

Abstract
This report describes the isolation, molecular characterization, and phylogenetic analysis of the first three complete genomes of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) isolated from three patients involved in the first outbreak of COVID‐19 in Lombardy, Italy. Early molecular epidemiological tracing suggests that SARS‐CoV‐2 was present in Italy weeks before the first reported cases of infection.
Keywords: complete genomes of SARS‐CoV‐2, COVID‐19, phylogenetic analysis
1. INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), a new coronavirus that causes severe respiratory diseases and is closely related to SARS‐CoV, was described for the first time in the city of Wuhan in the Hubei province of China in late December 2019 (https://www.who.int/csr/don/05‐january‐2020‐pneumonia‐of‐unkown‐cause‐china/en/). It belongs to the β‐coronavirus genus of the Coronaviridae family, and has 96% genomic identity with a previously detected SARS‐like bat coronavirus. 1 , 2 The virus subsequently spread and, on 30 January 2020, the World Health Organisation (WHO) declared it a public health emergency of international concern (https://www.who.int/news‐room/detail/30‐01‐2020‐statement‐on‐the‐second‐meeting‐of‐the‐international‐health‐regulations‐(2005)‐emergency‐committee‐regarding‐the‐outbreak‐of‐novel‐coronavirus‐(2019‐nCoV). On 26 February, the Director‐General of the WHO announced that the number of new cases of the disease, now officially known as COVID‐19, reported outside China since the day before had for the first time exceeded the number of new cases in China (https://www.who.int/dg/speeches/detail/who‐director‐general‐s‐opening‐remarks‐at‐the‐mission‐briefing‐on‐covid‐19‐‐‐26‐february‐2020). By 13 March 2020, a total of 137 445 cases and 5088 fatalities had been reported in 117 countries (https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6), giving rise to major concerns throughout the world, but particularly in South Korea, Iran, and Italy.
Following the detection of two imported cases involving Chinese travelers on January 31, the first cluster of 16 Italian cases was reported in the north‐Italian region of Lombardy on February 21, after which the number of new notified cases exponentially grew until, by 13 March, it had reached a total of 15 113 cases and 1016 deaths. Other confirmed cases of infection have subsequently been reported in a number of other Italian regions, such as the Veneto, Emilia‐Romagna, Piemonte, Liguria, and Marche. Simultaneously, various cases suspected to have been acquired in Italy have been described in a number of other countries.
We have now molecularly characterized and phylogenetically analyzed three complete genomes of SARS‐CoV‐2 isolated from three of the first 16 patients observed in Italy, none of whom reported a recent history of foreign travel.
2. PATIENTS AND METHODS
All of the data used in this study were previously anonymized as required by the Italian Data Protection Code (Legislative Decree 196/2003) and the general authorizations issued by the Data Protection Authority. Ethics Committee approval was deemed unnecessary because, under Italian law, it is only required in the case of prospective clinical trials of medical products for clinical use (Art. 6 and Art. 9 of Legislative Decree 211/2003). However, all of the patients gave their written informed consent to the medical procedures/interventions carried out for routine treatment purposes.
Clinical details of patients are described in Supporting Information Material.
After isolating the virus in Vero cells, SARS‐CoV‐2 RNA was extracted from the culture supernatant after 24 hours, and the full genome was obtained by amplifying 26 fragments using previously published specific primers. 3 The polymerase chain reaction products were used to prepare a library for Illumina deep sequencing using a Nextera XT DNA Sample Preparation and Index kit (Illumina, San Diego, CA) in accordance with the manufacturer's manual, and sequencing was carried out on a Illumina MiSeq platform using the 2 × 150 cycle paired‐end sequencing protocol. The results were mapped and aligned to the reference genome obtained from GISAID (https://www.gisaid.org/, accession ID: EPI_ISL_412973) using Geneious software, v. 9.1.5 (http://www.geneious.com). 4
The genomes obtained from the three patients were aligned with a total of 157 SARS‐CoV‐2 genomes obtained worldwide and publicly available at GISAID on 3 March 2020 (https://www.gisaid.org/), and with an additional Italian strain that became available during the study. Table S1 shows the accession IDs, and sampling dates and locations of the sequences included in the dataset.
A root‐to‐tip regression analysis was made using TempEst in order to investigate the temporal signal of the dataset. 5
The Hasegawa‐Kishino‐Yano model with a proportion of invariant sites (HKY+I) was selected as the simplest evolutionary model by means of JmodelTest, v. 2.1.7, 6 and the phylogenetic analysis was made using a Bayesian Markov Chain Monte Carlo method implemented in BEAST, v.1.8.4. 7
Two coalescent priors (constant population size and exponential growth) and strict vs relaxed molecular clock models were tested by means of path sampling (PS) and stepping stone (SS) sampling. 8 The substitution rate prior was set as a normal distribution with mean 2.2 × 10−6 substitutions/site/day, standard deviation = 1.1 × 10−6 (http://virological.org/t/phylodynamic‐analysis‐176‐genomes‐6‐mar‐2020/356). 9 The time of the most recent common ancestor (tMRCA) was calculated using days as the unit of time.
All of the genes were tested for selection pressure using Datamonkey (https://www.datamonkey.org/).
3. RESULTS
Root‐to‐tip regression analysis of the temporal signal from the dataset revealed a relatively weak association between genetic distances and sampling days (a correlation coefficient of 0.46 and a coefficient of determination [R 2] of 0.21) (Figure 1).
Figure 1.
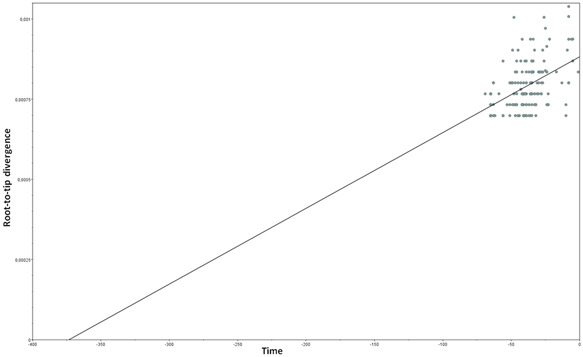
Root‐to‐tip regression analysis of the 161 SARS‐CoV‐2 sequences aligned
Comparison of the marginal likelihoods of the strict vs relaxed molecular clock and constant vs exponential coalescent models showed that the model best fitting the data was the exponential coalescent prior (PS BF exponential growth vs constant = 968.2; SS BF exponential growth vs constant = −967.5) under a log‐normal relaxed clock (PS BF strict vs relaxed clock = −2; SS BF strict vs relaxed clock = −1.6).
Figure 2 shows the obtained dated tree: isolates from China were intermixed throughout the tree, mainly in a basal position with respect to the other sequences. The three Italian genomes clustered in a single highly supported clade (clade A, highlighted in Figure 1; posterior probability, pp = 1) that also included two recently characterized genomes from Italy obtained from patients involved in the same outbreak in Lombardy, three isolates from Europe (two from Germany, one from Finland), and two Latin American sequences (one from Mexico and one from Brazil). One of the German isolates (from Bavaria, EPI_ISL_406862) was in the outgroup of the highly supported subclade (pp = 1) including all of the other strains, and the other German sequence shared a highly supported node (pp = 1) with the Mexican sequence.
Figure 2.
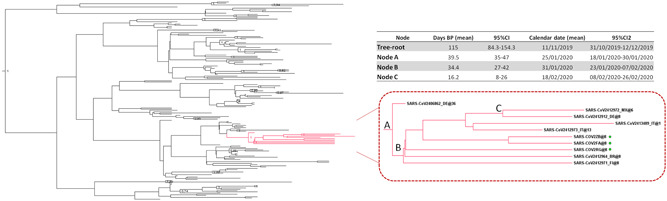
Dated tree of 161 SARS‐CoV‐2 sequences showing statistically significant support for clades along the branches (posterior probability >0.7). Clade A containing the Italian strains is highlighted in red. The patients characterized in this study are indicated by a symbol. The table shows the time of the most recent common ancestor (tMRCA) estimates and 95% high posterior density of the significant clade A nodes. SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2
Table in Figure 2 summarizes the estimated mean tMRCAs of the tree root and of the main significant clade. The estimated mean tMRCA of the tree root was 115 days before the present (95% high posterior density [HPD]: 84.3‐154.3), corresponding to 11 November 2019 (credibility interval: 31 October‐12 December). The estimated mean tMRCA of node A was 39.5 days before the present (95% HPD: 35‐47), corresponding to 25 January 2020 (credibility interval: 18‐30 January 2020), and the estimated mean tMRCA of node B was 34.4 days before the present (95% HPD: 27‐42), corresponding to 31 January 2020 (credibility interval: 23 January‐7 February). Finally, the estimated mean tMRCA of the internal node between the Mexican and the German isolates (node C) was 16.2 days before the present (95% HPD: 8‐26), corresponding to 18 February 2020 (credibility interval: 8‐26 February).
Comparison of the genetic distances estimated on the basis of the number of nucleotide substitutions indicated a mean 7.8 nucleotide substitutions between the isolates in clade A (range: 0‐24 nucleotide substitutions). Three sequences (two Italian and one Brazilian) were identical, whereas one Italian strain had a difference of as many as 18 nucleotides from the German outgroup sequence (EPI_ISL_406862). All of the sequences in clade A showed a D614G mutation in the S gene. No sites were identified as being under significant positive selection pressure.
4. DISCUSSION
The present phylogenetic analysis confirms that the common origin of the SARS‐CoV‐2 strains characterized so far was several weeks before the first cases of COVID‐19 pneumonia were described in China. 9 It also shows that the whole genomes of the three SARS‐CoV‐2 strains isolated from patients in northern Italy and characterized by us are closely related to each other, as well as to the other two published Italian sequences, and the German, Finnish, Mexican, and Brazilian sequences, all of which formed a highly supported clade.
The German sequence at the outgroup of the clade came from a COVID‐19 outbreak reported between 20 and 24 January and occurring after business meetings with a Shanghai business woman who tested positive after returning to China. 10 Our tMRCA estimate showed that the root of clade A was in the month of January 2020, a period compatible with this event. However, our data do not allow us to make any hypotheses concerning the possible routes followed by the virus to reach Italy because, given the limited number of sparsely sampled sequences in the tree, 11 it is impossible to infer the directionality of transmission, and this means that multiple independent importations to Europe cannot be excluded.
Our data suggest that SARS‐CoV‐2 virus entered northern Italy between the second half of January and early February 2020, which is weeks before the first Italian case of COVID‐19 was identified and therefore long before the current containment measures were taken.
Interestingly, although they were sampled in the same area on the same day, the genomes isolated from these three patients have a number of different, mainly synonymous substitutions. In particular, one patient living near the municipality in which the highest number of cases was recorded showed a high degree of genomic heterogeneity, thus suggesting considerable genetic drift.
In conclusion, our data show that the SARS‐CoV‐2 isolates infecting the Italian patients involved in the early epidemic in northern Italy and those isolated from other European and Latin American patients reporting contacts with Italy, are closely related to the strain isolated during one of the first European clusters observed in Bavaria in late January 2020. 12 On the basis of the phylogenetic analysis alone, we cannot exclude possibly multiple introductions in Germany and Italy from China (or other countries), but the epidemiological data showing that the first cases in Germany preceded the first cases in Italy by almost a month suggest that the strain entered Germany before Italy. Finally, as we have characterized only three genomes so far, we cannot exclude the presence of other different strains in Italy that may be the result of multiple introductions. Further epidemiological and molecular studies of a larger sample are needed to clarify these issues.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
AL, GZ, and MG conceived and designed the study. LM, AR, DB, SR, GR, SA, and MG were involved in patient care and the collection of biological materials. AL, AB, MT, AG, and CB performed the experiments. GZ, AL, and AB made the phylogenetic analyses. AL, GZ, AB, SR, and MG wrote the first draft of the manuscript. All of the authors contributed to revising the manuscript, and read and approved the submitted version.
Supporting information
Supporting information
ACKNOWLEDGMENT
We acknowledge the authors and the originating and submitting laboratories of the GISAID sequences.
Zehender G, Lai A, Bergna A, et al. Genomic characterization and phylogenetic analysis of SARS‐COV‐2 in Italy. J Med Virol. 2020;92:1637–1640. 10.1002/jmv.25794
Gianguglielmo Zehender and Alessia Lai contributed equally to this paper.
DATA AVAILABILITY STATEMENT
The sequencing data used for this study will be submitted to GISAID with accession IDs: EPI_ISL_417445, EPI_ISL_417446, EPI_ISL_417447.
REFERENCES
- 1. Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S. Full‐genome evolutionary analysis of the novel corona virus (2019‐nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol. 2020;79:104212. 10.1016/j.meegid.2020.104212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020:579. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kearse M, Moir R, Wilson A, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647‐1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path‐O‐Gen). Virus Evol. 2016;2(1):vew007. 10.1093/ve/vew007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Posada D. jModelTest: Phylogenetic model averaging. Mol Biol Evol. 2008;25(7):1253‐1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- 7. Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969‐1973. 10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baele G, Lemey P, Bedford T, Rambaut A, Suchard MA, Alekseyenko AV. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol Biol Evol. 2012;29(9):2157‐2167. 10.1093/molbev/mss084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai A, Bergna A, Acciarri C, Galli M, Zehender G. Early phylogenetic estimate of the effective reproduction number of SARS‐CoV‐2. J Med Virol. 2020. 10.1002/jmv.25723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rothe C, Schunk M, Sothmann P, et al. Transmission of 2019‐nCoV infection from an asymptomatic contact in Germany. N Engl J Med. 2020;382(10):970‐971. 10.1056/NEJMc2001468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS‐CoV‐2. Virological. 2020;89(ii):1‐7. 10.2106/JBJS.F.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spiteri G, Fielding J, Diercke M, et al. Review of “First cases of coronavirus disease 2019 (COVID‐19) in the WHO European Region, 24 January to 21 February 2020” One‐Minute Summary PHO Reviewer's Comments (2020). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The sequencing data used for this study will be submitted to GISAID with accession IDs: EPI_ISL_417445, EPI_ISL_417446, EPI_ISL_417447.