

Abstract
The current pandemic caused by the new coronavirus is a worldwide public health concern. To aboard this emergency, and like never before, scientific groups around the world have been working in a fast and coordinated way to get the maximum of information about this virus when it has been almost 3 months since the first cases were detected in Wuhan province in China. The complete genome sequences of around 450 isolates are available, and studies about similarities and differences among them and with the close related viruses that caused similar epidemics in this century. In this work, we studied the complete genome of the first four cases of the new coronavirus disease in Chile, from patients who traveled to Europe and Southeast Asia. Our findings reveal at least two different viral variants entries to Chilean territory, coming from Europe and Asia. We also sub‐classified the isolates into variants according to punctual mutations in the genome. Our work contributes to global information about transmission dynamics and the importance to take control measures to stop the spread of the infection.
Keywords: COVID‐19, phylogeny, SARS‐CoV‐2
Highlights
First genetic study of the SARS‐CoV‐2 isolates in Chile.
At least two different viral variants were identified from Chilean travelers.
S and G Chilean variants are associated to European and Asian viral isolates.
1. INTRODUCTION
Mankind is facing a new viral outbreak that originated in the Wuhan province, Hubei region in China. The new virus, a coronavirus named SARS‐CoV‐2, was reported in December 2019. Since then, it has reached over 110 countries and territories with more than 125 000 reported cases at the time of this report. 1 This new coronavirus disease (COVID‐19) has caused more than 3100 deaths, mainly in continental China and mostly on elderly people, who are affected by fever and serious respiratory diseases like pneumonia. 2 , 3 , 4
SARS‐CoV‐2 has a single‐stranded RNA genome and its length is similar to other related coronaviruses, with an extension near 29 890 bp (GenBank NC_045512.2). The most related genomes available in public databases were bat‐SL‐CoVZC45 (GenBank MG772933) with an 87.99% sequence identity and bat‐SL‐CoVZXC21 (GenBank MG772934) with an 87.23% sequence identity, 5 followed by the human viruses SARS‐CoV‐Tor2 (GenBank NC_004718) and MERS‐CoV (GenBank NC_019843) with a 79.0% and 51.8% of nucleotide identity, respectively. 6
The genome organization of SARS‐CoV‐2 was shown to be similar of the related bats and human coronavirus. The open reading frames (ORFs) from 5′ to 3′ is as follows: 5′ UTR; ORF1ab with 16 nonstructural proteins (nsp) 1 to 16 including RNA polymerase RNA‐dependent nsp12, Helicase nsp13 and 3′‐to‐5′ exonuclease nsp14, S surface spike protein, E envelope protein, M membrane protein, and N nucleocapsid protein. There are also at least six predicted ORFs as hypothetical proteins with no associated function. 4 , 6 , 7
Near 286 complete genomes of SARS‐CoV‐2 and related viruses, has been submitted to the GISAID database (www.gisaid.org/CoV2020) collecting genetic information of the outbreak worldwide. The genomic sequences of all SARS‐CoV‐2 viruses isolated from patients share a sequence identity about a 99.9%, 7 suggesting a recently zoonotic infection, originated most probably from bats. 5 , 6 , 8
As the information appears daily, new insights and concepts are being adopted and implemented. Recently Tang et al 9 and GISAD database in the SARS‐CoV‐2 portal defined three subtypes: S, G, and V, according to nucleotide variants that produce amino acid changes. These changes are located in ORF8 L84S; S (spike protein) D614G and nsp3 G251V, in the nucleotide position 28144, 23403, and 3471, respectively, for S, G, and V, according to the reference sequence NC_045512.2.
Chile was the fourth country in South America after Brazil, Ecuador, and Argentina to report COVID‐19 in the region. In this report, we present the sequence analysis for the first four complete genomes for SARS‐CoV‐2 isolates on Chilean patients. Also, a phylogenetic study was performed with worldwide SARS‐CoV‐2 sequences and the full genomes from Chilean isolates, to identify their genetic similarity.
2. MATERIALS AND METHODS
2.1. Epidemiological information
The four cases presented in this report have contracted the infection abroad, either in Southeast Asia or Europe. The first two cases (20‐18918, 20‐19303) correspond to a couple in their early thirties, who traveled from Chile to Barcelona, Spain, where they stayed between January 27th and January 30th. On January 31th they traveled to Singapore, between February 4th to February 12th they were in Indonesia, then they visited Malaysia on February 13th, and the Maldives on February 15th. Between February 21st and 24th they stayed in Madrid, from where they traveled back to Chile arriving in Santiago on February 25th. The man showed symptoms first and was diagnosed as the first SARS‐CoV‐2 case in Chile, followed by his spouse 1 day later. That same day, a third case (20‐19305) was confirmed. A 56‐year‐old woman who visited London between February 22nd and 23rd, Venice (February 23rd ‐25th), London (February 25th‐28th), Madrid (February 28th to March 3rd) when she returned to Santiago. On the next day (March 5th) the fourth case (20‐19731) was reported, a 40‐year‐old woman, who traveled to Milan between February 25th and 29th, traveling back to Chile.
2.2. Sample types, RNA extraction, and virus detection
Chilean law by the Supreme Decree 7/2019 mandates notification of communicable diseases and their surveillance. All cases showed mild symptoms, and throat swab specimens were collected. A volume of 140 µL of each sample was used for viral RNA extraction with QIAamp Viral Mini Kit (Cat. No. 52926; Qiagen) in a QIACube extractor. All suspicious cases were confirmed by real‐time reverse transcription‐polymerase chain reaction (RT‐PCR), using specific probe and primers, synthesized and purified in our facilities, targeting the RNA‐dependent RNA polymerase (RdRp) region of SARS‐CoV‐2, according to the guidelines suggested by the World Health Organization. 10 SuperScript III One‐Step RT‐PCR Platinum Taq DNA Polymerase (Cat. No. 12574026; Invitrogen) was used for real‐time RT‐PCR. Running method 55°C for 10 minutes, followed by 94°C for 2 minutes, and 45 cycles at 94°C for 15 seconds and 58°C for 30 seconds. Ct's under 35 were considered as positive cases.
2.3. Full viral genome amplification
From total RNA extraction we performed the first amplification round using SuperScript III One‐Step RT‐PCR Platinum Taq DNA Polymerase (Invitrogen) and six pair of specific primers to obtain six complementary DNA fragments around 5 Kbp each, followed by a second amplification round with 24 specific primers (Table S1) to generate two fragments from each first round products, each subfragment (a total of 12) are around 2.3 to 2.7 Kbp.
2.4. Library generation and sequencing
The 12 DNA fragments from full genome amplification were pooled, and libraries were prepared with the Nextera XT Library Prep Kit (Illumina, San Diego, CA), purified with Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) and quantified by Victor Nivo Fluorometer (PerkinElmer) using Quant‐it dsDNA HS Assay kit (Invitrogen). The resulting DNA libraries were sequenced on MiSeq (Illumina) using a 300‐cycle reagent kit. About 0.3 GB of data was obtained for each sample.
2.5. Phylogenetic analysis
The sequencing quality was analyzed with software Fastqc v0.11.8 and then, the reads were filtered and trimmed using BBDuk software considering a minimum read length of 36 bases and quality more than equal to 10. SARS‐CoV‐2 assembly was performed with IRMA v0.9.3 using as reference NCBI sequence ID NC_045512.2. Sequence alignment was performed with MAFFT. The phylogenetic tree was built with IQ‐TREE v1.6.12 considering a bootstrap of 1000. We consider 218 full complete genome sequences available in the GISAID platform plus the full genome sequences from the first four Chilean cases.
3. RESULTS
The first four cases in Chilean territory were reported between March 3rd and 5th. All of these persons reported travel to places where the presence of the virus was confirmed and with an increasing number of cases.
We identified the SNPs that generates amino acid changes in all four Chilean genomes (Table 1). For the first and second samples (couple) the sequences are identical and the SNPs generates nonsynonymous mutations in ORF1ab‐nsp14‐3′‐to‐5′ exonuclease (V290F), ORF8 (L84S) and N‐nucleocapsid phosphoprotein (D103Y). The third Chilean case present mutations in the ORF1ab‐transmembrane domain 2 TM2 (F308Y), ORF3a (G193V), ORF8 (L84S), and N‐nucleocapsid phosphoprotein (S197L). The fourth case present mutations in ORF1ab‐ nsp3‐papain‐like proteinase (A225V), ORF1ab‐nsp12‐RNA‐dependent RNA polymerase (P323L), S‐surface spike glycoprotein (D614G), and in the N‐nucleocapsid phosphoprotein twice (R203K, G204R).
Table 1.
Nucleotide substitutions for Chilean virus isolates compared to the reference strain NC_045512.2
| SARS‐CoV‐2 sample | Nucleotide position | Base change | Open reading frame | Amino acid substitution |
|---|---|---|---|---|
| 20‐18918 | 8782 | C→T | ORF1ab‐transmembrane domain 2 (TM2) | Silent (S) |
| 17470 | C→T | ORF1ab‐nsp13‐helicase (HEL) | Silent (L) | |
| 18907 | G→T | ORF1ab‐nsp14‐3′‐to‐5′ exonuclease | V290F | |
| 26088 | C→T | ORF3a | Silent (I) | |
| 28144 | T→C | ORF8 | L84S | |
| 28580 | G→T | N–Nucleocapsid phosphoprotein | D103Y | |
| 20‐19303 | 8782 | C→T | ORF1ab‐transmembrane domain 2 (TM2) | Silent (S) |
| 17470 | C→T | ORF1ab‐nsp13‐helicase (HEL) | Silent (L) | |
| 18907 | G→K | ORF1ab‐nsp14‐3′‐to‐5′ exonuclease | No change/V290F | |
| 26088 | C→T | ORF3a | Silent (I) | |
| 28144 | T→C | ORF8 | L84S | |
| 28580 | G→T | N‐Nucleocapsid phosphoprotein | D103Y | |
| 20‐19305 | 1884 | C→Y | ORF1ab–nsp2 | Silent/A540V |
| 8782 | C→T | ORF1ab‐transmembrane domain 2 (TM2) | Silent (S) | |
| 9477 | T→A | ORF1ab‐transmembrane domain 2 (TM2) | F308Y | |
| 14807 | C→T | ORF1ab‐nsp12‐RNA‐dependent RNA polymerase | Silent (Y) | |
| 25979 | G→T | ORF3a | G193V | |
| 28144 | T→C | ORF8 | L84S | |
| 28657 | C→T | N–Nucleocapsid phosphoprotein | Silent (D) | |
| 28863 | C→T | N–Nucleocapsid phosphoprotein | S197L | |
| 20‐19371 | 241 | C→T | 5′ UTR | … |
| 3037 | C→T | ORF1ab–nsp3–papain‐like proteinase | Silent (F) | |
| 3393 | C→T | ORF1ab–nsp3–papain‐like proteinase | A225V | |
| 14408 | C→T | ORF1ab‐nsp12‐RNA‐dependent RNA polymerase | P323L | |
| 23403 | A→G | S‐Surface Glycoprotein (Spike) | D614G | |
| 28881 | G→A | N–Nucleocapsid phosphoprotein | R203K | |
| 28882 | G→A | N–Nucleocapsid phosphoprotein | R203K | |
| 28883 | G→A | N–Nucleocapsid phosphoprotein | G204R |
According to prevalent SNPs, all genomes have been classified by amino acid changes in specific ORFs. For the Chilean strains, the first three cases (20‐18918, 20‐19303, 20‐19305) are classified as “S” type, meanwhile, the fourth case (20‐19731) is a “G” type, according to nucleotide substitutions in the positions 28 144 and 23 403, respectively.
A maximum‐likelihood phylogeny tree was constructed using 218 complete genome sequences plus the four Chilean cases. Our first two samples, the married couple, mapped together (100% nucleotide identity) and with strains from Wuhan, China and Taiwan. The third sample groups in a well‐defined clade with Spanish isolates. The fourth Chilean strain, groups in a European clade with samples from Switzerland, Netherlands, and Germany. In this same clade, we can identify one of the Brazilian and Mexican isolates, representing isolates from Latin America. In addition, the complete genomes were colored according to the variant groups, defined by specific mutations (Figure 1).
Figure 1.
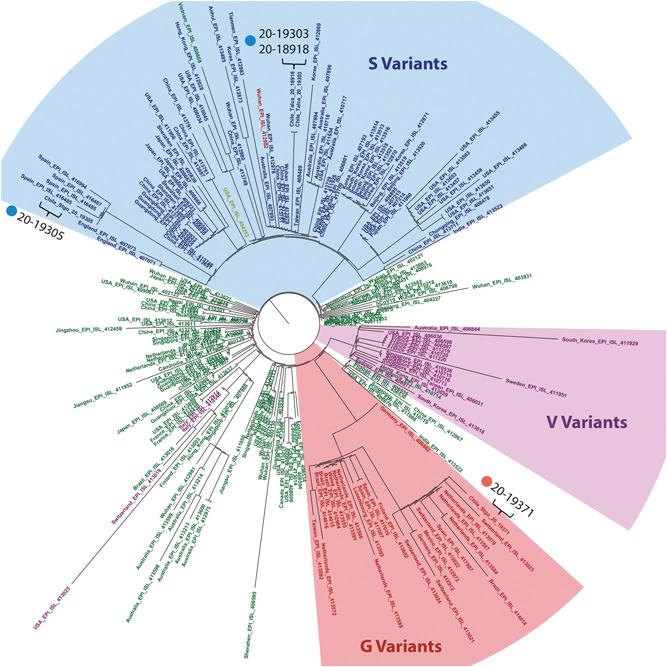
Phylogenetic tree with Maximum Composite Likelihood distance, representing 222 complete genomes including the four Chilean isolates. The name of the isolates were colored according to the variants as follows: S (blue), G (red), V (purple), unclassified variants (green), and the main clades were highlighted. Chilean strains are marked over the cladogram in the S and G variant clades
4. DISCUSSION
In this early stage of the epidemic, sharing data and information is crucial and the efforts of the scientist worldwide are admirable. After a few weeks since the outbreak started in Wuhan province, the full genome sequence of SARS‐CoV‐2 was available, and this information paved the way for the development of better detection protocols techniques, antiviral strategies, and phylogeny studies, among others scientific challenges. Here we report the first four cases of COVID‐19 and the complete genome sequencing for these strains. We developed an RT‐PCR based strategy to amplify the whole genome in two steps, followed by the library construction and further NGS using Illumina MiSeq. In less than 5 days since we detected the first case, the whole genomes for the first four cases were assembled. We implemented in early January the complete detection system by real‐time RT‐PCR for this new virus in the Public Health Institute of Chile, according to international guidelines.
The phylogenetic analysis plus the travel information of each patient, allows us to infer about the viral entries to Chilean territory. We detected two different viral variants entries to Chile, the S and G. For S variant, the viral genome distribution of Chilean isolates allows to associate these in two different clades, one related to the Wuhan province in China and Taiwan, and a second clade related with Spanish isolates, coincident with the patient's travel record. COVID‐19 cases in Spain started to be reported since February 1st with a very low number of cases for about a month, where the reported cases may be underestimated, and after a month the infected people started to raise over a hundred patients. 11 , 12 In the case of the G variant entry, the infected patient visited Europe and the complete sequence genome for this case, groups with isolates from the Netherlands, Switzerland, and Spain among other European countries (Figure 1). At the date of this report, we have detected more than 30 positive cases, mainly from Chilean travelers returning to the country and local transmission between their closest relatives.
At the beginning of the pandemic (until mid‐February), when 99% of the cases were focused in continental China, the death toll was about 2%. As the virus is spread by travelers, the number of cases and death occurrence has increased in other territories like South Korea, Iran, and Italy. In this last country the death toll up to date reaches the 6.2%, this number is still far from statistics of other related human coronavirus epidemics, like SARS (9.5%) and MERS (34.4%). 13 Until now, there is no enough evidence to relate specific mutation in the viral genome to a higher number of infected patients or even death, the main number of fatalities is still related to the elderly population.
In conclusion, our work presents the complete genome analysis for the first cases of COVID‐19 in Chile, detecting at least two different viral variants entries to Chilean territory. This information contributes to monitoring the spread of the infection and the surveillance for eventual recombination or genome mutations that the diversity of host, countries, weather conditions and other selective pressures that this new coronavirus could face. The globalization, increment of worldwide travelers and the high contagious rate of this virus require severe control measures to control infection dissemination.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
CAE participated in conceptualization, study design, interpreting the data analysis, methodology design, visualization, and wrote the whole manuscript. PB participated in methodology design and experimental assays, TP and TJ participated in data analysis and bioinformatics support. LJ, AL, and BG contributed to genome sequencing. AA, AW, LG, TC, and BP, participate in sample processing and real‐time RT‐PCR assays. US and FR participated in the critical review of the content. FJ contributed to the conceptualization, study design, supervision, critical review of the content, and approved the final version of the manuscript.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors are thankful to María Ibañez and Jorge Lobos for their valuable technical assistance.
Castillo AE, Parra B, Tapia P, et al. Phylogenetic analysis of the first four SARS‐CoV‐2 cases in Chile. J Med Virol. 2020;92:1562–1566. 10.1002/jmv.25797
REFERENCES
- 1. World Health Organization . Coronavirus disease Situation Report 53. World Health Organization. 2020;52. https://www.who.int/docs/default‐source/coronaviruse/20200312‐sitrep‐52‐covid‐19.pdf?sfvrsn=e2bfc9c0_2. Accessed March 25, 2020. [Google Scholar]
- 2. Parr J. Pneumonia in China: lack of information raises concerns among Hong Kong health workers. BMJ. 2020;368(January):m56. 10.1136/bmj.m56 [DOI] [PubMed] [Google Scholar]
- 3. Albarello F, Pianura E, Di Stefano F, et al. 2019‐Novel coronavirus severe adult respiratory distress syndrome in two cases in Italy: an uncommon radiological presentation. Int J Infect Dis. 2020;93(PG‐):192‐197. 10.1016/j.ijid.2020.02.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ren L‐L, Wang Y‐M, Wu Z‐Q, et al. Identification of a novel coronavirus causing severe pneumonia in human [published online ahead of print February 11, 2020]. Chin Med J (Engl). 1. 10.1097/cm9.0000000000000722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ceraolo C, Giorgi FM. Genomic variance of the 2019‐nCoV coronavirus. J Med Virol. 2020;92:1‐7. 10.1002/jmv.25700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benvenuto D, Giovanetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019‐new coronavirus epidemic: Evidence for virus evolution. J Med Virol. 2020;92:455‐459. 10.1002/jmv.25688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS‐CoV‐2 [published online ahead of print March 3, 2020]. Natl Sci Rev. 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Corman V, Bleicker T, Brünink S, et al. Diagnostic detection of 2019‐nCoV by real‐time RT‐RCR. Charité Virology, Berlin, Germany, 2020. https://www.who.int/docs/default‐source/coronaviruse/protocol‐v2‐1.pdf?sfvrsn=a9ef618c_2. Accessed March 25, 2020. [Google Scholar]
- 11. World Health Organization . Novel coronavirus situation report‐12. World Health Organization. 2020;2019. https://www.who.int/docs/default‐source/coronaviruse/situation‐reports/20200201‐sitrep‐12‐ncov.pdf?sfvrsn=273c5d35_2. Accessed March 25, 2020. [Google Scholar]
- 12. World Health Organization . Coronavirus disease situation report‐43. World Health Organization. 2020. 2019; 2633. https://www.who.int/emergencies/diseases/novel‐coronavirus‐2019. Accessed March 25, 2020. [Google Scholar]
- 13. Munster VJ, Koopmans M, van Doremalen N, van Riel D, de Wit E. A novel coronavirus emerging in China — key questions for impact assessment. N Engl J Med. 2020;382(8):692‐694. 10.1056/NEJMp2000929 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information