A. D., Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H., J. Med. Chem. 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Y. Li, J. Zhang, N. Wang, H. Li, Y. Shi, G. Guo, K. Liu, H. Zeng, Q. Zou, bioRxiv 2020, 2020.01.28.922922.
- 25.Z. Xu, C. Peng, Y. Shi, Z. Zhu, K. Mu, X. Wang, W. Zhu, bioRxiv 2020, 2020.01.27.921627.
- 26.X. Liu, X.-J. Wang, bioRxiv 2020, 2020.01.29.924100.
- 27.A. Zhavoronkov, V. Aladinskiy, A. Zhebrak, B. Zagribelnyy, V. Terentiev, D. S. Bezrukov, D. Polykovskiy, R. Shayakhmetov, A. Filimonov, P. Orekhov, Y. Yan, O. Popova, Q. Vanhaelen, A. Aliper, Y. Ivanenkov, 2020, DOI 10.26434/CHEMRXIV.11829102.V1.
- 28.H. Zhang, K. M. Saravanan, Y. Yang, Md. T. Hossain, J. Li, X. Ren, Y. Wei, Deep Learning Based Drug Screening for Novel Coronavirus 2019-NCov, Other, 2020. [DOI] [PMC free article] [PubMed]
- 29. Abuhammad A., Al-Aqtash R. A., Anson B. J., Mesecar A. D., Taha M. O., J. Mol. Recognit. 2017, 30, e2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berry M., Fielding B., Gamieldien J., Viruses 2015, 7, 6642–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lyu J., Wang S., Balius T. E., Singh I., Levit A., Moroz Y. S., O'Meara M. J., Che T., Algaa E., Tolmachova K., Tolmachev A. A., Shoichet B. K., Roth B. L., Irwin J. J., Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Liao L., Saravanan K. M., Yin P., Wei Y., PeerJ 2019, 7, e7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Torres P. H. M., Sodero A. C. R., Jofily P., F. P. Silva Jr , IJMS 2019, 20, 4574. [Google Scholar]
- 34. Ragoza M., Hochuli J., Idrobo E., Sunseri J., Koes D. R., J. Chem. Inf. Model. 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ashtawy H. M., Mahapatra N. R., IEEE/ACM Trans. Comput. Biol. Bioinf. 2015, 12, 335–347. [DOI] [PubMed] [Google Scholar]
- 36. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–49. [DOI] [PubMed] [Google Scholar]
- 37. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H., J. Med. Chem. 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turlington M., Chun A., Tomar S., Eggler A., Grum-Tokars V., Jacobs J., Daniels J. S., Dawson E., Saldanha A., Chase P., Baez-Santos Y. M., Lindsley C. W., Hodder P., Mesecar A., Stauffer S. R., Bioorg. Med. Chem. Lett. 2013, 23, 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mysinger M. M., Carchia M., Irwin J. J., Shoichet B. K., J. Med. Chem. 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.OpenEye Scientific Software, 2019.
- 41. Hawkins P. C. D., Skillman A. G., Warren G. L., Ellingson B. A., Stahl M. T., J. Chem. Inf. Model. 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Berman H. M., Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrödinger LLC, 2019.
- 44. Sterling T., Irwin J. J., J. Chem. Inf. Model. 2015, 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.X. Liu, B. Zhang, Z. Jin, H. Yang, Z. Rao, RCSB Protein Data Bank 2020, DOI 10.2210/PDB6LU7/PDB.
- 46. Liu T., Lin Y., Wen X., Jorissen R. N., Gilson M. K., Nucleic Acids Res. 2007, 35, D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang, Y. Duan, J. Yu, L. Wang, K. Yang, F. Liu, T. You, X. Liu, X. Yang, F. Bai1, H. Liu, X. Liu, L. W. Guddat, G. Xiao, C. Qin, Z. Shi, H. Jiang, Z. Rao, H. Yang, bioRxiv 2020, DOI 10.1101/2020.02.26.964882.
- 48. Shah F., Mukherjee P., Gut J., Legac J., Rosenthal P. J., Tekwani B. L., Avery M. A., J. Chem. Inf. Model. 2011, 51, 852–864. [DOI] [PubMed] [Google Scholar]
- 49. Das P. K., Puusepp L., Varghese F. S., Utt A., Ahola T., Kananovich D. G., Lopp M., Merits A., Karelson M., Antimicrob. Agents Chemother. 2016, 60, 7382–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pagadala N. S., Syed K., Tuszynski J., Biophys. Rev. Lett. 2017, 9, 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chaput L., Martinez-Sanz J., Saettel N., Mouawad L., J. Cheminf. 2016, 8, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.F. Gentile, V. Agrawal, M. Hsing, F. Ban, U. Norinder, M. E. Gleave, A. Cherkasov, bioRxiv 2019, 2019.12.15.877316.
- 53. Muramatsu T., Takemoto C., Kim Y.-T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S., Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kiemer L., Lund O., Brunak S., Blom N., BMC Bioinf. 2004, 5, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., Brown E. D., Chem. Biol. 2004, 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Anand K., Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 57. Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., Rao Z., Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bemis G. W., Murcko M. A., J. Med. Chem. 1996, 39, 2887–2893. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary