

Abstract
Antigenic drift of the hemagglutinin (HA) and neuraminidase (NA) proteins of the influenza virus cause a decrease in vaccine efficacy. Since the information about the evolution of these viruses in Saudi is deficient so we investigated the genetic diversity of circulating H1N1 viruses. Nasopharyngeal aspirates/swabs collected from 149 patients hospitalized with flu‐like symptoms during 2014 and 2015 were analyzed. Viral RNA extraction was followed by a reverse transcription‐polymerase chain reaction and genetic sequencing. We analyzed complete gene sequences of HA and NA from 80 positive isolates. Phylogenetic analysis of HA and NA genes of 80 isolates showed similar topologies and co‐circulation of clades 6b. Genetic diversity was observed among circulating viruses belonging to clade 6B.1A. The amino acid residues in the HA epitope domain were under purifying selection. Amino acid changes at key antigenic sites, such as position S101N, S179N (antigenic site‐Sa), I233T (antigenic site‐Sb) in the head domain might have resulted in antigenic drift and emergence of variant viruses. For NA protein, 36% isolates showed the presence of amino acid changes such as V13I (n = 29), I314M (n = 29) and 12% had I34V (n = 10). However, H257Y mutation responsible for resistance to neuraminidase inhibitors was missing. The presence of amino acid changes at key antigenic sites and their topologies with structural mapping of residues under purifying selection highlights the importance of antigenic drift and warrants further characterization of recently circulating viruses in view of vaccine effectiveness. The co‐circulation of several clades and the predominance of clade 6B.1 suggest multiple introductions in Saudi.
Keywords: Antigenic drift, H1N1, H1N1 viruses, hemagglutinin, influenza, neuraminidase, Saudi Arabia
Highlights
The presence of amino acid changes at key antigenic sites.
The topologies with structural mapping of residues under purifying selection highlights the importance of antigenic drift.
The co‐circulation of several clades and the predominance of clade 6B.1 suggest multiple introductions in Saudi.
Abbreviations
- A
alanine
- ARIs
acute respiratory infections
- D
aspartic acid
- E
glutamic acid
- H
histidine
- HA
hemagglutinin
- HAI Ab
hemagglutination inhibition antibodies
- I
isoleucine
- IRB
Institutional Review Board
- K
lysine
- KFMC
King Fahad Medical City
- N
asparagine
- NA
neuraminidase
- pdm09
pandemic strain of 2009
- Q
glutamine
- RT‐PCR
reverse transcription‐polymerase chain reaction
- S
serine
- T
threonine
- Y
tyrosine
1. INTRODUCTION
Influenza is an acute respiratory disease that occurs mostly during the winter season in temperate climates and throughout the year in the tropics. 1 Influenza A and B viruses are the major causes of influenza outbreaks. 2 Two to three different strains of influenza virus circulate during each influenza season. More than 1 billion cases of influenza occur globally every year, resulting in 3 to 5 million cases of severe illness and 300 000 to 500 000 deaths. The disease burden and epidemiologic pattern of influenza is due to the changing nature of the antigenic properties of influenza viruses. 3
Influenza A virus undergoes periodic modifications in the antigenic properties of its envelope glycoproteins; the hemagglutinin (HA) and the neuraminidase (NA). Three major hemagglutinin subtypes (H1, H2, and H3) and two neuraminidase subtypes (N1 and N2) have evolved to human species subtypes of influenza A virus. 4 Influenza virus genome have eight RNA segments and rate of reassortment among co‐infecting viruses can be high. 5 , 6 Antigenic shift leads to the emergence of new strains by reassortment between animal and human influenza viruses. The pandemics of 1908, 1957, 1968, and 2009 were caused by new viruses emerging as a result of antigenic shift. 7 , 8 Point mutations in the RNA segments coding for HA or NA are called antigenic drift. Antigenic drift causes less extensive and severe outbreaks. Between the years of antigenic shift, the variable extent and severity of influenza outbreaks is due to antigenic drift. 9
During World War I, Spanish flu pandemic killed 25 to 50 million people between 1918 and 1919 worldwide. 10 Spanish flu was one of the worst epidemics in the history of mankind killing healthy adults between 15 and 34 years. 11 The emergence of an antigenic shift in both hemagglutinin (H1) and neuraminidase (N1) proteins of the influenza A virus had extremely severe consequences. 12 The pathogenicity of the 1918 pandemic influenza virus was first demonstrated in a mouse model using reverse genetics. After infection in mice, the 1918 pandemic strain replicated efficiently producing 39 000 times more virus copies in the lungs compared to other H1N1 strains. 13 Since 1918, several influenza A virus pandemics have occurred. Genetic reassortment led to the emergence of the H2N2/1957 and H3N2/1968 pandemic influenza viruses. In 1957, the antigenic shift in the influenza A strain to H2 and N2 resulted in a severe pandemic causing approximately 1 million deaths worldwide. 14 The 1968 pandemic was less extensive due to an antigenic shift only in hemagglutinin gene (from H2N2 to H3N2). 15 The most recent emergence of the H1N1 influenza virus was in March 2009 in Mexico; however, the pandemic H1N1 virus circulation has continued globally ever since.
In June 2009, the pandemic alert level (phase 6) was declared by the World Health Organization (WHO). More than 214 countries reported the confirmed cases of pandemic H1N1 influenza A virus. 16 Influenza A (H1N1) 2009 pandemic strain represented a quadruple, genetic reassortment of two swine strains, a human strain, and an avian strain of influenza A viruses. 7 Centers for Disease Control and Prevention (CDC), USA reported approximately 61 million cases of pandemic H1N1 influenza between April 2009 and 10 April 2010, resulting in 274 000 hospitalizations and 12 470 deaths. 17 The 2009 H1N1 influenza pandemic was also associated with 100 000 to 400 000 deaths due to respiratory infection and 46 000 to 180 000 deaths due to cardiovascular infections. 18 , 19 The disease burden due to pandemic influenza A infection in the USA was 0.12 deaths per 100 000 individuals. 20 Severe pneumonia and acute respiratory distress syndrome caused most of the deaths. 21
The epidemiology of influenza A (H1N1) pdm09 with a molecular and phylogenetic analysis of HA gene during 2013/2014 influenza season in Canada, 22 2015/2016 season in Denmark, 23 2010 to 2014 in Dalian, 24 China, 25 Africa, 26 Europe, 27 , 28 India 29 confirmed the close match of the majority of circulating strains with the vaccine strains. The majority of influenza A (H1N1) pdm09 circulating in 2015/2016 belonged to the new genetic subgroup subclade 6B. 23 , 25 However, it also revealed a trend of strains to accumulate amino acid variations as a result of multiple re‐introductions and form new phylogenetic groups. Furthermore, a high number of the strains possessed the amino acid changes D97N (aspartic acid to asparagine), S185T (serine to threonine), K163Q (lysine to glutamine), A256T (alanine to threonine), and K283E (lysine to glutamic acid). 24 Influenza A (H1N1) 2009 pandemic viruses were initially reported in Saudi Arabia with the clinical features of the patients in 2010 from Saudi Arabia; the virus infection was confirmed by reverse transcription‐polymerase chain reaction (RT‐PCR). 30 , 31 Subsequent years have seen the studies reporting genetic diversity, phylogenetic relationships, and amino acid variations of HA and NA genes for influenza from the western region of Saudi Arabia. 32 , 33
Antigenic drift leads to the emergence of novel strains with a capacity to replicate efficiently in humans after infection or vaccination. Since a close antigenic match is required between circulating and vaccine strains for the efficacy of vaccines, and the disease burden is increased due to mismatches, therefore it is important to identify the mutations that affect the antibody response against natural infection or vaccination. 34 Especially the antigenic sites of HA and NA, which affect the receptor binding properties 35 and virulence 36 are crucial for understanding antigenic drift.
The current epidemiological and virological data on circulating influenza viruses in Saudi Arabia are quite deficient, and there is no influenza surveillance program in the country as well. 37 Most of the published work on H1N1 virus in Saudi Arabia deals with specimens originating in the western region during the 2009 influenza pandemic 33 , 37 , 39 and prevalence of different respiratory viruses including H1N1 38 , 40 or surveillance studies involving the pilgrims. 41 , 44 It is important to provide virological, molecular, and epidemiological information on circulating influenza strains, and its associated morbidity and mortality to make informed decisions and to undertake effective control measures to decrease the disease burden. Following the years of the pandemic, the virus undergoes antigenic drift and there is little published data about this phenomenon in Saudi Arabia. Therefore, we aimed to identify the circulation patterns, genetic diversity, structural and epidemiological characteristics of influenza A (H1N1) virus in Riyadh, Saudi Arabia from the flu season (2014‐2015).
2. MATERIAL AND METHODS
2.1. Ethical considerations
The institutional review board at King Fahad Medical City reviewed and approved the study protocol (IRB register number 16‐266).
2.2. Biosafety considerations
The handling of respiratory samples, as well as preparation of aliquots and viral RNA extraction, was executed using appropriate personal protective equipment in the biosafety level two‐laboratory (Research Laboratories, Research Center, King Fahad Medical City, Riyadh, Saudi Arabia).
2.3. Study specimens
The study specimens were obtained from a population (n = 148) admitted to King Fahad Medical City in Riyadh, Saudi Arabia. All samples testing positive for influenza (H1N1) virus locally by real‐time reverse transcription PCR (rRT‐PCR) assay and admitted to the hospital during 1 September 2014 to 16 December 2015 were used for this study. Written informed consents were obtained from the patients at the time of admission.
2.4. Laboratory investigations
Nasopharyngeal swabs and/or aspirates specimens were collected for laboratory‐based investigations. Demographic information, medical history, and outcome information were collected. Respiratory specimens were used for influenza H1N1 initial screening and further molecular testing.
2.4.1. RNA extraction and amplification
Total RNA was extracted using QIAamp Viral RNA Mini Kit (Qiagen, MA). Swabs were dissolved in the lysis buffer. Eight RNA segments of influenza A virus were simultaneously reverse‐transcribed (RT) and amplified by PCR using SuperScript III One Step RT‐PCR System (Life Technologies, CA) with the primer pairs as described previously 7 . RT‐PCR products were purified by ExoSAP‐IT Express PCR Product Cleanup Reagent (Applied Biosystems, Foster City, CA). High‐sensitivity microvolume nucleic acid quantitation was performed using the NanoDrop 3300 Fluorospectrometer. 45
2.4.2. Genetic sequencing and phylogenetic analyses
The nucleotide sequences of the amplified HA and NA fragments from 80 isolates with high copy numbers were determined using M13, sequencing primers and BigDye Terminator v3.1 Cycle Sequence kit according to the manufacturer's instructions (Applied Biosystems, San Diego). Removal of unincorporated ddNTP's (Dideoxynucleotides triphosphates), salts and dye blobs was performed with BigDye XTerminator Purification (Applied Biosystems). Capillary electrophoresis was performed in SeqStudio Sanger Sequencer (Applied Biosystems) and analysis of Sanger Sequencing data by assembly and editing was conducted by Sequencher 5.4.6 (Gene codes corporation, Ann Arbor, MI). Nucleotide sequences except primer regions were aligned with those retrieved from the Genbank Nucleotide Sequence Database. Inspection and manual modification and evolutionary analyses were conducted in Molecular Evolutionary Genetics Analysis Version X (MEGA X). Phylogenies were estimated using maximum likelihood methods implemented in MEGA, version X. 29 The maximum likelihood used Hasegawa‐Kishino‐Yano (HKY) model of nucleotide substitution with γ‐distributed rate variation (HKY + G) and bootstrapping (1000 replicates). The genomic data were analyzed and submitted to the public database (GenBank: https://www.ncbi.nlm.nih.gov/genbank) for HA under accession number (MK246015‐MK246094) and NA under accession number (MK228889‐MK228968).
2.4.3. Measurement of selection pressure
The selective pressure on encoding HA of influenza A(H1N1) was examined by calculating the ratio of synonymous and non‐synonymous substitutions (dN/dS, defined as ω) across lineage on a codon‐by‐codon basis. The individual site‐specific selection pressure and ω were estimated using the single likelihood ancestor counting (SLAC) and fixed effects likelihood (FEL) methods contained in the HYPHY package. 46 All analyses utilized the Datamonkey online tool (http://www.datamonkey.org). The value of ω was estimated based on the neighbor‐joining trees under the GTR substitution model. The significance level for a positively selected site by either SLAC/FEL or both methods was accepted at 0.1.
2.4.4. Prediction of glycosylation sites
The NetNGlyc 1.0 server was used to predict potential N‐linked glycosylation sites (amino acids Asn‐X‐Ser/Thr, whereby X is any amino acid except Asp or Pro). 47 A threshold value of >0.5 for the average potential score suggests glycosylation.
3. RESULTS
The evolutionary relationships between the vaccine and contemporary Saudi H1N1 strains were investigated to better understand what might have caused the surge in influenza cases in 2014/2015 season. We characterized the complete sequences of 80 isolates for HA and NA genes to understand their relationships from the topologies and structural homology.
To assess the evolution of the influenza A (H1N1) during the same period, representative circulating local strains from 2010 to 2015 seasons were also compared to the vaccine and reference sequences (Figure 1). There were distinct phylogenetic groups of A (H1N1)pdm09 strains between 2014 and 2015. All the influenza A (H1N1)pdm09 strains belonged to clade 6 viruses, where 92% (N = 73) grouped into sub‐clade 6b.1 and 8% (N = 7) grouped into clade 6b.2. Although both sub‐clades were related to the A/California/07/2009 vaccine strain (recommended every year since 2010‐2017) and shared >98.2% nucleotide and >97.4% amino acid sequence homology, they were considerably different from A/California/07/2009 in that they had several substitutions (Tables 1 and 2).
Figure 1.

Phylogenetic analysis of influenza A H1N1 HA and NA genes. The analysis involved 152 nucleotide sequences with a total of 1701 and 1410 positions for HA and NA, respectively. Sequences from 80 strains isolated in Saudi Arabia from 2014 to 2015 compared to the reference strains of known clades and previously reported strains from Saudi Arabia. The trees were generated using maximum likelihood by HKY+G model. Bootstrap values of 1000 replicates >70 are indicated at the nodes. The 2014 and 2015 isolates from Saudi Arabia characterized in this study are marked with closed circles, and representative sequences for all the known clades are marked with colored circles. Reference vaccine strain ((A/Michigan/45/2015) is marked with closed triangle. Only significant and signature amino acid substitutions are depicted on the tree. Substitutions shown in red color represent reversions. Scale bar represents approximately 0.2% nucleotide difference between close relatives
Table 1.
Mutations present in the HA protein of 2014 to 2015 circulating Saudi H1N1 strains compared to the vaccine strain
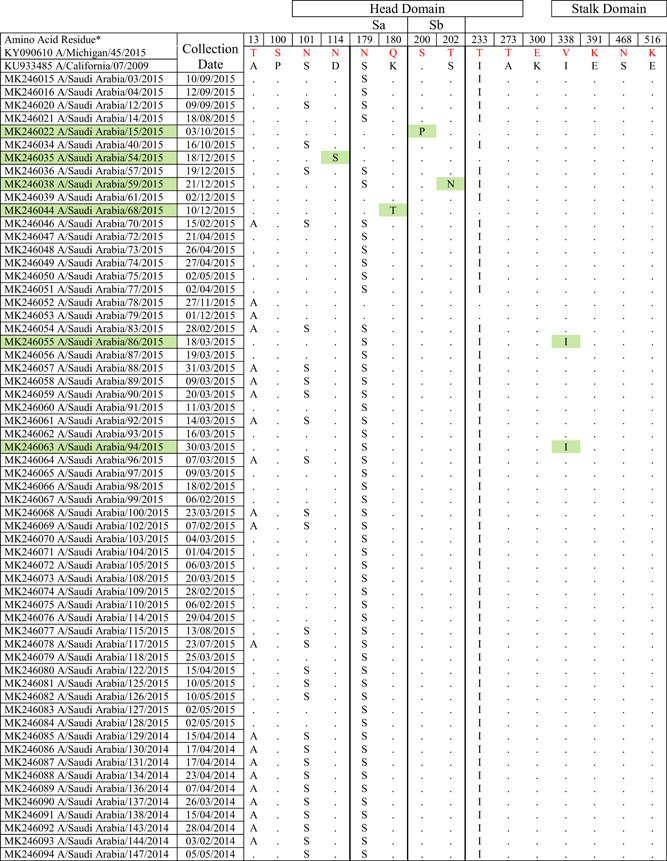
|
Note: Amino acid substitutions found in the HA protein of circulating H1N1 viruses compared to the HA proteins of A/Michigan/45/2015 and A/California/07/2009. All mutations are numbered by their position along the amino acid sequence relative to the methionine codon at the beginning of the N‐terminal signal peptide (position 1). Location of mutations with respect to head domain, stalk domain, and known Ab‐binding sites is indicated. Green colors indicate isolates with amino acid substitutions in Ab binding site, residues 180 in the Sa antigenic site; 200 and 202 in the Sb antigenic site.
Table 2.
Mutations present in the NA protein of 2014 to 2015 circulating Saudi H1N1 strains compared to the vaccine strain
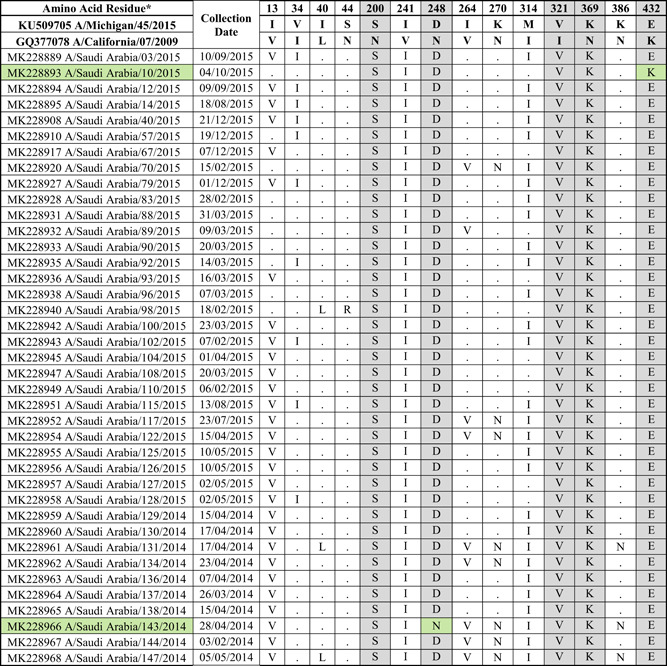
|
Note: Amino acid substitutions found in the NA protein of circulating H1N1 viruses compared to the NA proteins of A/Michigan/45/2015 and A/California/07/2009. All mutations are numbered by their position along the amino acid sequence relative to the methionine codon at the beginning of the N‐terminal signal peptide (position 1). The location of mutations with respect to known Ab‐binding sites is indicated. Gray color indicates mutations in known Ab‐binding sites. Green color indicates strains with no changes in the Ab‐binding sites.
Influenza A H1N1 pdm09 viruses were classified into eight major clades in which clade 6 is further sub‐classified into 6A, 6B, 6C, 6B.1, and 6B.2 based on HA gene diversity. All the isolates had hemagglutinin (HA) gene corresponding to clade 6B (monophyletic) represented by global circulating strains of H1N1pdm09 (Figure 1) defined by four major reversions A13T, S101N, S179N, and I223T. The WHO updated the H1N1 strain component of the flu vaccine in 2017 to 2018 season from A/Cal/07/09 to A/Michigan/45/2015. 44 Interestingly, the HA and NA proteins encoded by this strain do not contain all of the mutations found in the majority of 2014 to ‐2015 isolates described in this study.
The influenza A(H1N1)pdm09 sequences were characterized in a maximum likelihood phylogenetic tree with reference strains rooted from the previous vaccine strain, A/California/07/2009 (Figure 1). The influenza A(H1N1)pdm09 HA sequences characterized from the 2014 to 2015 season exhibited an overall protein homology of 98.5% to 99.5% (average 99.0%) compared to the 2017 to 2018 influenza vaccine component, A/Michigan/45/2015 virus. All influenza A(H1N1)pdm09 HA sequences contained mutations consistent with the dominating subgroup referred to as clade 6B and could all be further classified as subclade 6B.1 and 6B.2.
Saudi isolates were found to be clustering more closely to new vaccine strain (Michigan/45/2015) used for 2018/2019 season than the previously used vaccine strain (California/07/2009). Saudi isolates reported from 2013 clustered into clade 6c (Figure 1). Almost 92% of Saudi viruses from our study formed a subclass branch with high bootstrap value (91/100) designated as 6B.1 48 mainly defined by S179N and I223T reversions. The remaining seven strains formed another cluster (96/100 bootstrap value) with an S101N, T169V, S179N, I190V, I223T, and G508E amino acid substitutions (designated as sub‐clade 6B.2 in Figure 1).
HA protein surface structure is changed due to the gain or loss of N‐linked glycosylation sites. This gain can cause modulation of viral antigenicity by the masking effect on important antibody recognition sites. 49 Among the influenza A(H1N1)pdm09 HA sequences characterized in our study (Figure 2), one mutation, S179N (serine to asparagine), was observed that caused a predicted gain of a glycosylation motif. 50 Influenza viruses present in nasal and throat swabs from subjects infected during the 2014 to 2015 season encoded 42 amino acid changes in the HA protein compared to the strain A/Michigan/45/2015 (vaccine strain from 2017 to 2018) (Table 1). For H1N1 viruses from our study, the amino acid substitutions or mismatches were only considered if they occurred in five canonical H1N1 HA antigenic sites (Sa, Sb, Ca1, Ca2, Cb) composed of amino acid regions (H1N1 numbering): Sa: 141‐142, 170‐174, 176‐181; Sb: 201‐212; Ca1: 183‐187, 220‐222, 252‐254; Ca2: 154‐159, 238‐239; Cb: 87‐92. 51 Out of the 42 mutations, 7 substitution sites were located in the HA head domain while only 2 sites were located in the stalk domain (Table 1). Four mutations were found in previously characterized 52 Ab‐binding sites.
Figure 2.
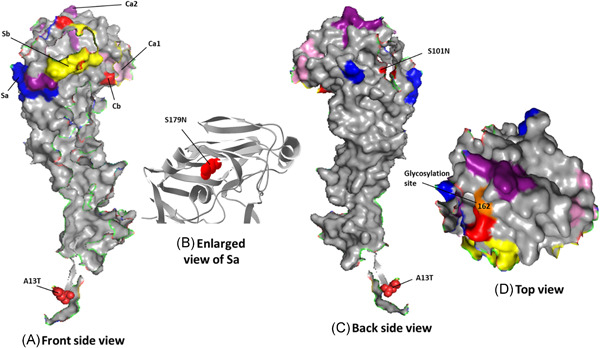
Amino acid changes present in the HA proteins of 2014 to 2015 circulating H1N1 strains compared to the vaccine strain. Three‐dimensional models of the A/Cal/04/09 HA1 (single chain) protein (PDB ID: 3LZG), showing the amino acid changes in circulating H1N1 viruses compared to that of the vaccine strain. Front and backside views of the HA1 monomer protein are shown. A, The HA antigenic sites Sa (dark blue), Sb (yellow), Ca1 (light pink), Ca2 (dark purple), and Cb (light red) are indicated. B, S179N amino acid change present in the antigenic site Sa of HA1 of H1N1 circulating strains compared to that of the H1N1 vaccine strain is shown in ribbon in red. C, The approximate location of HA amino acid change S220T, situated in the Ca1 antigenic site at the subunit interface, is indicated. (D) top view of head region of HA1 is showing the antigenic sites along with glycosylation site. The 3D model was customized using PyMOL
The circulating strains belonged to subclade 6B.1, defined by HA amino acid substitutions S101N, S179N, and I233T. The majority of the 2014/15 season Saudi isolates formed part of a large cluster possessing an S101N substitution in HA. Most of these isolates contained S179N (on the Sa antigenic site of HA resulting in a potential glycosylation site) and I233T amino acid substitutions in addition to the S101N substitution and formed a subclade subsequently designated as 6B.1. 48
As for the NA protein (sequenced from 80 nasopharyngeal aspirates and swabs), 14 amino acid changes were found when compared to the vaccine strain (Table 2). Five out of the fourteen changes (N200S, N248D, I321V, N369K, and K432E) were at previously characterized Ab‐binding sites. 53 Three of the mutations (K432E along the 430 loop, V241I, and N248D) were located near the catalytic site. All strains encoded the same NA sequence, with the exception of MK228966 (A/Saudi Arabia/143/2014) and MK228893 (A/Saudi Arabia/10/2015) viruses, which did not have the mutations N248D and K432E respectively. Altogether, these data indicate genetic variability in both, the HA and NA proteins of circulating H1N1 viruses from 2014 to 2015, compared to the vaccine strain.
Most antibodies elicited by influenza virus by natural infection and vaccination target the globular head domain of HA1, which is distal from the virus surface and readily accessible for immune recognition. Influenza virus HA, in turn, mutates to escape from pre‐existing immunity. This mutation‐based immune evasion process is known as antigenic drift. Early studies proposed five major antigenic sites in the HA1 globular head domain for H1, namely Sa, Sb, Ca1, Ca2, and Cb for H1 HA. 51 The locations of Sa, Sb, and Ca2 of H1 HA partially overlap with the RBS, whereas Ca1 and Cb of H1 HA are more distant from the RBS. These antigenic sites provide a structural framework to understand the evolutionary dynamics and constraints of the influenza virus in response to humoral immunity. 54 As shown on a 3D model of the HA protein (RCSB PDB ID: 3LZG), 55 five mutations were found in previously characterized 48 Ab‐binding sites: S179N and T180Q, in Sa; P200S and S202T, in Sb; and S220T, in Ca1 (Figure 2C and Table 1). 56
Further analyses of HA protein of study strains (Table 1) showed amino acid change at position 13 in the signal peptide of HA (A13T), all belonging to clade 1 i.e. (6B.1) while MK246055 (A/Saudi Arabia/86/2015) and MK246063 (A/Saudi Arabia/94/2015) had the I338V amino acid change in the stalk domain which is a characteristic marker for all clades, when compared to the clade 1 viruses. Amino acid changes were found at key antigenic sites, such as position S101N, S179N (antigenic site‐Sa), I233T (antigenic site‐Sb) in the head domain. Along with these substitutions, overall sequence diversity in HA was not high for 6B.1.
3.1. Selection pressure on A(H1N1)pdm09
We assumed that the influenza HA protein is subjected to selection pressure to evade the host cell recognition, the rate of change was assessed by the ω values in which ω < 1 meant that negative or purifying selection was present, ω = 1 when selection pressure was neutral, and ω > 1 when there was positive selection. 57 The analysis showed that the overall ω value of the coding HA1 regions of influenza A(H1N1)pdm09 was 0.23. Since the majority of residues (N = 42) in the HA1 domain showed ω < 1, this suggested that the amino acids in the HA epitope domain were under purifying selection. Although overall positive selection was not present, specific sites of positive selection were found using SLAC and FEL methods.
3.2. Prediction of glycosylation sites
The majority of influenza A(H1N1) strains possessed seven potential glycosylation sites in the HA1 domain at amino acid positions 10, 11, 23, 87, 162, 276, and 287, which were present in all but clade 6A. The mutation at N11S among clade 6A strains resulted in a loss of potential glycosylation site at this position. We did not find the D222G mutation commonly associated with increased virulence. 58
4. DISCUSSION
Surface proteins of influenza viruses are subjected to a high selective pressure because they have to avoid recognition by the host's immune system as well as improve virus fitness by modulating their activity according to the nature of the host. One of the main mechanisms of variation is antigenic drift, which is the introduction of mutations in known antigenic sites as a response to the recognition by the immune system. To describe the antigenic drift in the circulating H1N1 viruses from 2014 to 2015 season, we studied genetic diversity along with amino acid changes in HA and NA genes of 80 Saudi isolates. We found 42 and 14 amino acid changes in HA (Table 1 and Figure 2) and NA (Table 2) proteins respectively. Out of the 42 mutations, 7 substitution sites were located in the HA head domain while only 2 changes were located in the stalk domain (Table 1). Four amino acids were found in previously characterized 52 Ab‐binding sites. These changes were representative of those found in the HA or NA proteins of circulating viruses during 2014 to 2015 season (Figure 1). These amino acid changes remained present in the HA and NA proteins of the strains isolated during the subsequent season, further suggesting the stability of these changes.
Recent advancements in the method development have made phylogenetic inference quite important as a promising tool for evaluating the antigenic evolution of influenza viruses from the genetic data. Unlike the classical Hemagglutination Inhibition assay uses post‐infection animal and/or human antisera, phylogenetic analyses, on the other hand, can exploit easier to generate, and hence more richly available data to achieve high levels of prediction accuracy. 59 Despite good vaccine coverage, infected subjects continue to exhibit low Hemagglutination Inhibition Antibodies (HAI Ab) titers against the vaccine and circulating strains. 60 This suggests that poor responses to the H1N1 component of the vaccine as well as antigenic differences in the HA and NA proteins of currently circulating H1N1 viruses could be contributing to risk of infection even after vaccination.
After its emergence in 2009 and the following years, circulating influenza H1N1 viruses were closely related to the vaccine strain A/California/07/2009 and mostly characterized by a few amino acid changes. Based on HA protein, viruses along with their variants emerged from clades 3 and 4 and remained prevalent. In the first season following 2009 pandemic, H1N1 viruses diverged considerably leading to the formation of several clades with many genetic markers. During 2011 to 2012 season, six different clades namely 2, 3, 4, 5, 6, and 7 were in circulation throughout the world. Although, viruses belonging to clade 5 and 7 were few in circulation during that time but emergence and divergence of clade 6 viruses especially into 6b and 6c lead to the replacement of previous 6b strains in 2013 to 2014. 61 During 2015 to 2016 season, clade 6b viruses diverged further into 6b.1 and 6b.2 subclades after the introduction of new amino acid changes. These subclades remained dominant in Europe, North America but not in Asia, Africa or South America in 2015. 6b.1 became dominant all over the world in 2016 23 , 29 but we found that 6b.1 and 6b.2 were circulating in Saudi Arabia in 2015 (Table 1). The viruses belonging to clade 6b.1 were the most dominant compared to other regions at that time.
Although the rates of amino acid changes in the HA and NA genes in our study might not be very high than previous seasons 62 the on‐going genetic evolution that had been accumulated in circulating H1N1 strains since the WHO vaccine recommendation made for the season 2017 to 2018 might deepen the antigenic disparity between the vaccine and circulating H1N1 strains in the 2019 to 2020 season. 63 The phylogenetic clustering pattern of the HA and NA genes might substantiate the significance of the rise in influenza cases with regard to the comprehensive process of vaccine virus selection. The amino acid substitutions in the form of reversion mutations observed especially in the head domain of antigenic sites were quite striking. For 2019 to 2020, influenza vaccine composition for Northern Hemisphere was recommended by WHO. 64 Food and Drug Administration (FDA) along with Vaccines and Related Biologic Products Advisory Committee make the influenza vaccine composition recommendation in the United States of America (USA). 45 , 65 Both agencies recommend that influenza trivalent vaccines contain an A/Brisbane/02/2018 A(H1N1)pdm09‐like virus, an A/Kansas/14/2017 A(H3N2)‐like virus, and a B/Colorado/06/2017‐like (B/Victoria lineage) virus. The recommendation for quadrivalent vaccine included the trivalent vaccine viruses and a B/Phuket/3073/2013‐like (B/Yamagata lineage) virus. The A(H1N1)pdm09 and A(H3N2) recommendations are an update to the 2018‐2019 Northern Hemisphere vaccines. The A(H1N1)pdm09 component has been updated keeping in view of genetic and antigenic characterization data showing significantly reduced titers (eight‐fold or greater) to recent 6B.1A viruses, compared with the titers against the A/Michigan/45/2015 vaccine virus. 66 , 67 , 68
Most of the protective antibodies induced by vaccination or infection with influenza virus are specific to the HA protein. 69 , 70 As a result, most studies focus mainly on the antigenicity of the HA protein. However, protective antibodies specific to the NA protein are also elicited in animal models and humans, 71 , 73 and antigenic drift in the NA has been reported to occur. 74 , 75 There were 14 mutations in the NA protein encoded by Saudi viruses circulating during the 2014‐15 season when compared to the vaccine strain, 4 of which were located in previously identified antigenic sites (Table 2). 53 If the NAI Ab titers for the circulating strains are low than for the vaccine strain, it might suggest that there is antigenic variability in the NA protein expressed by currently circulating viruses. This, in turn, could lead to a decrease in the efficacy of the vaccine. These antigenic differences could be due to the amino acid changes in defined Ab binding sites (N200S, N248D, N369K, and K432E). 53 Alternatively, due to the poor understanding of the antigenicity of the NA protein, some other amino acid changes might also affect the Ab binding.
Amino acid changes at key antigenic sites in our study, such as position N179S, Q180T (antigenic site‐Sa), S200P, T202N (antigenic site‐Sb) along with N101S, N114S, and T233I in the head domain might have resulted in antigenic drift and emergence of variant viruses. H1 gene sequences in comparison to 2010‐2016 vaccine strain recently in India have shown mutations K166Q and S188T resulting in the emergence of variants. 76 Amino acid substitutions other than at position 166 in major antigenic site Sa can be selected by the immunological pressure of antibodies, that recognize an epitope including position 166. In combination with computational analyses and other methods, 77 in vitro selection of potential antigenic drift mutants could improve the selection of vaccine seed viruses. 78 Viral protein‐host interactions that are lost with the mutation are compensated by interactions with other amino acids, leading to a more stable HA. 79 The HA from influenza A(H1N1)pdm09 virus is steadily evolving towards a more stable and antigenically distinct protein. This trend indicates that current influenza vaccines are likely to have a decreased efficacy if the same strain is used in future vaccine formulations.
5. CONCLUSIONS
Between the years of antigenic shift, the variable extent and severity of influenza outbreaks are due to antigenic drift. Therefore, HA and NA complete gene sequencing data in this study provides a closer look into the epidemiological pattern of circulating influenza A (H1N1) viruses and the antigenic mismatches between circulating and vaccine strains. The potential amino acid changes responsible for antigenic drift found in this study would help further to measure the impact that influenza is having on hospitalizations and deaths in Saudi Arabia. These findings will direct further research for further vaccine effectiveness studies to aid in proper treatment options to reduce hospitalization due to these infections.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
AN: study design, experimental work, data collection and analysis, manuscript writing and review. KE: reviewed the experimental data, analyses and manuscript. BA: study design, data analysis, results discussion, manuscript review. AA: performed homology structural analysis of proteins. HA: data analysis, results discussion, manuscript review. SA: database work‐up and data collection. ME: results discussion, manuscript review. All authors read and approved the final manuscript.
ETHICS STATEMENT
All procedures performed in this study involving clinical specimen were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the Institutional Review Board at King Fahad Medical City (IRB Log No. 16‐266). Informed consent to participate was waived or not required since only remaining left‐over specimens were used for this study.
ACKNOWLEDGMENTS
The authors thank Dr Fatimah S Alhamlan and Lama Abdullah for their excellent technical support. They also thank KFMC diagnostic Laboratories and for their contribution in sample collection. This study was supported by the Intra‐mural Research Fund (IRF) from the King Fahad Medical City for funding this study (Grant No 017‐024).
Naeem A, Elbakkouri K, Alfaiz A, et al. Antigenic drift of hemagglutinin and neuraminidase in seasonal H1N1 influenza viruses from Saudi Arabia in 2014 to 2015. J Med Virol. 2020;92:3016–3027. 10.1002/jmv.25759
DATA AVAILABILITY STATEMENT
The clinical datasets used and/or analyzed during the current study are available from the corresponding author on request.
REFERENCES
- 1. World Health Organization . Influenza (Seasonal) Fact sheet N 211. November 2018. http://www.who.int/mediacentre/factsheets/fs211/en/. Accessed 24 June 2019
- 2. Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc London. 2001;356:1861‐1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klimov AI, Garten R, Russell C, et al. WHO recommendations for the viruses to be used in the 2012 Southern Hemisphere Influenza Vaccine: epidemiology, antigenic and genetic characteristics of influenza A(H1N1)pdm09, A(H3N2) and B influenza viruses collected from February to September 2011. Vaccine. 2012;30(45):6461‐6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tong S, Zhu X, Li Y, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9(10):e1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schäfer JR, Kawaoka Y, Bean WJ, Süss J, Senne D, Webster RG. Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology. 1993;194(2):781‐788. [DOI] [PubMed] [Google Scholar]
- 6. Webby RJ, Webster RG. Emergence of influenza A viruses. Philos Trans R Soc London, B: Biol Sci. 2001;356(1416):1817‐1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dawood FS, Jain S, Finelli L, et al. Emergence of a novel swine‐origin influenza A(H1N1) virus in humans. N Engl J Med. 2009;360(25):2605‐2615. [DOI] [PubMed] [Google Scholar]
- 8. Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of the early isolates of swine‐origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325(5937):197‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nelson MI, Holmes EC. The evolution of epidemic influenza. Nat Rev Genet. 2007;8:196‐205. [DOI] [PubMed] [Google Scholar]
- 10. Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918‐1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:105‐115. [DOI] [PubMed] [Google Scholar]
- 11. Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Infect Dis. 2007;195:1018‐1028. [DOI] [PubMed] [Google Scholar]
- 12. Taubenberger JK, Reid AH, Krafft AE, Bijwaard KE, Fanning TG. Initial genetic characterization of the 1918 “Spanish” influenza virus. Science. 1997;275:1793‐1796. [DOI] [PubMed] [Google Scholar]
- 13. Tumpey TM. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77‐80. [DOI] [PubMed] [Google Scholar]
- 14. Xu R, McBride R, Paulson JC, Basler CF, Wilson IA. Structure, receptor binding, and antigenicity of influenza virus hemagglutinins from the 1957 H2N2 pandemic. J Virol. 2010;84:1715‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Masurel N, Marine WM. Recycling of Asian and Hong Kong influenza A virus hemagglutinins in man. Am J Epidemiol. 1973;97:44‐49. [DOI] [PubMed] [Google Scholar]
- 16. World Health Organization . Pandemic (H1N1) 2009: update 112.2010. http://www.who.int/csr/don/2010_08_06/en/index.html. Accessed 24 June 2019
- 17. Shrestha SS, Swerdlow DL, Borse RH, et al. Estimating the burden of 2009 pandemic influenza A (H1N1) in the United States (April 2009‐April 2010). Clin Infect Dis. 2011;52:S75‐S82. [DOI] [PubMed] [Google Scholar]
- 18. Dawood FS, Iuliano AD, Reed C, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12:687‐695. [DOI] [PubMed] [Google Scholar]
- 19. Simonsen L, Spreeuwenberg P, Lustig R, et al. Global mortality estimates for the 2009 Influenza Pandemic from the GLaMOR project: a modeling study. PLoS Med. 2013;10:e1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fowlkes AL, Arguin P, Biggerstaff MS, et al. Epidemiology of 2009 pandemic influenza A (H1N1) deaths in the United States, April‐July 2009. Clin Infect Dis. 2011;52(Suppl 1):S60‐S68. [DOI] [PubMed] [Google Scholar]
- 21. Louie JK. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA. 2009;302:1896‐902. [DOI] [PubMed] [Google Scholar]
- 22. Skowronski D, Chambers C, Sabaiduc S, et al. Interim estimates of 2013/14 vaccine effectiveness against influenza A(H1N1)pdm09 from Canada s sentinel surveillance network. Euro Surveill. 2014;19(6):20698. [DOI] [PubMed] [Google Scholar]
- 23. Emborg HD, Krause TG, Nielsen L, et al. Influenza vaccine effectiveness in adults 65 years and older, Denmark, 2015/16—a rapid epidemiological and virological assessment. Euro Surveill. 2016;21(14):1560‐7917. [DOI] [PubMed] [Google Scholar]
- 24. Han Y, Sun N, Lv QY, Liu DH, Liu DP. Molecular epidemiology and phylogenetic analysis of HA gene of influenza A(H1N1)pdm09 strain during 2010‐2014 in Dalian, North China. Virus Genes. 2016;52(5):606‐612. [DOI] [PubMed] [Google Scholar]
- 25. Fang Q, Gao Y, Chen M, Guo X, Yang X, Wei L. Molecular epidemiology and evolution of influenza A and B viruses during winter 2013‐2014 in Beijing. China. Arch Virol. 2015;160(4):1083‐1095. [DOI] [PubMed] [Google Scholar]
- 26. Jones AH, Ampofo W, Akuffo R, et al. Sentinel surveillance for influenza among severe acute respiratory infection and acute febrile illness inpatients at three hospitals in Ghana. Influenza Other Respir Viruses. 2016;10(5):367‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mugosa B, Vujosevic D, Ciccozzi M, et al. Genetic diversity of the haemagglutinin (HA) of human influenza A (H1N1) virus in Montenegro: focus on its origin and evolution. J Med Virol. 2016;88(11):1905‐1913. [DOI] [PubMed] [Google Scholar]
- 28. Antón A, Marcos MA, Torner N, et al. Virological surveillance of influenza and other respiratory viruses during six consecutive seasons from 2006 to 2012 in Catalonia, Spain. Clin Microbiol Infect. 2016;22(6):564.e1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parida M, Dash PK, Kumar JS, et al. Emergence of influenza A (H1N1)pdm09 genogroup 6B and drug resistant virus, India, January to May 2015. Euro Surveill. 2016;21(5):6‐11. [DOI] [PubMed] [Google Scholar]
- 30. Alarabiyaonline . Detection of the first case of swine flu in Saudi Arabia Filipina nurse. 2010. http://www.alarabiya.net/articles/2009/06/03/74702.html. Accessed 24 June 2019
- 31. Agha A, Alrawi A, Cesar V, Munayco K, Bella A. Characteristics of patients hospitalized with 2009 H1N1 influenza in a tertiary carehospital in southern Saudi Arabia. Mediterr J Hematol Infect Dis. 2012;4:4. e2012002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. The Extinction Protocol . H1N1 outbreak reported in Saudi Arabia. 2012. http://theextinctionprotocol.wordpress.com/2012/11/05/h1n1‐outbreak‐reported‐in‐saudi‐arabia/. Accessed 24 June 2019
- 33. Uthman NA, Sohrab SS, Kamal IH, et al. Genetic diversity of the pandemic influenza A (H1N1) virus in Saudi Arabia. J Infect Dev Ctries. 2014;8(12):1563‐1573. [DOI] [PubMed] [Google Scholar]
- 34. Ushirogawa H, Naito T, Tokunaga H, et al. Re‐emergence of H3N2 strains carrying potential neutralizing mutations at the N‐linked glycosylation site at the hemagglutinin head, post the 2009 H1N1 pandemic. BMC Infect Dis. 2016;8(16):380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang CC, Chen JR, Tseng YC, et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc Natl Acad Sci U S A. 2009;106(43):18137‐18142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tate MD, Job ER, Brooks AG, Reading PC. Glycosylation of the hemagglutinin modulates the sensitivity of H3N2 influenza viruses to innate proteins in airway secretions and virulence in mice. Virology. 2011;413(1):84‐92. [DOI] [PubMed] [Google Scholar]
- 37. Hashem AM. Influenza immunization and surveillance in Saudi Arabia. Ann Thorac Med. 2016;11(2):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hussain R, Al‐Omar I, Memish Z. The diagnostic challenge of pandemic H1N1 2009 virus in a dengue‐endemic region: a case report of combined infection in Jeddah, Kingdom of Saudi Arabia. J Infect Public Health. 2012;5(2):199‐202. [DOI] [PubMed] [Google Scholar]
- 39. Masoodi TA, Shaik NA, Shafi G, Munshi A, Ahamed AK, Masoodi ZA. Comparative analysis of hemagglutinin of 2009 H1N1 influenza A pandemic indicates its evolution to 1918H1N1 pandemic. Gene. 2012;491(2):200‐204. [DOI] [PubMed] [Google Scholar]
- 40. Alsufiani HA, Aljabri HA, Morsy MM, et al. Clinical and laboratory characteristics of childhood pandemic 2009 H1N1 Influenza. Experience from the western region of Saudi Arabia. Saudi Med J. 2014;35(6):624‐625. [PubMed] [Google Scholar]
- 41. Ziyaeyan M, Alborzi A, Jamalidoust M, et al. Pandemic 2009 influenza A (H1N1) infection among 2009 Hajj Pilgrims from Southern Iran: a real‐time RT‐PCR‐based study. Influenza Other Respir Viruses. 2012;6(6):e80‐e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Memish ZA, Assiri AM, Hussain R, Alomar I, Stephens G. Detection of respiratory viruses among pilgrims in Saudi Arabia during the time of a declared influenza A(H1N1) pandemic. J Travel Med. 2012;19(1):15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ashshi A, Azhar E, Johargy A, et al. Demographic distribution and transmission potential of influenza A and 2009 pandemic influenza A H1N1 in pilgrims. J Infect Dev Ctries. 2014;8(9):1169‐1175. [DOI] [PubMed] [Google Scholar]
- 44. Atabani SF, Wilson S, Overton‐Lewis C, et al. Active screening and surveillance in the United Kingdom for Middle East respiratory syndrome coronavirus in returning travellers and pilgrims from the Middle East: a prospective descriptive study for the period 2013‐2015. Int J Infect Dis. 2016;47:10‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Desjardins P, Conklin D. NanoDrop microvolume quantitation of nucleic acids. J Vis Exp. 2010;45:2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delport W, Poon AF, Frost SD, Kosakovsky Pond SL. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26(19):2455‐2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gupta R, Jung E, Brunak S. Prediction of N‐glycosylation sites in human proteins. 2004. Database: NetNGlyc 1.0. http://www.cbs.dtu.dk/services/NetNGlyc/
- 48. Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell. 1982;31:417‐427. [DOI] [PubMed] [Google Scholar]
- 49. Katz J, Shu Y, McCauley J, et al. Recommended composition of influenza virus vaccines for use in the 2017–2018 northern hemisphere influenza season. Bulletin World Health Organization. 2017:7. [Google Scholar]
- 50. Kongchanagul A, Suptawiwat O, Kanrai P, Uiprasertkul M, Puthavathana P, Auewarakul P. Positive selection at the receptor‐binding site of hemagglutinin H5 in viral sequences derived from human tissues. J Gen Virol. 2008;89(8):1805‐1810. [DOI] [PubMed] [Google Scholar]
- 51. Skowronski DM, Janjua NZ, De Serres G, et al. Low 2012‐13 influenza vaccine effectiveness associated with mutation in the egg‐adapted H3N2 vaccine strain not antigenic drift in circulating viruses. PLoS One. 2014;9(3), e92153. 10.1371/journal.pone.0092153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wolf YI, Viboud C, Holmes EC, Koonin EV, Lipman DJ. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol Direct. 2006;1:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu X, Liu F, Zeng H, et al. Evaluation of the antigenic relatedness and cross‐protective immunity of the neuraminidase between human influenza A (H1N1) virus and highly pathogenic avian influenza A (H5N1) virus. Virology. 2014;454‐455:169‐175. [DOI] [PubMed] [Google Scholar]
- 54. Wu NC, Wilson IA. A perspective on the structural and functional constraints for immune evasion: insights from influenza virus. J Mol Biol. 2017;429(17):2694‐2709. 10.1016/j.jmb.2017.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE Jr., Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328:357‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krause JC, Tumpey TM, Huffman CJ, et al. Naturally occurring human monoclonal antibodies neutralize both 1918 and 2009 pandemic influenza A (H1N1) viruses. J Virol. 2010;84:3127‐3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4(12):e1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chutinimitkul S, Herfst S, Steel J, et al. Virulence‐associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J Virol. 2010;84(22):11802‐11813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Broberg E, Melidou A, Prosenc K, Bragstad K, Hungnes O. Predominance of influenza A(H1N1)pdm09 virus genetic subclade 6B.1 and influenza B/Victoria lineage viruses at the start of the 2015/16 influenza season in Europe. Euro Surveill. 2016;21:30184. [DOI] [PubMed] [Google Scholar]
- 60. Abusrewil S, Algeer A, Aljifri A, et al. Influenza surveillance in Middle East, North, East and South Africa: report of the 8th MENA Influenza Stakeholders Network. Influenza Other Respir Viruses. 2019;13(3):298‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wedde M, Biere B, Wolff T, Schweiger B. Evolution of the hemagglutinin expressed by human influenza A(H1N1)pdm09 and A(H3N2) viruses circulating between 2008‐2009 and 2013‐2014 in Germany. Int J Med Microbiol. 2015;305:762‐775. [DOI] [PubMed] [Google Scholar]
- 62. Neher RA, Bedford T. nextflu: real‐time tracking of seasonal influenza virus evolution in humans. Bioinformatics. 2015;31(21):3546‐3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Westgeest KB, Russell CA, Lin X, et al. Genome‐wide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J Virol. 2014;88(5):2844‐2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim JI, Lee I, Park S, et al. Phylogenetic relationships of the HA and NA genes between vaccine and seasonal influenza A(H3N2) strains in Korea. PLoS One. 2017;12(3):e0172059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. World Health Organization . Recommended composition of influenza virus vaccines for use in the 2019‐2020 northern hemisphere influenza season. Geneva, Switzerland: World Health Organization. 2019. https://www.who.int/influenza/vaccines/virus/recommendations/2019_20_north/en/. Accessed 25 June 2019. [Google Scholar]
- 66. Herzallah HK, Bubshait SA, Antony AK, Al‐Otaibi ST. Incidence of influenza A H1N1 2009 infection in Eastern Saudi Arabian hospitals. Saudi Med J. 2011;32(6):598‐602. [PubMed] [Google Scholar]
- 67. Food and Drug Administration Center for Biologics Evaluation and Research . Vaccines and Related Biological Products Advisory Committee meeting announcement, 6‐7 March 2019. Silver Spring, MD: Department of Health and Human Services, Food and Drug Administration. 2019. https://www.fda.gov/advisory‐committees/advisory‐committee‐calendar/vaccines‐and‐related‐biological‐products‐advisory‐committee‐march‐6‐7‐2019‐meeting‐announcement. Accessed 25 June 2019. [Google Scholar]
- 68. Food and Drug Administration Center for Biologics Evaluation and Research . Vaccines and Related Biological Products Advisory Committee meeting announcement, 22 March 2019. Silver Spring, MD: Department of Health and Human Services, Food and Drug Administration. 2019. https://www.fda.gov/advisory‐committees/advisory‐committee‐calendar/vaccines‐and‐related‐biological‐products‐advisory‐committee‐march‐22‐2019‐meeting‐announcement. Accessed 25 June 2019. [Google Scholar]
- 69. Virelizier JL. Host defenses against influenza virus: the role of anti‐hemagglutinin antibody. J Immunol. 1975;115:434‐439. [PubMed] [Google Scholar]
- 70. Nicholls JM, Chan RW, Russell RJ, Air GM, Peiris JS. Evolving complexities of influenza virus and its receptors. Trends Microbiol. 2008;16:149‐157. [DOI] [PubMed] [Google Scholar]
- 71. Monto AS, Petrie JG, Cross RT, et al. Antibody to influenza virus neuraminidase: an independent correlate of protection. J Infect Dis. 2015;212:1191‐1199. [DOI] [PubMed] [Google Scholar]
- 72. Wohlbold TJ, Krammer F. In the shadow of hemagglutinin: a growing interest in influenza viral neuraminidase and its role as a vaccine antigen. Viruses. 2014;6:2465‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Couch RB, Atmar RL, Franco LM, et al. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J Infect Dis. 2013;207:974‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kilbourne ED, Johansson BE, Grajower B. Independent and disparate evolution in nature of influenza A virus hemagglutinin and neuraminidase glycoproteins. Proc Natl Acad Sci U S A. 1990;87:786‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Laver WG, Air GM, Webster RG, Markoff LJ. Amino acid sequence changes in antigenic variants of type A influenza virus N2 neuraminidase. Virology. 1982;122:450‐460. [DOI] [PubMed] [Google Scholar]
- 76. Jagadesh A, Krishnan A, Nair S, Sivadas S, Arunkumar G. Genetic characterization of hemagglutinin (HA) gene of influenza A viruses circulating in Southwest India during 2017 season. Virus Genes. 2019;55:458‐464. 10.1007/s11262-019-01675-x [DOI] [PubMed] [Google Scholar]
- 77. Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the evolution of human influenza A. Science. 1999;286:1921‐1925. [DOI] [PubMed] [Google Scholar]
- 78. Yasuhara A, Yamayoshi S, Soni P, et al. Diversity of antigenic mutants of influenza A(H1N1)pdm09 virus escaped from human monoclonal antibodies. Sci Rep. 2017;7(1):17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Castelán‐Vega JA, Magaña‐Hernández A, Jiménez‐Alberto A, Ribas‐Aparicio RM. The hemagglutinin of the influenza A(H1N1)pdm09 is mutating towards stability. Adv Appl Bioinform Chem. 2014;7:37‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The clinical datasets used and/or analyzed during the current study are available from the corresponding author on request.