

Abstract
Starting around December 2019, an epidemic of pneumonia, which was named COVID‐19 by the World Health Organization, broke out in Wuhan, China, and is spreading throughout the world. A new coronavirus, named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) by the Coronavirus Study Group of the International Committee on Taxonomy of Viruses was soon found to be the cause. At present, the sensitivity of clinical nucleic acid detection is limited, and it is still unclear whether it is related to genetic variation. In this study, we retrieved 95 full‐length genomic sequences of SARAS‐CoV‐2 strains from the National Center for Biotechnology Information and GISAID databases, established the reference sequence by conducting multiple sequence alignment and phylogenetic analyses, and analyzed sequence variations along the SARS‐CoV‐2 genome. The homology among all viral strains was generally high, among them, 99.99% (99.91%‐100%) at the nucleotide level and 99.99% (99.79%‐100%) at the amino acid level. Although overall variation in open‐reading frame (ORF) regions is low, 13 variation sites in 1a, 1b, S, 3a, M, 8, and N regions were identified, among which positions nt28144 in ORF 8 and nt8782 in ORF 1a showed mutation rate of 30.53% (29/95) and 29.47% (28/95), respectively. These findings suggested that there may be selective mutations in SARS‐COV‐2, and it is necessary to avoid certain regions when designing primers and probes. Establishment of the reference sequence for SARS‐CoV‐2 could benefit not only biological study of this virus but also diagnosis, clinical monitoring and intervention of SARS‐CoV‐2 infection in the future.
Keywords: homology, nucleotide, reference sequence, SARS‐CoV‐2, variation
Highlights
In this study, we retrieved 95 full‐length genomic sequences of SARAS‐CoV‐2 strains, established the reference sequence, and analyzed sequencevariations along the SARS‐CoV‐2 genome. The homology among all viral strains was generally high. Although overall variation in ORF regions is low, 13 variation sites in 1a, 1b, S, 3a, M, 8, and N regions were identified, among which positions nt28144 in region 8 and nt8782 in ORF 1a showed mutation rate of 30.53% and 29.47% respectively.
1. INTRODUCTION
Cases of pneumonia with unknown cause emerged in Wuhan, China in December 2019. 1 Epidemic investigation and gene sequencing revealed that a novel coronavirus was the etiologic agent. The virus was tentatively named 2019‐nCoV but officially named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) later by the Coronavirus Study Group of the International Committee on Taxonomy of Viruses, and the disease caused by this virus was name COVID‐19 by the World Health Organization. 2 , 3 Up to March 8 (24:00 GMT), 80 735 confirmed cases, have been diagnosed in Mainland China, causing 3119 deaths. 4 Cases have also been reported from 101 countries or areas including Thailand, Japan, Korea, Australia, France, and the United States. 5 Family clustering of infection, 3000 cases of healthcare personnel infection, and other evidence together have provided strong supporting evidence for human‐to‐human transmission of SARS‐CoV‐2 infection with a basic reproduction number (R0) of 2‐4. 6 , 7 , 8 SARS‐CoV‐2 has a high transmissibility and can have a long incubation time before manifesting symptoms including fever, coughing, shortness of breath, and diarrhea. SARS‐CoV‐2 infection can be symptom‐free in some patients, but may cause multiple organ failures in lung, heart, and liver in some other patients. The mortality rate of SARS‐CoV‐2 infection is about 3%. 1 , 9 , 10
The Yongzhen Zhang team in China was the first group to determine the full‐length genomic sequence of the SARS‐CoV‐2 virus. 11 The genome is arranged in the order of a 5′‐untranslated region (UTR)‐replicase complex (orf 1ab)‐structural proteins (Spike(S)‐Envelope(E)‐Membrane (M)‐Nucleocapsid (N))−3′‐UTR and nonstructural open‐reading frames (ORFs). Before the emergence of SARS‐CoV‐2, six human coronaviruses including α coronaviruses 229E and NL63, β coronavirus OC43 and HKU1, Middle East respiratory syndrome coronavirus (MERSr‐CoV) and SARS‐associated coronavirus (SARSr‐CoV) had been identified. Among them, MERSr‐CoV and SARSr‐CoV can be transmitted from human to human, and were highly pathogenic resulting in high mortality. 12 , 13 , 14
So far, scientists from different countries have obtained and uploaded more than 100 full‐length or partial genomic sequences for SARS‐CoV‐2. Some companies have developed rapid nucleic acid detection kits based on these sequences. However, significant differences in the sensitivity and specificity among these kits have been found from clinical application of these kits. In addition, a standardized quantitative detection method is still lacking. Missed diagnosis and misdiagnosis are currently not uncommon due to these reasons. 15 , 16 , 17
To provide template sequence for proper design of polymerase chain reaction (PCR) primers and probes to minimize false negative results, and to obtain reliable sequence information for molecular and immunological studies on and vaccine development for SARS‐CoV‐2 virus, we retrieved from the National Center for Biotechnology Information (NCBI) and GISAID websites, full‐length sequences from different regions of the world, established the reference sequence for SARS‐CoV‐2 by homology and phylogenetic tree analyses, analyzed mutations at different locations, and conducted preliminary bioinformatics analyses for the reference sequence.
2. MATERIALS AND METHODS
2.1. Sources and selection of sequences
The NCBI (http://www.ncbi.nlm.nih.gov/genbank/) and GISAID (https://www.gisaid.org/) databases up to 14 Feb, 2020 were searched by using keywords “novel, coronavirus, complete, Wuhan” or “2019‐nCoV.” The inclusion criteria included the length of the full‐length sequence, which was 25000 to 32000 bp, and was verified to be human SARS‐CoV‐2 sequence. Repetitively submitted sequences and sequences with too many undetermined nucleotides were excluded from this study. Sequences were classified as stage 1 and 2 based on the time sequences retrieved. Sequences obtained on Feb 6, 2020 went into stage 1, while sequences retrieved from Feb 6 to 14 were classified as stage 2 (Figure 1 and Table S1).
Figure 1.
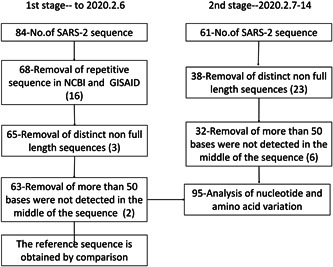
Flow chart of severe acute respiratory syndrome coronavirus 2 (SARS‐COV‐2) sequence data collection
2.2. Establishment of reference sequence
Homology analysis and sequence alignment were conducted for all stage 1 sequences by using Primer 7.0 and Mega (7.0.14). The reference sequence was conducted by selecting the most common nucleotide in each position. The reliability of the reference sequence was confirmed by comparing it with stage 2 sequences.
2.3. Phylogenetic analysis
The ClustalW program of the MEGA software (7.0.14) was used to conduct multiple sequence alignment and the phylogenetic tree was constructed by using a maximum likelihood approach based on stage 1 sequences. Related coronaviral sequences were used as references (229E(KY369908), NL63(MK334046), OC43(MG197709), HKU1(KT779555), MERSr‐CoV(KJ813439), SARSr‐CoV(AY278488), bat coronavirus(MN996532)).
2.4. Analyses of nucleotide and amino acid sequence variation
Primer 7.0 was used to compare the reference nucleotide sequence to those of related human isolates and analyze the variation at different locations. Sequence comparison and variation analysis were also conducted at the amino acid level.
2.5. Analysis of published primer sequences
Sequences of PCR primers/probes from published articles were aligned with our reference sequence to analyze sequence variation. Whether these primer/probe sequences overlapped with variation sites was also examined.
3. RESULTS
3.1. Information about sequences included
A total of 145 sequences were obtained from databases. These sequences were examined and 50 sequences were excluded from the study according to predetermined criteria. As a result, 95 sequences, among which 63 were obtained from stage 1 and 32 from stage 2, were used for analyses (Figure 1). These sequences were reported from China, America, Australia, Thailand, the United Kingdom, Germany, France, Finland, Korea, Japan, and Singapore, etc (Table S1).
3.2. Establishment of reference sequence
The reference sequence was constructed by nucleotide sequence alignment (accession number: EPI_ISL_412026). The reference sequence composed of 29 870 nucleotides and 9744 amino acids. The length and start and end locations of individual encoding regions are shown in Table 1.
Table 1.
Comparison of homology among open‐reading frames of SARS‐COV‐2 isolate strains
| Region (ORF) | Nucleotide (nt) | Amino acid (aa) | ||||
|---|---|---|---|---|---|---|
| Start and end | Length | Homology (%) a | Start and end | Length | Homology (%) a | |
| Full length | 1‐29870 | 29 870 | 99.99 (99.91‐100) | 1‐9744 | 9744 | 99.99 (99.79‐100) |
| 1ab | 266‐21555 | 21 306 | 100 (99.91‐100) | 1‐7096 | 7096 | 100 (99.80‐100) |
| 1a | 266‐13483 | 13 218 | 99.99 (99.88‐100) | 1‐4401 | 4401 | 100 (99.73‐100) |
| 1b | 13468‐21555 | 8088 | 100 (99.93‐100) | 4402‐7096 | 2695 | 100 (99.85‐100) |
| S | 21563‐25384 | 3822 | 100 (99.82‐100) | 7097‐8369 | 1273 | 100 (99.53‐100) |
| 3a | 25393‐26220 | 828 | 100 (99.76‐100) | 8370‐8644 | 275 | 100 (99.27‐100) |
| E | 26245‐26472 | 228 | 100 (100‐100) | 8645‐8719 | 75 | 100 (100‐100) |
| M | 26523‐27191 | 669 | 100 (99.70‐100) | 8720‐8941 | 222 | 100 (99.95‐100) |
| 6 | 27202‐27387 | 186 | 100 (100‐100) | 8942‐9002 | 61 | 100 (100‐100) |
| 7a | 27394‐27759 | 366 | 100 (99.73‐100) | 9003‐9123 | 121 | 100 (99.17‐100) |
| 7b | 27756‐27887 | 132 | 100 (100‐100) | 9124‐9166 | 43 | 100 (100‐100) |
| 8 | 27894‐28259 | 366 | 100 (99.45‐100) | 9167‐9287 | 121 | 100 (98.35‐100) |
| N | 28274‐29533 | 1260 | 100 (99.84‐100) | 9288‐9706 | 419 | 100 (99.76‐100) |
| 10 | 29558‐29674 | 117 | 100 (99.15‐100) | 9707‐9744 | 38 | 100 (97.37‐100) |
Abbreviations: E, Envelope; M, Membrane; N, Nucleoprotein; ORF, open‐reading frame; S, Spike; SARS‐COV‐2, severe acute respiratory syndrome coronavirus 2; 1ab, open‐reading frames 1ab; 1a, open‐reading frames 1a; 1b, open‐reading frames 1b; 3a, open‐reading frames 3a; 6, open‐reading frames 6; 7a, open‐reading frames 7a; 7b, open‐reading frames 7b; 8, open‐reading frames 8; 10, open‐reading frames 10.
Median (min‐max).
3.3. Phylogenetic analysis
The phylogenetic tree was constructed using sequences from database search stage 1 and other coronaviruses. While different types of coronaviruses showed scattered distribution, all SARS‐CoV‐2 strains clustered together tightly. Importantly, the reference sequence was located in the middle of the SARS‐CoV‐2 cluster, demonstrating good representativeness of the reference sequence constructed (Figure S1).
3.4. Homology analyses
Comparison of the reference sequence with those of respective isolates showed that the overall homology among full‐length genomic sequences was 99.99% (99.91%‐100%), and 15 (15.79%) clinical isolates were identical to the reference strain. With respect to individual encoding regions, the homology in open‐reading frame (ORF) 1a was 99.99% (99.88%‐100%); the homology among the majority of other regions was 100%; no variation was found in E, 6, and 7b regions. At the amino acid level, the homology among full‐length sequences was 99.99% (99.79%‐100%), with homology among most isolates in each region being 100% (Table 1).
3.5. Variation analyses at the nucleotide and amino acids levels
Sequence alignment found that mutations at both nucleotide and amino acid levels were relatively rare. However, mutations did exist. Mutations which occurred in ≥3 strains were found in these locations: 1a (nt2662,8782,11083), 1b (nt17373,18060), S (nt21707,24034), 3a (nt26144), M (nt26729), 8 (nt28077,28144), N (nt28854,29095). Strikingly, position 8:nt28144 showed the highest mutation rate of 30.53% (29/95), where T was replaced with C. Similarly, position 1a:nt8782 had a mutation rate of 29.47% (28/95), where C was replaced mostly by T. At the amino acid level, mutations which occurred in ≥3 strains were found in these locations: 1a (aa3606), S (aa49,860), 3a (aa251), 8 (aa62,84), N (aa194) (Table 2, Figures 2 and 3, and Table S2). In addition, six deletion mutations were found in five isolated strains. These mutations resulted in four different truncations in amino acid sequence (1/3/6/8 aa). Furthermore, two deletion mutations were found in the 5′ and 3′ nonencoding regions, respectively (Table S3).
Table 2.
The major locus of nucleotide or amino acid variation in SARS‐CoV‐2 isolate strains (≥3/95)
| Regions (ORF) | Nucleotide mutations | Amino acid mutations | ||||
|---|---|---|---|---|---|---|
| site | No. | Type | Site | No. | Type | |
| 1a | 2662 | 3 | C→T | 3606 | 6 | L→F |
| 8782 | 28 | C→T/Y | ||||
| 11083 | 6 | G→T | ||||
| 1b | 17373 | 3 | C→T | |||
| 18060 | 3 | C→T | ||||
| S | 21707 | 4 | C→T | 49 | 4 | H→Y |
| 24034 | 7 | C→T/Y | 860 | 3 | V→Q | |
| 3a | 26144 | 6 | G→T | 251 | 6 | G→V |
| M | 26729 | 5 | T→C/Y | |||
| 8 | 28077 | 5 | G→C/S | 62 | 5 | V→L |
| 28144 | 29 | T→C/Y | 84 | 29 | L→S | |
| N | 28854 | 6 | C→T/Y | 194 | 6 | S→L |
| 29095 | 11 | C→T | ||||
Abbreviations: M, Membrane; N, Nucleoprotein; ORF, open‐reading frame; S, Spike; SARS‐COV‐2, severe acute respiratory syndrome coronavirus 2; 1a, open‐reading frames 1a; 1b, open‐reading frames 1b; 3a, open‐reading frames 3a; 8, open‐reading frames 8.
Figure 2.

Distribution of the number of mutant bases or amino acids in each SARS‐COV‐2 isolate strain. A, Full‐length and partial regions (1ab, 1a, 1b, S, E, M, N) nucleotides. B, Partial regions (5NCR, 3a, 6, 7a, 7b, 8, 10) nucleotides. C, Partial regions (1ab, 1a, 1b, S, E, M, N) amino acids. D, Partial regions (5‐untranslated region, 3a, 6, 7a, 7b, 8, 10) amino acids. SARS‐COV‐2, severe acute respiratory syndrome coronavirus 2
Figure 3.

Common sites and frequency of mutation in SARS‐COV‐2 isolate strains (≥5/95). A, Nucleotides. B, Amino acids. SARS‐COV‐2, severe acute respiratory syndrome coronavirus 2
3.6. Analysis of published primer sequences
Sequence alignment revealed differences between some primer/probe sequences and the reference sequence (Table 3). In a newly published article, 7 there are site differences between the primers from ORF1b and the reference sequence. In another publication, 16 the primer pair and probe sequences were derived from the N region, and also have site differences from the reference sequence. Apparently, these published primer pairs/probes are not likely to work well with the majority of viral isolates.
Table 3.
Differences between published primer/probe sequences and reference or clinical isolates
| Target gene | Direction | Primer (5′−3′) | Location | Reference strain | Clinical isolates | References |
|---|---|---|---|---|---|---|
| ORF1b | Forward | CAAGTGGGGTAAGGCTAGACTTT | 14961‐14983 | TAAATGGGGTAAGGCTAGACTTT | 95 | Lancet (https://doi.org/10.1016/S0140‐6736(20)30154‐9) |
| Reverse | ACTTAGGATAATCCCAACCCAT | 15283‐15304 | ATTTAGGATAATCCCAACCCAT | 95 | ||
| S | Forward | CCTACTAAATTAAATGATCTCTGCTTTACT | 22712‐22741 | No difference | No difference | |
| Reverse | CAAGCTATAACGCAGCCTGTA | 22849‐22869 | No difference | No difference | ||
| ORF1b | Forward | TGGGGYTTTACRGGTAACCT | 18778‐18797 | No difference | No difference | Clinical Chemistry (https://doi.org/10.1093/clinchem/hvaa029) |
| Reverse | AACRCGCTTAACAAAGCACTC | 18889‐18909 | No difference | No difference | ||
| Probe | TAGTTGTGATGCWATCATGACTAG | 18849‐18872 | No difference | No difference | ||
| N | Forward | TAATCAGACAAGGAACTGATTA | 29145‐29166 | No difference | No difference | |
| Reverse | CGAAGGTGTGACTTCCATG | 29236‐29254 | No difference | No difference | ||
| Probe | GCAAATTGTGCAATTTGCGG | 29179‐29198 | GCAAATTGCACAATTTGCCC | 95 | ||
| ORF1b | Forward | GTGARATGGTCATGTGTGGCGG | 15431‐15452 | No difference | No difference | Euro Surveill (https://doi.org/10.2807/1560‐7917.ES.2020.25.3.2000045) |
| Probe2 | CAGGTGGAACCTCATCAGGAGATGC | 15470‐15494 | No difference | No difference | ||
| Probe1 | CCAGGTGGWACRTCATCMGGTGATGC | 15469‐15494 | CCAGGTGGAACCTCATCAGGAGATGC | 95 | ||
| Reverse | CARATGTTAAASACACTATTAGCATA | 15505‐15530 | No difference | No difference | ||
| E | Forward | ACAGGTACGTTAATAGTTAATAGCGT | 26269‐26294 | No difference | No difference | |
| Probe | ACACTAGCCATCCTTACTGCGCTTCG | 26332‐26357 | No difference | No difference | ||
| Reverse | ATATTGCAGCAGTACGCACACA | 26360‐26381 | No difference | No difference | ||
| N | Forward | CACATTGGCACCCGCAATC | 28706‐28724 | No difference | 1 | |
| Probe | ACTTCCTCAAGGAACAACATTGCCA | 28753‐28777 | No difference | No difference | ||
| Reverse | GAGGAACGAGAAGAGGCTTG | 28814‐28833 | No difference | No difference |
Note: Italics indicates the location of the difference.
Abbreviations: N, Nucleoprotein; S, Spike; 1b, open‐reading frames 1b.
4. DISCUSSION
SARS‐CoV‐2 virus belongs to β‐coronavirus. An enveloped virus with a diameter of 60 to 140 nm, SARS‐CoV‐2 is round or oval‐shaped with some polymorphism. 3 The genomic characteristics of SARS‐CoV‐2 virus are significantly different from those of MERs‐CoV or SARs‐CoV. This study showed that its homology with the Bat coronavirus isolate RaTG13 strain (MN996532) was 96%, but has no more than 80% homology with other isolates of bat SARS‐like coronavirus (Table S4), which is similar to the results from other studies. 18 Recent studies have shown that the homology with a coronavirus strain isolated from pangolin was 99%, suggesting that SARS‐CoV‐2 might have originated from bat and pangolin might have served as the intermediate host between bat and human. 19 , 20 However, further research is needed to confirm these assumptions.
The study on genomic variation of SARS‐CoV‐2 is very important for investigation of pathogenesis, disease course, prevention, and treatment of SARS‐CoV‐2 infection. Establishment of the reference sequence for this virus is a fundamental work which will facilitate viral detection, functional analysis, vaccine design, epidemic investigation, evaluation of drug efficacy, among others.
Based on more than 3000 hepatitis B virus (HBV) sequences reported from different countries, our team has divided HBV isolates into 31 different HBV subtypes established by selecting nucleotide with the highest frequency in each position. Using infectious plasmids constructed based on A2, B2, C2, and D1 subtype‐specific reference sequences, in vitro and in vivo studies have confirmed complete biological functions of these reference sequences. 21 , 22
Using the same approaches, in this study, the reference sequence for SARS‐CoV‐2 was constructed based on genomic sequence of 63 isolated strains. The genome size of the reference strain was 29870 bp. The reference sequence was identical to the genomic sequence of 15 strains (15.79%) isolated from clinical samples, suggesting the reference strain would display full biological functions and pathogenicity. Since the sequences retrieved later (Feb 7‐14) showed high homology with the reference sequence (>99.9%), there was no need to adjust the reference sequence.
As a typical RNA virus, the evolution rate of coronavirus could be 10−4 substitute per bp per year, and mutation could occur during each replication cycle. 12 However, phylogenetic analysis and sequence alignment showed that the homology among different isolates was extremely high. Compared to the reference sequence, the homology of the vast majority of isolates was above 99.99% at both the nucleotide and amino acid levels. In fact, the homology in most encoding regions was 100%. The lowest homology was found in strain EPI_ISL_406592 from Shenzhen, China (99.91%, nt) and strain EPI_ISL_408485 from Beijing, China (99.79%, aa). Overall, results from our analyses suggest that the virus in this epidemic might originate from the same animal species, and caused widespread infection in a short period of time. 2 , 23
Although sequence variation among SARS‐CoV‐2 isolates was low, and sequence analysis showed a rather random distribution of mutations, we did find mutation hot spots in this study. Nucleic acid detection is currently the gold standard method for diagnosis of COVID‐19. However, the sensitivity of this method is not high in clinical application. 24 Sampling could be one of the reasons accounting for the low sensitivity, the reagents used for detection could be another critical reason. In this study, sequence from 95 strains were examined and 12 locations where mutations occurred in ≥3 strains were found. Importantly, among these locations, mutations were found at nt8782 of ORF 1a, nt28144 of ORF 8, and nt29095 of N region in 28, 29, and 11 strains, respectively. Therefore, data from this study showed that, while designing PCR primers and probes, sequences in these locations should be avoided, and attention must be paid to locations listed in Table S2 and S3 to avoid false negative results. Furthermore, some published primer/probe sequences were compared to the reference sequence established and differences were found. This might partially explain why false negative results in nucleic acid detection of SARS‐CoV‐2 for diagnosis of COVID‐19 is currently not uncommon. Sequence variation information obtained and SARS‐CoV‐2 reference sequence established in this study can provide reliable guidance for the design of primers/probes with maximal sensitivity for detection of SARS‐CoV‐2 nucleic acid.
SARS‐CoV‐2 encodes four important viral proteins including S, E, M, and N proteins. 2 This study shows that SARS‐COV‐2 is relatively conserved, especially in the E, 6, 7b regions where no mutation was found. Hotspot mutations in ORFs 1a, S, 8, and the N region will cause changes in the amino acid sequences of these proteins, and the effects of these mutations on viral replication, transmission and the induced immune responses need to be further investigated. The significance of these variations is unclear, and may be directed mutations adapted to the environment.
A potential shortcoming of this study is that, since all sequences used in this study were retrieved from databases, the accuracy of sequences could not be verified. Although the sequence included in this study is still small, it basically includes most of SARS‐CoV‐2 complete viral sequences that have been published worldwide and is widely distributed, which should be able to represent the characteristics of the virus.
In summary, in this study SARS‐CoV‐2 genomic sequences that are available from the NCBI and GISAID databases so far were analyzed and the reference sequence for this virus was established. The variations in individual coding regions at both the nucleotide and amino acid level were further analyzed and part of the reasons why the sensitivity of current nucleic acid detection methods is far from ideal was revealed. Establishment of the reference sequence for SARS‐CoV‐2 could benefit not only biological study of this virus but also diagnosis, clinical monitoring and intervention of SARS‐CoV‐2 infection in the future.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
ZL, ZC, XH, TH, CW, and ZZ collect and analyze data. ZZ, CW, and JL wrote the manuscript. JL participated in the coordination of the study and manuscript modification. ZZ conceived the project. All authors contributed, read, and approved the manuscript.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENT
The authors are very grateful to Mr. Kaiyang Wu for editing software for nucleotide sequence homology comparison and alignment. This study was supported by the Anhui Provincial Natural Science Foundation of China (grant number: 1608085MH162) and Emergency Science and Technology Plan for Novel Coronavirus Infection of Anqing (grant number: 2020Z1001). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Wang C, Liu Z, Chen Z, et al. The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. J Med Virol. 2020;92:667–674. 10.1002/jmv.25762
REFERENCES
- 1. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu R, Zhao X, Li J, et al. Genomic characterization and epidemiology of 2019 novel coronavirus: implications of virus origins and receptor binding. Lancet. 2020;395:565‐574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382:727‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. National Health Commission of the People's Republic of China . http://www.nhc.gov.cn (Assessed on March 8th, 2020). [DOI] [PMC free article] [PubMed]
- 5. WHO main website . https://www.who.int (accessed March 8th, 2020).
- 6. The Novel Coronavirus Pneumonia Emergency Response Epidemiology Team . The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID‐19) in china. Chin J Epidemiol. 2020;41:145‐151. [PMC free article] [PubMed] [Google Scholar]
- 7. Chan JFW, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person‐to‐person transmission: a study of a family cluster. Lancet. 2020;395:514‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus‐infected pneumonia. N Engl J Med. 2020. 10.1056/NEJMoa2001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guan WJ, Ni Z, Hu Y, et al. Clinical characteristics of 2019 novel coronavirus infection in China. medRxiv. 2020:20020974. 10.1101/2020.02.06.20020974 [DOI] [Google Scholar]
- 11. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24:490‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20:660‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chan JFW, Lau SKP, To KKW, Cheng VCC, Woo PCY, Yuen K. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS‐like disease. Clin Microbiol Rev. 2015;28:465‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang N, Wang L, Deng X. Recent advances in the detection of respiratory virus infection in humans. J Med Virol. 2020;92(4):408‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chu D, Pan Y, Cheng S, et al. Molecular diagnosis of a novel coronavirus (2019‐nCoV) causing an outbreak of pneumonia. Clin Chem. 2020:hvaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Corman VM, Landt O, Kaiser M, et al. Detection of 2019 novel coronavirus (2019‐nCoV) by real‐time RT‐PCR. Euro Surveill. 2020;25:2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou P, Yang X, Wang X, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiao KP, Zhai JP, Feng YY, et al. Isolation and characterization of 2019‐nCoV‐like coronavirus from Malayan pangolins. bioRxiv. 2020:951335. 10.1101/2020.02.17.951335 [DOI] [Google Scholar]
- 20. Lam T, Shum M, Zhu HZ, et al. Identification of 2019‐nCoV related coronaviruses in Malayan pangolins in southern China. bioRxiv. 2020:945485. 10.1101/2020.02.13.945485 [DOI] [Google Scholar]
- 21. Chen L, Shi Y, Yang W, et al. Differences in Cpg island distribution between subgenotypes of the hepatitis B virus genotype. Med Sci Monitor. 2018;24:6781‐6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Xia J, Sun B, et al. In vitro and in vivo replication of a chemically synthesized consensus genome of hepatitis B virus genotype B. J Virol Methods. 2015;213:57‐64. [DOI] [PubMed] [Google Scholar]
- 23. Chan JFW, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infec. 2020;9:221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang S, Bo K, Ma JL, et al. A deep learning algorithm using CT images to screen for Corona Virus Disease (COVID‐19). medRxiv. 2020:20023028. 10.1101/2020.02.14.20023028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information