

Abstract
Last December 2019, a new virus, named novel Coronavirus (COVID‐2019) causing many cases of severe pneumonia was reported in Wuhan, China. The virus knowledge is limited and especially about COVID‐2019 pathogenesis. The Open Reading Frame 1ab (ORF1ab) of COVID‐2019 has been analyzed to evidence the presence of mutation caused by selective pressure on the virus. For selective pressure analysis fast‐unconstrained Bayesian approximation (FUBAR) was used. Homology modelling has been performed by SwissModel and HHPred servers. The presence of transmembrane helical segments in Coronavirus ORF1ab non structural protein 2 (nsp2) and nsp3 was tested by TMHMM, MEMSAT, and MEMPACK tools. Three‐dimensional structures have been analyzed and displayed using PyMOL. FUBAR analysis revealed the presence of potential sites under positive selective pressure (P < .05). Position 723 in the COVID‐2019 has a serine instead a glycine residue, while at aminoacidic position 1010 a proline instead an isoleucine. Significant (P < .05) pervasive negative selection in 2416 sites (55%) was found. The positive selective pressure could account for some clinical features of this virus compared with severe acute respiratory syndrome (SARS) and Bat SARS‐like CoV. The stabilizing mutation falling in the endosome‐associated‐protein‐like domain of the nsp2 protein could account for COVID‐2019 high ability of contagious, while the destabilizing mutation in nsp3 proteins could suggest a potential mechanism differentiating COVID‐2019 from SARS. These data could be helpful for further investigation aimed to identify potential therapeutic targets or vaccine strategy, especially in the actual moment when the epidemic is ongoing and the scientific community is trying to enrich knowledge about this new viral pathogen.
Keywords: epidemiology, infection, pandemics, pathogenesis, protein‐protein interaction analysis, research and analysis methods
Highlights
Analysis of selective pressure on COVID‐19 ORF1ab has been analyzed.
Aminoacid position 501, 723 and 1010 have changed than SARs virus and Bat‐SARS‐like coronaviirus.
Stabilizing mutation at nsp2 protein could explain why COVID‐19 is more contagious than SARS.
Destabilizing mutaion of nsp3 protein could explain the difference oberved between SARS and COVID‐19.
1. INTRODUCTION
A novel Coronavirus, the COVID‐2019, first appeared in Wuhan, China, last December 2019, spreading in other provinces/regions of China and in many countries other continents. 1 The epidemic originated probably from bat after viral mutation in the spike glycoprotein, as recently suggested, 2 began human‐to‐human transmission. The rapid spread of epidemic generated fear leading China authorities to restrict people movement to and from Wuhan in China, where the first start of epidemic was reported. As of 12 February 2020, 45 206 cases have been documented with 44 687 cases in Mainland China, including 1117 deaths and 5123 recovered https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6. The emergence of such a novel, highly virulent pathogen warrants rapid investigation of its etiology and evolution to control the impact on human health.
Knowledge about COVID‐2019 is still incomplete, many questions have raised and many answers are needed first of all regarding its pathogenicity, its ability to change, how many people will get sick from each infected person, the so‐called R0 and when infection will be preventable or treatable. 3 In the last period where many researchers are intensively studying the mechanism of COVID‐2019 replication, pathogenicity, and therapeutic strategies, the present study has been realized. The aim was to provide information about how quickly the virus could potentially increase its genetic variability, with important implications for disease progression and drug or vaccine development. At this aim the Open Reading Frame 1ab (ORF1ab) of COVID‐2019 has been analyzed to evidence the presence of mutation caused by selective pressure on the virus and their influence on viral ability to infect human host promoting epidemic spread.
2. MATERIALS and METHODS
2.1. Sequence dataset
The ORF1ab of 15 COVID‐2019 sequences have been downloaded from GISAID (https://www.gisaid.org/) and GenBank (http://www.ncbi.nlm.nih.gov/genbank/) databanks. A dataset has been built using the five sequences of the severe acute respiratory syndrome (SARS) virus and five sequences from Bat SARS‐like virus sharing the highest sequence similarity to the COVID‐2019 sequence (Table 1). The pairwise percentage of similarity has been calculated using Basic Local Alignment Search Tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi); duplicated sequences have been removed from the dataset. The 25 sequences have been aligned using a multiple sequence alignment multiple alignment using fast fourier transform online tool 4 and manually edited using Bioedit program v7.0.5. 5
Table 1.
Accession numbers, virus type, and the archive where they have been taken from
| Accession number | Virus | Sequences archive |
|---|---|---|
| EPI_ISL_403933 | 2019‐nCoV | GISAID |
| EPI_ISL_403934 | 2019‐nCoV | GISAID |
| EPI_ISL_403936 | 2019‐nCoV | GISAID |
| EPI_ISL_403962 | 2019‐nCoV | GISAID |
| EPI_ISL_402132 | 2019‐nCoV | GISAID |
| EPI_ISL_402130 | 2019‐nCoV | GISAID |
| EPI_ISL_404895 | 2019‐nCoV | GISAID |
| EPI_ISL_404253 | 2019‐nCoV | GISAID |
| EPI_ISL_402125 | 2019‐nCoV | GISAID |
| EPI_ISL_402124 | 2019‐nCoV | GISAID |
| EPI_ISL_403930 | 2019‐nCoV | GISAID |
| EPI_ISL_402120 | 2019‐nCoV | GISAID |
| EPI_ISL_402129 | 2019‐nCoV | GISAID |
| EPI_ISL_404228 | 2019‐nCoV | GISAID |
| EPI_ISL_403931 | 2019‐nCoV | GISAID |
| MG772933.1 | Bat SARS‐like | GeneBank |
| KY417146.1 | Bat SARS‐like | GeneBank |
| KT444582.1 | Bat SARS‐like | GeneBank |
| KY417147.1 | Bat SARS‐like | GeneBank |
| DQ084199.1 | Bat SARS‐like | GeneBank |
| AY559093.1 | SARS | GeneBank |
| JX163925.1 | SARS | GeneBank |
| GU553365.1 | SARS | GeneBank |
| JQ316196.1 | SARS | GeneBank |
| AY714217.1 | SARS | GeneBank |
Abbreviations: 2019‐nCoV, novel coronavirus; SARS, severe acute respiratory syndrome.
2.2. Selective pressure analysis
The selective pressure analysis was focused on the polyprotein ORF1ab because it differs from the most similar bat Coronavirus (QHR63299) for only 103 amino acid residues, 64 of them are conservative changes. In particular, non structural protein 2 (nsp2) differs from bat Coronavirus for 11 residues while nsp3 for 64 residues of which 44 are conservative changes.
Adaptive Evolution Server (http://www.datamonkey.org/) was used to find eventual sites under of positive or negative selection pressure. At this purpose the following tests has been used: fast‐unconstrained Bayesian approximation (FUBAR). 6 These tests allowed to infer the site‐specific pervasive selection, the episodic diversifying selection across the region of interest and to identify episodic selection at individual sites. 7 Statistically significant positive or negative selection was based on P < .05.
2.3. Structural modelling
Homology modelling has been attempted with SwissModel 8 and HHPred 9 servers. Models for ORF1ab nsp2 and nsp3 proteins available at the I‐Tasser web site (corresponding to codes QHD43415_2 and QHD43415_3) 10 have been considered. PDB Proteins structurally close to the target have been evaluated using the TM‐score 11 while the RAMPAGE 12 online tool has been used to assess the folding quality of the model.
To test for the presence of transmembrane helical segments in Coronavirus ORF1ab nsp2 and nsp3, TMHMM, 13 MEMSAT, 14 and MEMPACK 15 online tools have been used. Three‐dimensional structures have been analyzed and displayed using PyMOL. 16
3. RESULTS
3.1. Selective pressure analysis
Regarding the FUBAR analysis performed on the ORF1ab region, the presence of potential sites under positive selective pressure have been found (P < .05), in particular: on the amino acidic position 501 the COVID‐2019 has a glutamine residue, the Bat SARS‐like coronavirus has a threonine residue and the SARS virus has an alanine residue. At position 723 in the COVID‐2019 there is a serine residue while the Bat SARS‐like virus and the SARS virus have a glycine residue. On the aminoacidic position 1010, the COVID‐2019 has a proline residue, the Bat SARS‐like coronavirus has a histidine residue and the SARS virus has an isoleucine residue. Significant (P < .05) pervasive negative selection in 2416 sites (55%) has been evidenced and confirmed by FUBAR analysis.
3.2. Structural modelling
To map the structural variability of the ORF1ab region of the virus and its sites under selection pressure, homology modelling has been attempted. Unfortunately, neither SwissModel nor HHPred found suitable templates for the amino acid region containing the sites under selective pressure. For that reason, the corresponding models available on the I‐Tasser web site has been used. Moreover, some regions of the nsp2 and nsp3 proteins structurally homologous to other known viral proteins have been identified through HHpred analysis and have been mapped within the ORF1ab nsp2 and nsp3 sequences (Figure 1).
Figure 1.
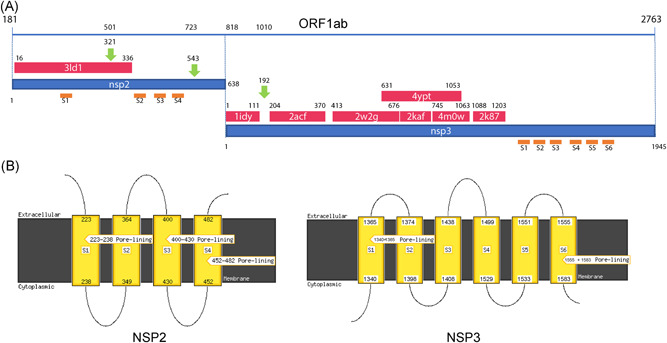
A, HHpred mapping of the homologous structures onto the ORF1ab sequence shown as a blue line on the top of the panel. Numbering above the line refers to the entire ORF. Red and Blue strips represent the PDB homologous structures and the nsp2 and nsp3 sequences, respectively. PDB codes are reported within the corresponding red stripes. Numbering below the blue line is relative to each single nsp. Orange lines indicate approximately the positions of the transmembrane helices predicted by MEMSAT. Label refers to panel B. B, diagram of the topology of predicted transmembrane helices. Number refer to the corresponding nsp sequences. nsp, non structural protein; ORF, open reading frame
The results of the analysis suggest the presence of a segment within the nsp2 and the nsp3 regions that has no evident homologous structures. In an attempt to structurally characterize as far as possible these regions, TMHMM, MEMSAT, and MEMPACK analyses have been utilized and have shown the presence of several potential trans‐membrane helices (Figure 1). In particular, our transmembrane helices were predicted by MEMSAT in nsp2 while six helices were predicted by MEMSTA and TMHMM in nsp3 (Figure 1).
Referring to the amino acids under positive selective pressure found using the FUBAR analysis: the amino acid in position 501 (position 321 of the nsp2 protein), the corresponding site in the Bat SARS‐like coronavirus has an apolar amino acid while the SARS and COVID‐2019 has a polar amino acid. It can be speculated, that due to its side chain length, polarity, and potential to form H‐bonds the glutamine amino acid may confer higher stability to the protein. The mutations fall within the protein nsp2 on the region homologous to the endosome‐associated protein similar to the avian infectious bronchitis virus (PDB 3ld1) that plays a key‐role in the viral pathogenicity. (Figure 2) In the nsp2 structure model available at the I‐Tasser site, this position appears to be exposed to the solvent.
Figure 2.
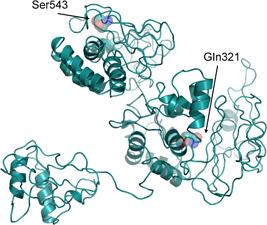
I‐Tasser model of the COVID‐2019 nsp2. Residues under positive selective pressure with a P < .05 are shown as sticks and transparent spheres and are marked by the corresponding labels. COVID‐2019, novel Coronavirus; nsp2, non structural protein‐2
As for the residue in position 723 (543 in the nsp3 protein), the COVID‐2019 sequence displays a Ser replacing for Gly in Bat SARS‐like and SARS coronaviruses. In this case, it may be argued that this substitution could increase local stiffness of the polypeptide chain both for steric effect (at variance with Ser, Gly has no side chain) and for ability of Ser side chain to form H‐bonds. Moreover, Ser can act as a nucleophile in determined structural environments, such as those of enzyme active sites. Within the I‐Tasser model, this position is predicted to have a low solvent accessibility (Figure 2).
Regarding the amino acid in position 1010 (corresponding to position 192 of the nsp3 protein), the homologous region of the Bat SARS‐like coronavirus and SARS virus have a polar and an apolar amino acid, respectively, while the COVID‐2019 has proline. In this case, it may be speculated that due to the steric bulge and stiffness of the proline, the molecular structure of the COVID‐2019 may undergo a local conformation perturbation compared with the proteins of the other two viruses. In Nsp3, the mutation falls near the protein similar to a phosphatase present also in the SARS coronavirus (PDB code 2acf) playing a key‐role in the replication process of the virus in infected cells 17 (Figure 3). In the I‐Tasser model, the position is partially accessible to the solvent. It should be emphasized that all these considerations are speculative and they need to be substantiated by the availability of the experimental crystallographic structure of the corresponding proteins.
Figure 3.
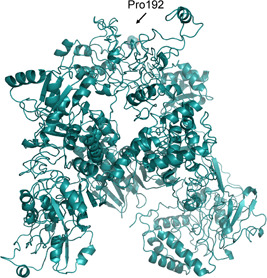
I‐Tasser model of the COVID‐2019 nsp3. The residue under positive selective pressure with a P < .05 is shown as sticks and transparent spheres and is marked by the corresponding label. COVID‐2019, novel Coronavirus; nsp, non structural protein
4. DISCUSSION
The COVID‐2019 ongoing epidemic is worrying worldwide for its high contagiosity. From its first appearance in Wuhan, China, about 1 month ago, the virus infected thousands people with new cases number rapidly growing every day. For this acceleration in human‐to‐human transmission in China but with evident spreading also in other countries, World Health Organization declared the epidemic a global health emergency. 18 , 19
Many questions are open and need an answer, of these the most frequent is how much this virus can be dangerous and how much it differs from SARS virus which epidemic scared all the world some years ago. In this study some interesting findings have been evidenced to support and fill gaps in knowledge about the new COVID‐2019 that is still causing infection all over the world. 20 , 21
The positive selective pressure in this protein could justify some clinical features of this virus compared with SARS and Bat SARS‐like CoV. 22 First which are the probably most common sites undergoing to an aminoacidic change, providing an insight of some important proteins of the COVID‐2019 that are involved in the mechanism of viral entry and viral replication. This data can contribute for a better understanding of how this virus acts in its pathogenicity. Furthermore, to identify a potential molecular target is fundamental to follow the molecular evolution of the virus suggesting some interesting sites for potential therapy or vaccine.
The structural similarity of the region in which falls the positive selective pressure as so as the stabilizing mutation falling in the endosome‐associated‐protein‐like domain of the nsp2 protein, could explain why this virus is more contagious than SARS. The destabilizing mutation happening near the phosphatase domain of the nsp3 proteins could suggest a potential mechanism differentiating COVID‐2019 from SARS.
The results of this study could fill some gaps about COVID‐2019 knowledge especially in the actual moment when the epidemic is ongoing and the scientific community is trying to enrich knowledge about this new viral pathogen. During epidemic, all strength has to be done to enforce virus fight. This can be achieved by understanding the main drivers for pathogen appearance, spreading, and supremacy on human defense.
Angeletti S, Benvenuto D, Bianchi M, Giovanetti M, Pascarella S, Ciccozzi M. COVID‐2019: The role of the nsp2 and nsp3 in its pathogenesis. J Med Virol. 2020;92:584–588. 10.1002/jmv.25719
Silvia Angelettie, Domenico Benvenuto, Stefano Pascarella, and Massimo Ciccozzi contributed equally to this work.
REFERENCES
- 1. Hui DS, I Azhar E, Madani TA, et al. The continuing 2019‐nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int J Infect Dis. 2020;91:264‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benvenuto D, Giovannetti M, Ciccozzi A, Spoto S, Angeletti S, Ciccozzi M. The 2019‐new coronavirus epidemic: evidence for virus evolution. J Med Virol. 2020;92(4):455‐459. 10.1002/jmv.25688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu J, Zheng X, Tong Q, et al. Overlapping and discrete aspects of the pathology and pathogenesis of the emerging human pathogenic coronaviruses SARS‐CoV, MERS‐CoV, and 2019‐nCoV. J Med Virol. 2020. 10.1002/jmv.25709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019;20(4):1160‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hall TA. BioEdit A user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95‐98. [Google Scholar]
- 6. Murrell B, Moola S, Mabona A, et al. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol Biol Evol. 2013;30(5):1196‐1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, Kosakovsky Pond SL. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012;8(7):e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296‐W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zimmermann L, Stephens A, Nam SZ, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430(15):2237‐2243. [DOI] [PubMed] [Google Scholar]
- 10. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER suite: Protein structure and function prediction. Nature Methods. 2015;12(1):7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702‐710. [DOI] [PubMed] [Google Scholar]
- 12. Lovell SC. Structure validation by Calpha geometry: phi, psi, and Cbeta deviation. Proteins. 2003;50(3):437‐450. [DOI] [PubMed] [Google Scholar]
- 13. Moller S, Croning MDR, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17(7):646‐653. [DOI] [PubMed] [Google Scholar]
- 14. Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nugent T, Ward S, Jones DT. The MEMPACK alpha‐helical transmembrane protein structure prediction server. Bioinformatics. 2011;27(10):1438‐1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schrödinger LLC. The {PyMOL} Molecular Graphics System. Version 1.80 LLC, New York, NY. 2015.
- 17. Saikatendu KS, Joseph JS, Subramanian V, et al. Structural basis of severe acute respiratory syndrome coronavirus ADP‐ribose‐1''‐phosphate dephosphorylation by a conserved domain of nsP3. Structure. 2005;13(11):1665‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):P497‐P506. 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhu N, Zhang D, Wang W, et al. China novel coronavirus investigating and research team. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. The Lancet . Emerging understandings of 2019‐nCoV. Lancet. 2020;395(10221):311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giovanetti M, Benvenuto D, Angeletti S, Ciccozzi M. The first two cases of 2019‐nCoV in Italy: where they come from? J Med Virol. 2020. 10.1002/jmv.25699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guarner J. Three emerging coronaviruses in two decades. Am J Clin Pathol. 2020:aqaa029. 10.1093/ajcp/aqaa029 [DOI] [PMC free article] [PubMed] [Google Scholar]