

Abstract
To investigate the evolutionary history of the recent outbreak of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) in China, a total of 70 genomes of virus strains from China and elsewhere with sampling dates between 24 December 2019 and 3 February 2020 were analyzed. To explore the potential intermediate animal host of the SARS‐CoV‐2 virus, we reanalyzed virome data sets from pangolins and representative SARS‐related coronaviruses isolates from bats, with particular attention paid to the spike glycoprotein gene. We performed phylogenetic, split network, transmission network, likelihood‐mapping, and comparative analyses of the genomes. Based on Bayesian time‐scaled phylogenetic analysis using the tip‐dating method, we estimated the time to the most recent common ancestor and evolutionary rate of SARS‐CoV‐2, which ranged from 22 to 24 November 2019 and 1.19 to 1.31 × 10−3 substitutions per site per year, respectively. Our results also revealed that the BetaCoV/bat/Yunnan/RaTG13/2013 virus was more similar to the SARS‐CoV‐2 virus than the coronavirus obtained from the two pangolin samples (SRR10168377 and SRR10168378). We also identified a unique peptide (PRRA) insertion in the human SARS‐CoV‐2 virus, which may be involved in the proteolytic cleavage of the spike protein by cellular proteases, and thus could impact host range and transmissibility. Interestingly, the coronavirus carried by pangolins did not have the RRAR motif. Therefore, we concluded that the human SARS‐CoV‐2 virus, which is responsible for the recent outbreak of COVID‐19, did not come directly from pangolins.
Keywords: COVID‐19, cross‐species transmission, evolutionary rate, potential intermediate animal host, SARS‐CoV‐2, TMRCA
Highlights
We identified a unique peptide (PRRA) insertion in the human SARS‐CoV‐2 virus, which may be involved in the proteolytic cleavage of the spike protein by cellular proteases, and thus could impact host range and transmissibility.
1. INTRODUCTION
On 11 February 2020, the International Committee on Taxonomy of Viruses officially renamed the novel coronavirus (ie, previously 2019‐nCoV) responsible for the current outbreak of COVID‐19, severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). This was chosen based on analysis of the new coronavirus's evolutionary history and associated pathogen (ie, SARS‐CoV). The virus, which emerged in December 2019 in the Chinese city of Wuhan, causes a respiratory illness called COVID‐19, which can spread from person to person. 1 , 2 As of 21 February 2020, there have been 76 288 cases of SARS‐CoV‐2 confirmed in mainland China, including 11 477 serious, 2 345 deaths, and 20 659 discharged, as well as 68 cases in Hong Kong, 10 in Macao, and 26 in Taiwan. More than 1300 cases have also been confirmed in at least 27 other countries on four continents. World Health Organization (WHO) officials outlined their top research priorities for controlling the outbreak of the coronavirus‐associated disease known as COVID‐19 and highlighted the importance of developing candidate therapeutics and easy‐to‐apply diagnostics for identifying active, asymptomatic, and resolved infections. Of note, the Coronaviridae family not only includes SARS‐CoV‐2, but also SARS‐CoV, Middle East respiratory syndrome coronavirus (MERS‐CoV), and common cold viruses (eg, 229E, OC43, NL63, and HKU1). 3 The SARS‐CoV pathogen was responsible for >8 000 cases and 774 deaths in 37 countries during the 2002 to 2003 SARS outbreak, 4 , 5 , 6 and the MERS‐CoV pathogen was responsible for 2 494 cases and 858 deaths in 27 countries during the 2012 MERS outbreak. 7 , 8
Coronaviruses are known to circulate in mammals and birds. Previous studies revealed that both SARS‐CoV and MERS‐CoV are zoonotic in origin, originally coming from bats, 9 , 10 , 11 , 12 with SARS‐CoV spreading from bats to palm civets to humans, 13 , 14 , 15 and MERS‐CoV spreading from bats to camels to humans. 16 , 17 Recent research has also reported that the SARS‐CoV‐2 virus likely originated in bats, a proposal based on the similarity of its genetic sequence to that of other known coronaviruses. 18 However, like SARS‐CoV, MERS‐CoV, and many other coronaviruses, the SARS‐CoV‐2 virus may have been transmitted to humans by an intermediate animal host. 19 Therefore, the identity of the animal source of SARS‐CoV‐2 remains a key and urgent question. Furthermore, to stem future outbreaks of this type and preventing the transmission of zoonotic diseases to humans should be a top research priority.
The existence of an intermediate animal host of SARS‐CoV‐2 between a probable bat reservoir and humans is still under investigation. The discovery of a virus closely related to the newly emerged SARS‐CoV‐2 in a data set from pangolins sampled more than a year ago illustrates that the sampling of other mammals handled or consumed by humans could uncover even more closely related viruses. 20
During a press conference on 7 February 2020, two researchers (Shen Yongyi and Xiao Lihua) from the South China Agricultural University in Guangzhou identified the pangolin as a potential source of the SARS‐CoV‐2 virus based on genetic comparison of coronaviruses taken from pangolins and from humans infected during the recent outbreak. By analyzing more than 1 000 metagenomic samples and using molecular biology testing, they found that the positive rate of β coronavirus in pangolins was 70% and that the genome sequence of an isolated virus strain was 99% similar to that of the SARS‐CoV‐2 virus. Thus, whether pangolins acted as a direct intermediate animal host of the SARS‐CoV‐2 virus is worth further investigation.
In the present study, we performed analyses of the transmission dynamics and evolutionary history of the virus based on 70 genomes of SARS‐CoV‐2 strains sampled from Australia (n = 4), Belgium (n = 1), China (Hubei Province, n = 19; Guangdong Province, n = 16; Zhejiang Province, n = 4; Taiwan, n = 1), Finland (n = 1), France (n = 4), Germany (n = 1), Japan (n = 1), Korea (n = 1), Singapore (n = 3), Thailand (n = 2), United Kingdom (n = 2), and United States (n = 10) with sampling dates between 24 December 2019 and 3 February 2020. We reanalyzed two of the 21 pangolin metagenome samples from previously published data 20 and compared the amino acid sequences of the S protein of SARS‐CoV‐2 and SARS‐related coronaviruses. These analyses should extend our understanding of the origins and dynamics, cross‐species transmission, and subsequent host adaptation of the SARS‐CoV‐2 outbreak in China and elsewhere.
2. MATERIALS AND METHODS
2.1. Collation of SARS‐CoV‐2 genome data sets
As of 9 February 2020, 73 genomes of SARS‐CoV‐2 strains obtained from humans have been released on GISAID (http://gisaid.org/). 21 The BetaCoV/Wuhan/IPBCAMS‐WH‐02/2019 (EPI_ISL_403931), BetaCoV/Shenzhen/SZTH‐001/2020 (EPI_ISL_406592), and BetaCoV/Shenzhen/SZTH‐004/2020 (EPI_ISL_406595) samples show evidence of sequencing artefacts due to the appearance of clustered spurious single nucleotide polymorphisms (SNPs) and were thus excluded from this study. The final data set (“dataset_70”) included 70 genomes of SARS‐CoV‐2 strains from Australia (n = 4), Belgium (n = 1), China (n = 40), Finland (n = 1), France (n = 4), Germany (n = 1), Japan (n = 1), Korea (n = 1), Singapore (n = 3), Thailand (n = 2), UK (n = 2), and USA (n = 10) with sampling dates between 24 December 2019 and 3 February 2020. Of the 40 samples collected from China, 19 were from Hubei Province, 16 were from Guangdong Province, 4 were from Zhejiang Province, and 1 was from Taiwan (Table S1).
To investigate the potential intermediate hosts of SARS‐CoV‐2 (between originating animal and human hosts), two samples (SRR10168377 and SRR10168378) obtained from previously reported Malayan pangolin (Manis javanica) viral metagenomic sequencing data (Bio Project PRJNA573298) were downloaded from the NCBI SRA public database. 20 After assembly, the SRR10168377 and SRR10168378 genomes were 16 999 and 6 392 bp in length, respectively. We defined another data set (“dataset_6”) composed of six genome sequences of coronavirus strains. BetaCoV/Wuhan‐Hu‐1/2019 (EPI_ISL_402125) was grouped as “Clade A,” one (BetaCoV/bat/Yunnan/RaTG13/2013; EPI_ISL_402131) and two (bat‐SL‐CoVZC45; MG772933 and bat‐SL‐CoVZXC21; MG772934) SARS‐related coronaviruses were grouped as “Clade B” and “Clade D,” respectively. The two assembled genomes from SRR10168377 and SRR10168377 were grouped into “Clade C.” The two data sets (“dataset_70” and “dataset_6”) were aligned using MAFFT v7.222 22 and then manually curated using BioEdit v7.2.5. 23
2.2. Recombination and phylogenetic analyses
To assess the recombination of “dataset_70,” we employed the pairwise homoplasy index (PHI) to measure the similarity between closely linked sites using SplitsTree v4.15.1. 24 The best‐fit nucleotide substitution models for the two data sets were identified according to the Bayesian information criterion (BIC) method with 3 (24 candidate models) or 11 (88 candidate models) substitution schemes in jModelTest v2.1.10. 25 To evaluate the phylogenetic signals of “dataset_70” and “dataset_6,” we performed likelihood‐mapping analysis 26 using TREE‐PUZZLE v5.3, 27 with 25 000 to 175 000 randomly chosen quartets for the two data sets. For “dataset_70,” split network analysis was performed using Kishino‐Yano‐85 distance transformation with the NeighborNet method, which can be loosely thought of as a “hybrid” between the neighbor‐joining (NJ) and split decomposition methods, implemented in TREE‐PUZZLE v5.3. For “dataset_70,” NJ 28 phylogenetic trees were constructed using the Kimura 2‐parameter method 29 implemented in MEGA v7.0.26. 30 For “dataset_6,” NJ 28 phylogenetic trees were constructed using the Maximum Composite likelihood (MCL) method, 31 and rate variation among sites was modeled with a gamma distribution (shape parameter = 4) in MEGA v7.0.26. 30 For “dataset_70,” maximum‐likelihood (ML) phylogenies were reconstructed using the Hasegawa‐Kishino‐Yano (HKY) 29 nucleotide substitution model in PhyML v3.1. 32 For “dataset_6,” ML phylogenies were reconstructed using the general time reversible 33 nucleotide substitution model with gamma‐distributed rate variation among sites (GTR + G) model in PhyML v3.1. 32 For all NJ and ML phylogenies of the two data sets, bootstrap support values were calculated with 1 000 replicates 34 and trees were midpoint rooted. For “dataset_70,” regression analyses were used to determine the correlations among sampling dates and root‐to‐tip genetic divergences of the respective ML phylogenies with TempEst v1.5. 35 We also estimated the evolutionary rate and time to the most recent common ancestor (TMRCA) for “dataset_70” using ML dating in the TreeTime package. 36
2.3. Reconstruction of time‐scaled phylogenies
To reconstruct the evolutionary history of SARS‐CoV‐2, Bayesian inference through a Markov chain Monte Carlo (MCMC) framework was implemented in BEAST v1.8.4, 37 with the BEAGLE v2.1.2 library program 38 used for computational enhancement. We used two schemes to set the time‐scale prior for each data set: that is, constrained evolutionary rate method with a log‐normal prior (mean = 1.0 × 10−3 substitutions per site per year; 95% Bayesian credible interval [BCI]: 1.854 × 10−4 to 4 × 10−3 substitutions per site per year) placed on the evolutionary rate parameter, as per previous studies, 39 , 40 , 41 and the tip‐dating method, for which the overall estimated evolutionary rate was given an uninformative continuous‐time Markov chain (CTMC) reference prior. We ran Bayesian phylogenetic analyses using various clock model combinations (ie, strict clock and uncorrelated log‐normal relaxed clock 42 ) and coalescent tree priors (ie, constant size and exponential growth). To ensure adequate mixing of model parameters, MCMC chains were run for 100 million steps with sampling every 10 000 steps from the posterior distribution. Convergence was evaluated by calculating the effective sample sizes of the parameters using Tracer v1.7.1. 43 All parameters had an effective sample size >200, indicative of sufficient sampling. Trees were summarized as maximum‐clade credibility (MCC) trees using TreeAnnotator v1.8.4 after discarding the first 10% as burn‐in, and then visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree).
2.4. Transmission network reconstruction
The HIV TRAnsmission Cluster Engine (HIV‐TRACE; www.hivtrace.org) 44 was employed to infer transmission network clusters for SARS‐CoV‐2 “dataset_70.” All pairwise distances were calculated and the putative linkages between each pair of genomes were considered whenever their divergence was ≤0.0001 (0.01%) or ≤0.00001 (0.001%) substitutions/site (TN93 substitution model). Multiple linkages were then combined into putative transmission clusters. Clusters that comprised of only two linked nodes were identified as dyads. This approach detects transmission clusters in which the clustering strains are genetically similar, implying a direct or indirect epidemiological connection.
2.5. Similarity plot analysis
To investigate the putative parents of SARS‐CoV‐2, we performed similarity plot analysis based on the Kimura two‐parameter method 29 with a window size of 200 bp and step size of 20 bp using SimPlot v.3.5.14. 45 We divided “dataset_6” into four clades (ie, Clade A, Clade B, Clade C, and Clade D), with Clade A designated as the query group.
3. RESULTS
3.1. Demographic characteristics of SARS‐CoV‐2
“Dataset_70” included 70 genomes of SARS‐CoV‐2 strains sampled from Australia (n = 4), Belgium (n = 1), China (Hubei Province, n = 19; Guangdong Province, n = 16; Zhejiang Province, n = 4; Taiwan, n = 1), Finland (n = 1), France (n = 4), Germany (n = 1), Japan (n = 1), Korea (n = 1), Singapore (n = 3), Thailand (n = 2), UK (n = 2), and USA (n = 10) with sampling dates between 24 December 2019 and 3 February 2020 (Table S1). The samples were primarily from China (57.14%) and Hubei Province (27.14%), the Chinese Province was acknowledged as the original epicenter of the SARS‐CoV‐2 outbreak.
3.2. Tree‐like signals and phylogenetic analyses
For “dataset_70” and “dataset_6,” HKY and GTR + G were the models of best‐fit, respectively, across the two different substitution schemes (ie, 24 and 88 candidate models) according to the BIC method, and were thus used in subsequent likelihood‐mapping and phylogenetic analyses for the two data sets. The PHI tests of “dataset_70” did not find statistically significant evidence of recombination (P = 1.0). Likelihood‐mapping analysis of “dataset_70” revealed that 69.7% of the quartets were distributed in the center of the triangle, indicating a strong star‐like topology signal reflecting a novel virus, which may be due to exponential epidemic spread (Figure 1A). Likewise, 25.9% of the quartets from “dataset_6” were distributed in the center of the triangle, indicating a strong phylogenetic signal (Figure 1B). The split network generated for “dataset_70” using the NeighborNet method was highly unresolved, and the phylogenetic relationship of “dataset_70” was probably best represented by a network rather than a tree (Figure 1C). The existence of polytomies indicated—in contrast to that expected in a strictly bifurcating tree—an explosive, star‐like evolution of SARS‐CoV‐2. Both the NJ and ML phylogenetic analyses of SARS‐CoV‐2 “dataset_70” also showed star‐like topologies, in accordance with the likelihood‐mapping results (Figures 2 and S1). The ML phylogenetic tree showed greater star‐like topology than the NJ phylogenetic tree, indicating that the ML method was more reasonable for “dataset_70.” Root‐to‐tip regression analyses between genetic divergence and sampling date using the best‐fitting root showed that “dataset_70” had a minor strong positive temporal signal (R 2 = 0.0808; correlation coefficient = 0.2843) (Figure 3). This result suggests a minor clock‐like pattern of molecular evolution, with an estimated substitution rate of 3.3452 × 10−4 substitutions per site per year and TMRCA occurring on 19 October 2019. ML dating analyses between genetic divergence and sampling date also showed that “dataset_70” had a minor strong positive temporal signal (R 2 = 0.08) (Figure S2). The estimated evolutionary rate and TMRCA were 3.34 × 10−4 substitutions per site per year and 19 October 2019, respectively, in accordance with the root‐to‐tip regression results. Based on Bayesian time‐scaled phylogenetic analysis using the constrained evolutionary rate method with a log‐normal prior (mean = 1.0 × 10−3 substitutions per site per year; 95% BCI: 1.854 × 10−4 to 4 × 10−3 substitutions per site per year) placed on the evolutionary rate parameter, the estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 from “dataset_70” ranged from 21 May 2019 to 13 October 2019 (95% BCI: 27 and 30 January 2020) and from 1.57 × 10−4 to 1.06 × 10−3 substitutions per site per year (95% BCI: 1.08 × 10−4 to 3.10 × 10−3), respectively (Table 1). Furthermore, based on Bayesian time‐scaled phylogenetic analysis using the tip‐dating method, the estimated TMRCA dates and evolutionary rates from “dataset_70” ranged from 22 to 24 November 2019 (95% BCI: 23 October 2019 and 16 December 2019) and from 1.19 × 10−3 to 1.31 × 10−3 substitutions per site per year (95% BCI: 6.22 × 10−4 to 1.96 × 10−3), respectively (Table 1). Thus, the estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 from “dataset_70” were consistent among the different clock models (strict and relaxed) but were distinct among the different dating methods (constrained‐dating and tip‐dating). The estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 from “dataset_70” using the tip‐dating method exhibited much narrower 95% BCIs than the constrained‐dating method. In addition, the estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 from “dataset_70” were consistent between the different coalescent tree models (ie, constant and exponential) when using the tip‐dating method but were distinct when using the constrained‐dating method. For each data set, we employed the HKY nucleotide substitution model, as well as a constant size coalescent tree prior and strict molecular clock model to estimate the TMRCA. The estimates of the MCC phylogenetic relationships among the SARS‐CoV‐2 genomes from the Bayesian coalescent framework using the tip‐dating method, as well as the constant size coalescent tree prior and strict molecular clock, are displayed in Figure 4. As shown, eight phylogenetic clusters (number of sequences 2 to 7; posterior probability 0.99 to 1.0) were identified.
Figure 1.
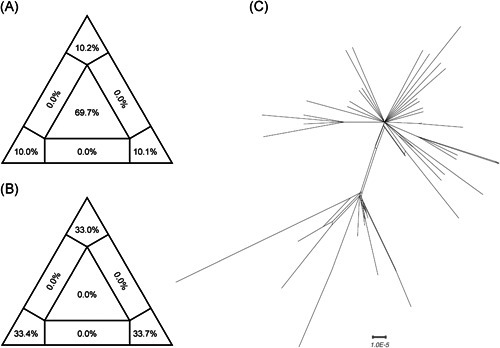
Likelihood‐mapping and split network analyses of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Likelihoods of three tree topologies for each possible quartet (or for a random sample of quartets) are denoted by data points in an equilateral triangle. Distribution of points in seven areas of the triangle reflects tree‐likeness of data. Specifically, three corners represent fully resolved tree topologies; center represents an unresolved (star) phylogeny; and sides represent support for conflicting tree topologies. Results of likelihood‐mapping analyses of two data sets (“dataset_70,” A; and “dataset_6,” B) and split network analyses of “dataset_70” (C) are shown
Figure 2.
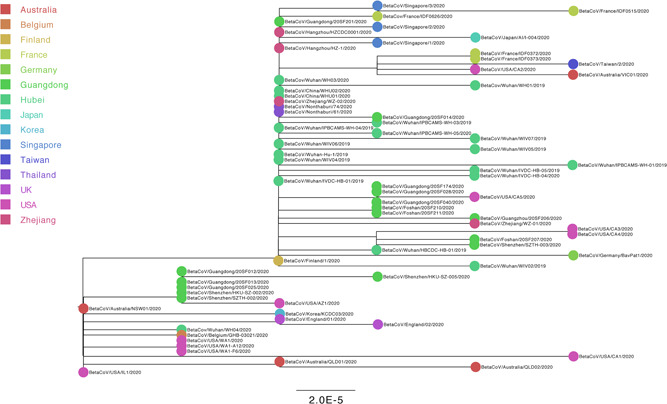
Estimated maximum‐likelihood phylogenies of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Colors indicate different sampling locations. Tree is midpoint rooted. Results of maximum‐likelihood phylogenetic analyses of “dataset_70” are shown
Figure 3.
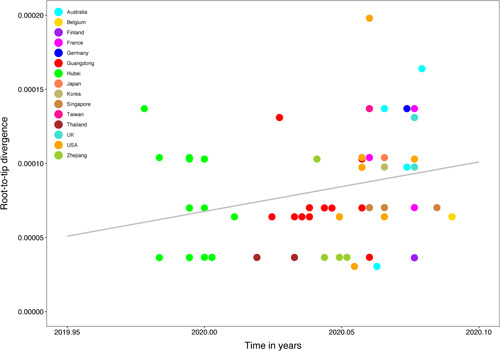
Regression of root‐to‐tip genetic distance against year of sampling for severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Colors indicate different sampling locations. Gray indicates linear regression line. Results of linear regression analyses of “dataset_70” are shown
Table 1.
Bayesian phylogenetic estimates of evolutionary parameters for genome sequences of 2019‐novel coronavirus (2019‐nCoV) under different clock models and coalescent tree priors
| Clock model | Clock prior | Coalescent tree prior | Substitution rate (substitutions per site per year) | Clade A MRCA | ||||
|---|---|---|---|---|---|---|---|---|
| Mean | Lower 95% HPD | Upper 95% HPD | Mean | Lower 95% HPD | Upper 95% HPD | |||
| Strict | Constraint‐dating | Constant | 1.06E‐03 | 1.24E‐04 | 3.10E‐03 | 2019‐10‐13 | 2019‐04‐05 | 2020‐01‐30 |
| Exponential | 1.58E‐04 | 1.10E‐04 | 2.27E‐04 | 2019‐05‐23 | 2019‐01‐31 | 2019‐09‐06 | ||
| Tip‐dating | Constant | 1.24E‐03 | 6.74E‐04 | 1.82E‐03 | 2019‐11‐24 | 2019‐10‐29 | 2019‐12‐15 | |
| Exponential | 1.19E‐03 | 6.22E‐g04 | 1.81E‐03 | 2019‐11‐22 | 2019‐10‐23 | 2019‐12‐15 | ||
| Relaxed | Constraint‐dating | Constant | 1.01E‐03 | 1.11E‐04 | 3.01E‐03 | 2019‐09‐29 | 2019‐03‐05 | 2020‐01‐29 |
| Exponential | 1.57E‐04 | 1.08E‐04 | 2.26E‐04 | 2019‐05‐21 | 2019‐01‐27 | 2019‐09‐05 | ||
| Tip‐dating | Constant | 1.31E‐03 | 7.40E‐04 | 1.96E‐03 | 2019‐11‐24 | 2019‐10‐25 | 2019‐12‐16 | |
| Exponential | 1.28E‐03 | 6.46E‐04 | 1.92E‐03 | 2019‐11‐23 | 2019‐10‐24 | 2019‐12‐16 | ||
Abbreviations: 2019‐nCoV, 2019‐novel coronavirus; HPD, highest posterior density; MRCA, most recent common ancestor.
Figure 4.
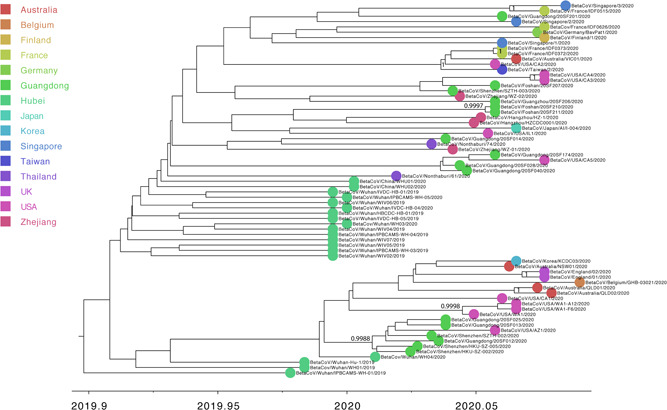
Estimated maximum‐clade‐credibility tree of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) using tip‐dating method. Colors indicate different sampling locations. Nodes are labeled with posterior probability values. Estimated maximum‐clade credibility (MCC) tree of “dataset_70” are shown
3.3. Transmission network analysis
We considered individuals as genetically linked when the genetic distance between SARS‐CoV‐2 strains was <0.01% substitutions/site. Based on this, we identified one large transmission cluster that included 66 of 70 (94.29%) genomes, thus suggesting low genetic divergence for “dataset_70” (Figure S3). We also considered individuals as genetically linked when the genetic distance between SARS‐CoV‐2 strains was <0.001% substitutions/site. Based on this, we identified 6 transmission clusters that included 37 of 70 (52.86%) genomes for “dataset_70” (Figure 5). Clusters ranged in size from 2 to 23 genomes.
Figure 5.
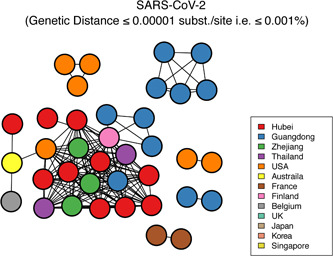
Transmission clusters of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Structure of inferred SARS‐CoV‐2 transmission clusters from “dataset_70” using genetic distances of <0.001% substitutions/site is shown. Nodes (circles) represent connected individuals in overall network, and putative transmission linkages are represented by edges (lines). Nodes are color‐coded by sampling locations
3.4. Potential intermediate host analyses for SARS‐CoV‐2
The NJ and ML phylogenetic topologies of “dataset_6” were consistent with each other (Figure S4), indicating that the use of a small number of sequences could show similar topological results. Homology plot analysis of “dataset_6” also revealed that BetaCoV/bat/Yunnan/RaTG13/2013 was more similar to the SARS‐CoV‐2 virus than the coronavirus obtained from the two pangolin samples (SRR10168377 and SRR10168378), consistent with phylogenetic analysis (Figure S5). Of note, “Clade D” (bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21) had higher similarity to the SARS‐CoV‐2 virus in the first 12 000 bp region of the full alignment than to the pangolin coronavirus (Figure S5). We also found that a unique peptide (PRRA) insertion region in the spike protein at the junction of S1 and S2 junction in the human SARS‐CoV‐2 virus (“Clade A”) induced a furin cleavage motif (RRAR), which could be a predicted polybasic cleavage site, and thus a unique feature of SARS‐CoV‐2, in comparison with the other three clades (“Clade B,” “Clade C,” and “Clade D”) (Figure S6).
4. DISCUSSION
On the basis of “dataset_70,” our likelihood‐mapping analysis confirmed additional tree‐like signals over time compared with our previous results. 46 , 47 This result implies increasing genetic divergence of SARS‐CoV‐2 in human hosts (Figure 1A), consistent with the findings of our earlier studies. 46 , 47 Split network analysis for SARS‐CoV‐2 “dataset_70” using the NeighborNet method was highly unresolved, indicating an explosive, star‐like evolution of SARS‐CoV‐2, and recent and rapid human‐to‐human transmission (Figure 1C). These results are consistent with the ML phylogenetic analyses, which showed polytomy topology from “dataset_70” (Figure 2). However, NJ phylogenetic analyses showed a more bifurcating tree topology compared with the ML phylogenetic analyses (Figure S1). This is a good example showing the differences between NJ and ML phylogenetic construction methods. “Dataset_70” had a minor strong positive temporal signal based on root‐to‐tip regression and ML dating analyses (Figures 3 and S2), with the estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 found to be nearly identical using both analyses (Table 1). The estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 were very similar across different clock models and coalescent tree priors using the tip‐dating method. The estimated TMRCA dates and evolutionary rates for SARS‐CoV‐2 were also very similar across different clock models using the constrained‐dating method, but highly distinct across the different coalescent tree priors (Table 1). The TMRCA estimated by the tip‐dating method was relatively narrower than that determined by the constrained‐dating method, consistent with our previous studies. 46 , 47 Bayesian analyses with the tip‐dating method using a strict clock as well as constant size coalescent tree prior indicated that SARS‐CoV‐2 is evolving at a rate of 1.24 × 10−3 substitutions per site per year (Table 1), in accordance with our prior research 46 , 47 and similar to that found for other human coronaviruses. 41 Our results also suggest that the virus originated on 24 November 2019, which is in further agreement with our earlier studies. 46 , 47 We identified eight phylogenetic clusters (number of sequences 2 to 7) with posterior probabilities between 0.99 and 1.0 using Bayesian inference (Figure 4). We also identified six transmission clusters (number of sequences 2 to 23) when the genetic distance between the SARS‐CoV‐2 strains was <0.001% substitutions/site (Figure 5). However, our conclusions should be considered preliminary and explained with caution due to the limited number of SARS‐CoV‐2 genome sequences presented in this study. As more genome sequences become available, there may be stronger among‐lineage rate variation over time as to warrant using a relaxed clock model, but we anticipate that the evolutionary rates and TMRCA dates will be broadly similar to those estimated here. As the number of substitutions is still small, it is tempting to speculate that sequencing errors could have a considerable impact on the evolutionary rate and TMRCA date estimates. We removed three SARS‐CoV‐2 genome sequences (ie, BetaCoV/Wuhan/IPBCAMS‐WH‐02/2019, EPI_ISL_403931; BetaCoV/Shenzhen/SZTH‐001/2020, EPI_ISL_406592; BetaCoV/Shenzhen/SZTH‐004/2020, EPI_ISL_406595) with potential sequencing errors, but these may have less impact on the above estimates when more substitutions of SARS‐CoV‐2 are accumulated over time. We also expect that as more SARS‐CoV‐2 genome sequences become available, the estimated 95% BCIs of the evolutionary rates and TMRCA dates will be narrower.
We found that the Pangolin‐CoV virus from the two pangolin samples was clustered with the SARS‐CoV‐2 virus with 100% bootstrap support; however, BetaCoV/bat/Yunnan/RaTG13/2013 was more similar to the SARS‐CoV‐2 virus than to the pangolin coronavirus and the human SARS‐CoV‐2 virus (“Clade A”) showed a unique peptide (PRRA) insertion not found in the other three clades (“Clade B,” “Clade C,” “Clade D”). This insertion constitutes an RRAR motif in the spike protein at the junction of S1 and S2 junction in the human SARS‐CoV‐2 virus, after considering the next amino acid (R) of the unique peptide (PRRA) (Figure S6). Of note, the highly favored motifs for furin cleavage are Arg‐X‐(Arg/Lys)‐Arg (RXRR or RXKR), and the minimal motifs for furin cleavage can be RXXR. 48 We also note that some of the other coronaviruses have a furin motif in almost the same location in their spike proteins. 49 , 50 Lentiviruses have an RKXR (R, arginine; K, lysine; X, any amino acid) site between gp120 and gp41, cleaved by furin to convert gp160 into subunits. 51 , 52 , 53 , 54 Therefore, it is tempting to speculate that cleavage or lack of cleavage of the spike protein at this site could significantly impact host range and transmissibility. Taken together, the pangolin coronavirus samples (SRR10168377 and SRR10168378) were less similar to the SARS‐CoV‐2 virus than to the BetaCoV/bat/Yunnan/RaTG13/2013 virus and did not have the RRAR motif. Therefore, we concluded that the human SARS‐CoV‐2 virus, which is responsible for the current outbreak of COVID‐19, did not come directly from pangolins. However, due to the limited viral metagenomic data obtained from pangolins, we cannot exclude that other pangolins from China may contain coronaviruses that exhibit greater similarity to the SARS‐CoV‐2 virus.
In conclusion, our results emphasize the importance of further epidemiological investigations, genomic data surveillance, and experimental studies of the role of the unique furin cleavage motif (RRAR) of SARS‐CoV‐2 in the spike protein at the junctions of S1 and S2. Such work could positively impact public health in terms of guiding prevention efforts to reduce SARS‐CoV‐2 transmission in real time, and to stem future outbreaks of zoonotic diseases.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
XL conceived and designed the study and drafted the manuscript. XL, BF, and AC analyzed the data. XL, QN, JZ, QZ, YL, BF, and AC interpreted the data and provided critical comments. All authors reviewed and approved the final manuscript.
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
{kind=link}
Supporting information
Supporting information
ACKNOWLEDGMENTS
This study was supported by a grant from the National Natural Science Foundation of China (No. 31470268) to Prof. Yi Li. This study was sponsored by the K.C. Wong Magna Fund in Ningbo University. We gratefully acknowledge the authors and Originating and Submitting Laboratories for their sequences and meta‐data shared through GISAID, 21 on which this study is based.
Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2. J Med Virol. 2020;92:602–611. 10.1002/jmv.25731
Xingguang Li, Junjie Zai, and Qiang Zhao contributed equally to this study.
Contributor Information
Xingguang Li, Email: xingguanglee@hotmail.com.
Yi Li, Email: yujp@wh.iov.cn.
Brian T. Foley, Email: Btf@lanl.gov.
Antoine Chaillon, Email: achaillon@health.ucsd.edu.
REFERENCES
- 1. Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. Lancet. 2020;395:514‐523. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus‐infected pneumonia. N Engl J Med. 2020:NEJMoa2001316. 10.1056/NEJMoa2001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490‐502. 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967‐1976. 10.1056/NEJMoa030747 [DOI] [PubMed] [Google Scholar]
- 5. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953‐1966. 10.1056/NEJMoa030781 [DOI] [PubMed] [Google Scholar]
- 6. Zhong N, Zheng B, Li Y, et al. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362:1353‐1358. 10.1016/s0140-6736(03)14630-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814‐1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 8. de Groot RJ, Baker SC, Baric RS, et al. Middle East respiratory syndrome coronavirus (MERS‐CoV): announcement of the Coronavirus Study Group. J Virol. 2013;87:7790‐7792. 10.1128/JVI.01244-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lau SKP, Li KSM, Huang Y, et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome‐related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self‐limiting infection that allows recombination events. J Virol. 2010;84:2808‐2819. 10.1128/JVI.02219-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guan Y, Zheng BJ, He YQ, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276‐278. 10.1126/science.1087139 [DOI] [PubMed] [Google Scholar]
- 11. Lau SKP, Woo PCY, Li KSM, et al. Severe acute respiratory syndrome coronavirus‐like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040‐14045. 10.1073/pnas.0506735102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li W, Shi Z, Yu M, et al. Bats are natural reservoirs of SARS‐like coronaviruses. Science. 2005;310:676‐679. 10.1126/science.1118391 [DOI] [PubMed] [Google Scholar]
- 13. Song HD, Tu CC, Zhang GW, et al. Cross‐host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. 2005;102:2430‐2435. 10.1073/pnas.0409608102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chinese SMEC. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666‐1669. 10.1126/science.1092002 [DOI] [PubMed] [Google Scholar]
- 15. Wang M, Yan M, Xu H, et al. SARS‐CoV infection in a restaurant from palm civet. Emerg Infect Dis. 2005;11:1860‐1865. 10.3201/eid1112.041293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Müller MA, Corman VM, Jores J, et al. MERS coronavirus neutralizing antibodies in camels, Eastern Africa, 1983‐1997. Emerg Infect Dis. 2014;20:2093‐2095. 10.3201/eid2012.141026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chu DKW, Poon LLM, Gomaa MM, et al. MERS coronaviruses in dromedary camels, Egypt. Emerg Infect Dis. 2014;20:1049‐1053. 10.3201/eid2006.140299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou P, Yang X‐L, Wang X‐G, et al. Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. Preprint at BioRxiv. 2020. 10.1101/2020.01.22.914952 [DOI] [Google Scholar]
- 19. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu P, Chen W, Chen JP. Viral metagenomics revealed sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses. 2019;11:979. 10.3390/v11110979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elbe S, Buckland‐Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 2017;1:33‐46. 10.1002/gch2.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall TA. BioEdit: a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95‐98. https://doi.org/citeulike‐article‐id:691774 [Google Scholar]
- 24. Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254‐267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- 25. Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9:772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt HA, von Haeseler A. Maximum‐likelihood analysis using TREE‐PUZZLE. Curr Protoc Bioinformatics. 2007;6:6. 10.1002/0471250953.bi0606s17 Chapter 6, Unit 6 [DOI] [PubMed] [Google Scholar]
- 27. Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE‐PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502‐504. 10.1093/bioinformatics/18.3.502 [DOI] [PubMed] [Google Scholar]
- 28. Saitou N, Nei M. The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406‐425. 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- 29. Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111‐120. 10.1007/bf01731581 [DOI] [PubMed] [Google Scholar]
- 30. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870‐1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor‐joining method. Proc Natl Acad Sci USA. 2004;101:11030‐11035. 10.1073/pnas.0404206101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum‐likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307‐321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 33. Lanave C, Preparata G, Saccone C, Serio G. A new method for calculating evolutionary substitution rates. J Mol Evol. 1984;20:86‐93. 10.1007/bf02101990 [DOI] [PubMed] [Google Scholar]
- 34. Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783‐791. 10.1111/j.1558-5646.1985.tb00420.x [DOI] [PubMed] [Google Scholar]
- 35. Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evol. 2016;2:vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sagulenko P, Puller V, Neher RA. TreeTime: maximum‐likelihood phylodynamic analysis. Virus Evol. 2018;4:vex042. 10.1093/ve/vex042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969‐1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suchard MA, Rambaut A. Many‐core algorithms for statistical phylogenetics. Bioinformatics. 2009;25:1370‐1376. 10.1093/bioinformatics/btp244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Z, Li H, Wu X, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. 2004;4:21. 10.1186/1471-2148-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cotten M, Watson SJ, Kellam P, et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382:1993‐2002. 10.1016/S0140-6736(13)61887-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cotten M, Watson SJ, Zumla AI, et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio. 2014;5, 10.1128/mBio.01062-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. 10.1371/journal.pbio.0040088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol. 2018;67:901‐904. 10.1093/sysbio/syy032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kosakovsky Pond SL, Weaver S, Leigh Brown AJ, Wertheim JO. HIV‐TRACE (TRAnsmission Cluster Engine): a tool for large scale molecular epidemiology of HIV‐1 and other rapidly evolving pathogens. Mol Biol Evol. 2018;35:1812‐1819. 10.1093/molbev/msy016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lole KS, Bollinger RC, Paranjape RS, et al. Full‐length human immunodeficiency virus type 1 genomes from subtype C‐infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73:152‐160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li X, Wang W, Zhao X, et al. Transmission dynamics and evolutionary history of 2019‐nCoV. J Med Virol. 2020:jmv.25701. 10.1002/jmv.25701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X, Zai J, Wang X, Li Y. Potential of large 'first generation' human‐to‐human transmission of 2019‐nCoV. J Med Virol. 2020;92:448‐454. 10.1002/jmv.25693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li W, Wicht O, van Kuppeveld FJM, He Q, Rottier PJM, Bosch BJ. A single point mutation creating a furin cleavage site in the spike protein renders porcine epidemic diarrhea coronavirus trypsin independent for cell entry and fusion. J Virol. 2015;89:8077‐8081. 10.1128/JVI.00356-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jaimes JA, Millet JK, Goldstein ME, Whittaker GR, Straus MR. A fluorogenic peptide cleavage assay to screen for proteolytic activity: applications for coronavirus spike protein activation. J Vis Exp. 2019. 10.3791/58892 [DOI] [PubMed] [Google Scholar]
- 50. Kleine‐Weber H, Elzayat MT, Hoffmann M, Pohlmann S. Functional analysis of potential cleavage sites in the MERS‐coronavirus spike protein. Sci Rep. 2018;8:16597. 10.1038/s41598-018-34859-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Falcigno L, Oliva R, D'Auria G, et al. Structural investigation of the HIV‐1 envelope glycoprotein gp160 cleavage site 3: role of site‐specific mutations. ChemBioChem. 2004;5:1653‐1661. 10.1002/cbic.200400181 [DOI] [PubMed] [Google Scholar]
- 52. Moulard M, Decroly E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim Biophys Acta. 2000;1469:121‐132. 10.1016/s0304-4157(00)00014-9 [DOI] [PubMed] [Google Scholar]
- 53. Moulard M, Hallenberger S, Garten W, Klenk HD. Processing and routage of HIV glycoproteins by furin to the cell surface. Virus Res. 1999;60:55‐65. 10.1016/s0168-1702(99)00002-7 [DOI] [PubMed] [Google Scholar]
- 54. Decroly E, Vandenbranden M, Ruysschaert JM, et al. The convertases furin and PC1 can both cleave the human immunodeficiency virus (HIV)‐1 envelope glycoprotein gp160 into gp120 (HIV‐1 SU) and gp41 (HIV‐I TM). J Biol Chem. 1994;269:12240‐12247. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information
Supporting information