

Abstract
Epithelioid hemangioendothelioma is a low grade malignant vascular tumor with an intermediate clinical behavior between benign hemangiomas and high grade angiosarcomas. Pathologic or molecular factors to predict this clinical heterogeneity are not well defined. A WWTR1-CAMTA1 fusion is present in most classic epithelioid hemangioendothelioma, regardless of their clinical behavior, suggesting that additional genetic abnormalities might be responsible in driving a more aggressive biology. A small subset of cases show distinct morphology and are characterized genetically by a YAP1-TFE3 fusion. Two histologic grades have been described in classic epithelioid hemangioendothelioma of the soft tissue. However, proposed criteria do not apply to other clinical presentations and have not been assessed in the YAP1-TFE3 positive tumors. Furthermore, no previous studies have compared the survival of these 2 molecular subsets. In this study we investigate the clinicopathologic and molecular findings of a large cohort of 93 translocation-positive epithelioid hemangioendothelioma managed at our institution. Patient characteristics, histologic features, treatment outcomes, and genetic abnormalities were investigated and these factors were correlated with overall survival. In 18 patients (15 with WWTR1-CAMTA1 and 3 with YAP1-TFE3) Memorial Sloan Kettering-IMPACT targeted DNA sequencing was performed to identify secondary genetic alterations showing more than half of tumors had a genetic alteration beyond the disease-defining gene fusion. Patients with conventional epithelioid hemangioendothelioma with WWTR1-CAMTA1 fusion had a less favorable outcome compared to the YAP1-TFE3 subset, the 5-year-overall survival being 59% versus 86%, respectively. Soft tissue epithelioid hemangioendothelioma were frequently solitary, followed an uneventful clinical course being often managed with curative surgery. Multifocality, pleural involvement, lymph node or distant metastases had a significantly worse outcome. Patients with pleural disease or lymph node metastases had an aggressive clinical course akin to high grade sarcomas, with 22% and 30%, respectively, alive at 5 years, compared to >70% survival rate in patients lacking these 2 adverse factors.
Keywords: epithelioid hemangioendothelioma, fusion, CAMTA1, WWTR1, YAP1, TFE3, survival
INTRODUCTION
Epithelioid hemangioendothelioma is a rare vascular malignancy arising at various anatomic sites, showing a significant heterogeneity in clinical presentation and prognosis. Its clinical behavior is intermediate in severity between the benign nature of hemangiomas and highly aggressive angiosarcomas [1, 2]. Epithelioid hemangioendothelioma affects women more than men and generally has a peak incidence in the fourth or fifth decade [3, 4]. While it can arise in any part of the body, epithelioid hemangioendothelioma has a predilection for the lung, liver, and soft tissue [4, 5]. Prior studies report that 20 to 30% of patients develop metastatic disease, with a 5-year overall survival rate of greater than 70% [4, 6, 7].
Epithelioid hemangioendothelioma has distinctive morphologic features, composed of relatively monomorphic epithelioid cells arranged in cords and nests within a myxohyaline stroma. In contrast to most other epithelioid vascular tumors, epithelioid hemangioendothelioma lacks overt evidence of vasoformation [3]. Instead, cells show intracytoplasmic lumina represented by variable number of vacuoles, so called blister-cells. The genetic hallmark of epithelioid hemangioendothelioma is a recurrent WWTR1-CAMTA1 gene fusion, present in the overwhelming majority of cases [8, 9]. However, a small subset are characterized by a distinct morphology and a YAP1-TFE3 gene fusion. These tumors are composed of well-formed vascular channels, cells with abundant eosinophilic cytoplasm, and strong nuclear expression of TFE3 on immunohistochemistry [10]. Both WWTR1 (TAZ) and YAP1 represent transcriptional co-activators downstream of the Hippo pathway, being expressed in the endothelial lineage and likely providing a strong promoter for the oncogenic activation of both CAMTA1 and TFE3 [10, 11]. Despite some overlap in clinical presentation, it remains unclear if the YAP1-TFE3 molecular subset should be regarded as a member of the epithelioid hemangioendothelioma family. In this study we investigate a large cohort of molecularly confirmed epithelioid hemangioendothelioma with either WWTR1-CAMTA1 or YAP1-TFE3 fusion diagnosed or treated at Memorial Sloan Kettering Cancer Center, to identify clinicopathologic factors with prognostic significance and to outline treatment patterns and clinical outcomes.
MATERIALS AND METHODS
Patient and Tumor Characteristics
Approval for this retrospective study was obtained from the Memorial Sloan Kettering Cancer Center Institutional Review Board. The medical record and personal consultation files of the senior author were searched for patients diagnosed with epithelioid hemangioendothelioma. All cases included in the study tested positive for either the WWTR1-CAMTA1 or YAP1-TFE3 fusion by fluorescence in situ hybridization (FISH). Hematoxylin and eosin–stained slides and previously performed immunohistochemical stains were reviewed and histological grade was reassessed in each case.
The medical records were reviewed to identify date of diagnosis, patient age at the time of diagnosis, location of the primary tumor(s), other distant metastatic foci, and date of last follow-up or death. Tumors were labeled as unifocal if only a solitary lesion was present. Multifocal disease was designated if multiple tumors were present within one organ. The presence of distant metastases were defined as tumor involvement outside the organ of origin. For example, if a patient presented with multifocal pulmonary nodules and one bone lesion, the latter was designated as distant metastasis. In most cases (including the multifocal lung and/or liver epithelioid hemangioendotheliomas) an exhaustive cross-sectional imaging and PET-CT scans were performed to exclude other primary sites (such as soft tissues), but were not found.
Patients with epithelioid hemangioendothelioma often present with multiple involved organs, commonly in the lung and liver. In such cases, the site of origin could not be determined and were designated as multifocal metastatic tumors. Lung tumors were classified as solitary nodular, multifocal nodular – so called ‘intravascular bronchiolo-alveolar tumors’, pleural-based, or mixed patterns (parenchymal and pleural), based on the pathologic and radiologic findings. Histologic grading was defined based on a combination of factors including: moderate to marked nuclear pleomorphism, increased mitotic figures of >2 mitotic figures /10 high power fields and presence of necrosis. If two or more of the above features were present the tumor was designated as malignant or grade II. Tumors lacking nuclear pleomorphism, increased mitotic activity, or necrosis were designated as grade I. The date of last follow-up was defined as the date of last contact at our institution or documentation from an outside institution indicating vital status. The follow-up clinical data retrieved included: modality of initial therapy, recurrence status and time on each line of systemic treatment. Some cases were pathologic consultation requests from outside institutions and follow-up was obtained by contacting the individual pathologists, but in some patients the clinical information available remained limited. The hospital database of tumors with IMPACT and/or Archer testing was searched to identify epithelioid hemangioendothelioma samples with next generation sequencing or RNA sequencing data, respectively.
Immunohistochemistry
Immunohistochemical stains available were reviewed, including CD31 and ERG (Ventana; prediluted) to confirm endothelial differentiation. Most cases were also tested for epithelial markers such as AE1/AE3 (Dako; 1:400) and CAM5.2 (Becton Dickinson; 1:50), as part of the initial workup when vascular differentiation was not evident. All YAP1-TFE3 positive epithelioid hemangioendotheliomas were also tested for TFE3 immunoreactivity (Ventana; prediluted).
FISH
FISH on interphase nuclei from paraffin-embedded 4-mm-thick sections was performed applying custom probes using bacterial artificial chromosomes (BAC) for WWTR1, CAMTA1, TFE3 and YAP1. BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu). (Supplem Table 1). The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (Oakland, CA) (http://bacpac.chori.org). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described [10]. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were excluded.
Memorial Sloan Kettering-IMPACT and Archer Assay
The IMPACT next generation sequencing platform has been described in detail previously [12]. It is an FDA-approved hybridization capture-based genomic sequencing assay performed in a Clinical Laboratory Improvement Ammendments-certified laboratory that examines all exons and selected introns of 310 to 468 cancer-associated genes, depending on the version. Tumor samples lacking oncogenic alterations by IMPACT are nominated for targeted RNA sequencing (Archer FusionPlex Custom Solid Panel) to assess for gene fusions. The detailed procedure of Anchored Multiplex PCR RNA sequencing assay has been previously described [13]. Unidirectional gene-specific primers were designed to target specific exons in 62 genes known to be involved in oncogenic fusions in solid tumors. Genomic alterations detected on IMPACT are annoteded according to the OncoKB database [14], a precision oncology knowledge base denoting the oncogenic effects and predictive significance of molecular alterations. Genomic data and OncoKB annotations were visualized in cBioPortal for Cancer Genomics [15, 16].
Statistical analysis
Statistical analysis was performed on SPSS software 22.0 (IBM Corporation, New York, NY, U.S.). The associations between the clinical variables and matched groups were evaluated by Fisher’s exact test. The overall survival time was measured in months from the date of diagnosis to the date of death. Kaplan-Meier estimate was used to calculate the overall survival. The statistical significance of different clinicopathologic variables (gender, age, primary location, presence of metastasis at diagnosis, pleural or lymph node involvement) in relation to survival was assessed by log-rank analysis. Prognostic variables that were significant on univariate analyses were subsequently subjected to multivariate analyses using the Cox proportional hazards model. A p<0.05 was considered as significant for all statistical analyses.
RESULTS
Patients Demographics and Clinical Presentation
A total of 83 patients with WWTR1-CAMTA1 gene fusion positive epithelioid hemangioendothelioma were diagnosed at Memorial Sloan Kettering Cancer Center between July 1992 and November 2018. Patient and tumor characteristics are described in Table 1. There was a slight female predominance (n = 43; 52%) and age at diagnosis ranged between 18 and 80 years, with a median age of 51. Histologic grade for the classic epithelioid hemangioendothelioma variant was nearly split between grade I and grade II (48% and 52%, respectively). The majority of patients (n=61; 73%) had multifocal primary disease at the time of diagnosis and 70% (n=58) developed distant metastases. Primary lung epithelioid hemangioendothelioma without evidence of distant disease was the most common clinical presentation (n=31; 37%). In combination with other metastatic foci, such as the liver or bone, 49 cases (59%) involved the lung at diagnosis.
Table 1. Clinical and Pathologic Features of Patients with Molecularly Confirmed Epithelioid Hemangioendothelioma.
| WWTR1-CAMTA1 | YAP1-TFE3 | ||||
|---|---|---|---|---|---|
| N | % | N | % | ||
| Total | 83 | 10 | |||
| Sex | F | 43 | 52 | 6 | 60 |
| M | 40 | 48 | 4 | 40 | |
| Age | <50 | 41 | 49 | 9 | 90 |
| >50 | 42 | 51 | 1 | 10 | |
| Primary Site | Lung | 31 | 37 | 2 | 20 |
| ST | 17 | 20 | 3 | 30 | |
| Liver | 13 | 16 | 0 | 0 | |
| Lung/liver | 13 | 16 | 0 | 0 | |
| Lung/bone | 5 | 6 | 0 | 0 | |
| Bone | 3 | 4 | 0 | 0 | |
| Peritoneal | 1 | 1 | 0 | 0 | |
| Liver, lung | 0 | 0 | 2 | 20 | |
| Liver, lung, bone | 0 | 0 | 2 | 20 | |
| Liver, lung, ST | 0 | 0 | 1 | 10 | |
| Pattern of Pulmonary Spread | Multifocal nodular | 23 | 28 | 5 | 50 |
| Mixed | 17 | 20 | 0 | 0 | |
| Pleural | 7 | 8 | 0 | 0 | |
| Nodular | 2 | 2 | 1 | 10 | |
| Unknown | 0 | 0 | 1 | 10 | |
| Focality | Multifocal | 59 | 71 | 7 | 70 |
| Unifocal | 24 | 29 | 2 | 20 | |
| Unknown | 0 | 0 | 1 | 10 | |
| Metastasis | N | 25 | 30 | 3 | 30 |
| Y | 58 | 70 | 7 | 70 | |
| Type of metastasis | Distant | 29 | 35 | 6 | 60 |
| Regional | 29 | 35 | 1 | 10 | |
| Unknown | 25 | 30 | 3 | 30 | |
| LN | N | 63 | 76 | 4 | 40 |
| Y | 20 | 24 | 4 | 40 | |
| Unknown | 0 | 0 | 2 | 20 | |
| Pleural disease at diagnosis | N | 59 | 71 | 8 | 80 |
| Y | 24 | 29 | 1 | 10 | |
| Unknown | 0 | 0 | 1 | 10 | |
| Pleural involvement | N | 54 | 65 | 8 | 80 |
| Y | 29 | 35 | 1 | 10 | |
| Unknown | 0 | 0 | 1 | 10 | |
| Grade | I | 40 | 48 | 7 | 70 |
| II | 43 | 52 | 3 | 30 | |
ST = soft tissue, Y = yes, N= no
The pathologic and/or radiologic pattern of lung involvement varied: most patients (n=23; 47%) had multiple pulmonary nodules or masses scattered throughout the lung parenchyma. In 35% of cases (n=17), parenchymal nodules and the pleural surface were involved in a mixed pattern. The remaining cases either involved the pleura only (n=7; 14%), or rarely (1; 2%), manifested as a solitary pulmonary nodule. Among the entire WWTR1-CAMTA1 positive cohort, 29 patients (35%) had pleural involvement, which most commonly (83% of pleural cases) was present at diagnosis.
Among patients with unifocal disease, the soft tissues were the most common site of primary disease (n=16; 73%), followed by bone (3; 14%), liver (2; 9%), and lung (1; 5%). All patients (n=3) with primary bone tumors had unifocal disease. A total of 17 patients (20% of the whole WWTR1-CAMTA1-positive cohort) had primary soft tissue tumors, all but one had solitary disease. Other relatively common presentations included liver-contained disease (n=13; 16%) or synchronous lung and liver involvement (n=13; 16%). Nearly one quarter of patients (n=20; 24%) developed lymph node metastases. Other metastatic foci in descending order of frequency included soft tissue, bone, lung and/or pleura, lymph nodes, visceral sites, and brain.
Ten patients who were diagnosed with epithelioid hemangioendothelioma between May 1991 and December 2018 were positive for a YAP1-TFE3 gene fusion. The median age at the time of diagnosis in this cohort of patients was 34 years (range: 28–62), six of whom were female. Primary tumors originated in the soft tissue (n=3), lung (2), or liver and lung (2) with synchronous bone (2) or soft tissue involvement (1). Two of three patients with unifocal disease at diagnosis had disease originating in soft tissue. The other patient with soft tissue disease presented with locally destructive disease of the head and neck, involving the calvarium. Six of the ten patients had confirmed distant metastases at diagnosis. One patient had pleural involvement and 4 developed lymph node metastases. Eight of ten YAP1-TFE3 patients were alive at the last follow-up; one died of disease after 17 years of follow-up and the other died of acute myeloid leukemia 24 years after initial diagnosis.
Pathologic characteristics of various anatomic sites and histologic grading
Thoracic epithelioid hemangioendothelioma. Out of 49 patients with lung involvement, one patient presented with a solitary lung nodule; the remaining showed either multiple nodules, pleural involvement or a mixed pattern (Fig 1,2). The patients with ‘intravascular bronchiolo-alveolar tumors’-type involvement showed multiple to miliary parenchymal nodules, documented radiographically or on pathology reports. Microscopically, the tumor was present within the alveolar spaces as tumor plugs retaining the alveolar architecture, or forming confluent sheets with complete obliteration of the alveolar framework (Fig 3). Some of these lesions showed significant nuclear pleomorphism, vascular invasion and necrosis in keeping with a malignant /grade II disease (Fig 2,3). There were 2 clinical presentations of pleural involvement: pleural-based disease without parenchymal invasion and mixed pleural-parenchymal involvement (Fig 1,4). The degree of pleural involvement was quite variable, ranging from localized to a segment or lobe, to diffuse, encasing the entire lung as a thick fibrous shell, reminiscent of mesothelioma. Indeed, one of the patients required pneumenectomy due to its diffuse extent of disease (Fig 4). Occasionally, the pleural involvement extended into the lung fissures, sparing the parenchyma (Fig 4). Microscopically, the pleural epithelioid hemangioendothelioma had distinct morphologic features compared to other anatomic sites, showing epithelioid to spindled endothelial cells embedded within a prominent desmoplastic stroma (Fig 4). In fact, a significant number of pleural epithelioid hemangioendothelioma had a predominant spindle cell phenotype, either arranged in short fascicles or haphazard growth, with variable degree of nuclear pleomorphism and hyperchromasia. These features posed diagnostic challenges, particularly in small samples, mimicking either inflammatory fibrous proliferations or sarcomatoid mesothelioma. Furthermore, the positivity for cytokeratin in isolation was also a pitfall, suggesting a metastatic carcinoma or mesothelioma. Thus, the molecular demonstration of WWTR1-CAMTA1 fusion in these cases was paramount in confirming the correct diagnosis of epithelioid hemangioendothelioma.
Figure 1.
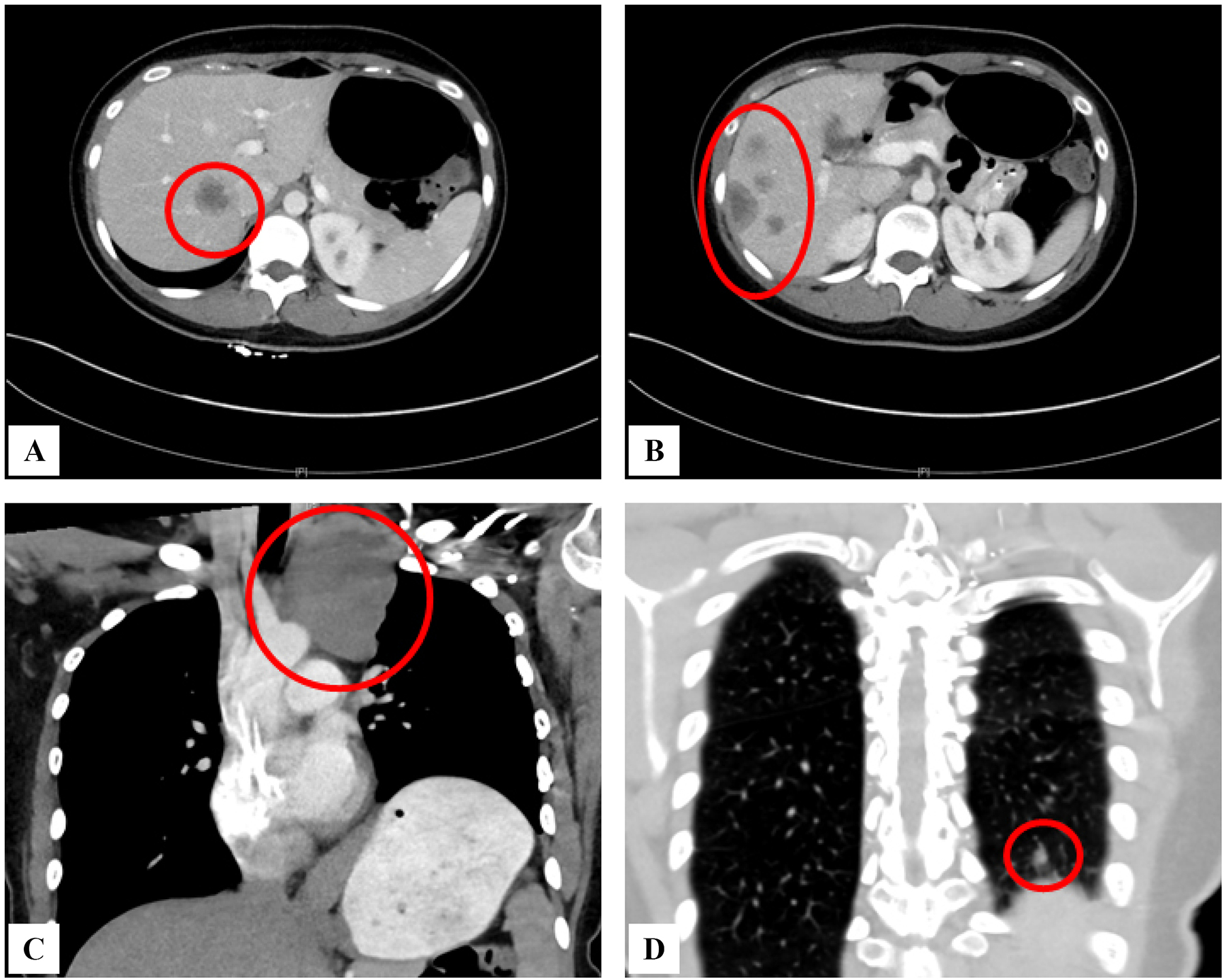
Radiographic images illustrating a multifocal metastatic epithelioid hemangioendothelioma patient with parenchymal, pleural and liver involvement.
Figure 2.
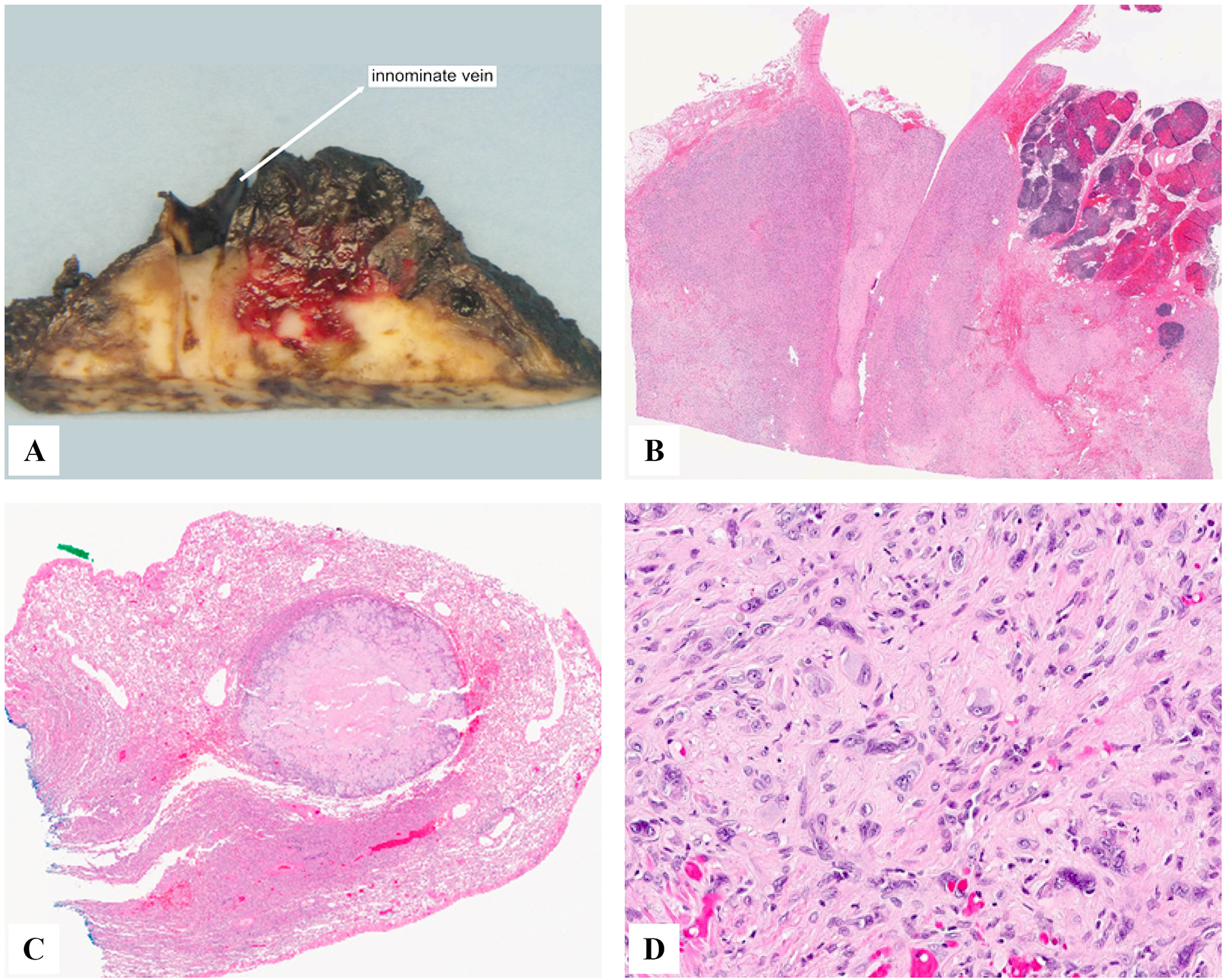
Pathologic examination of same patient as above showing intravascular growth involving the innominate vein (A,B), surrounding mediastinal soft tissue, thymus and pleura (B), and a well-defined intra-pulmonary nodule (C). The pleural disease showed increased nuclear pleomorphism and mitotic activity and a mixed epithelioid and sarcomatoid growth (D), in keeping with a histologic grade II.
Figure 3.
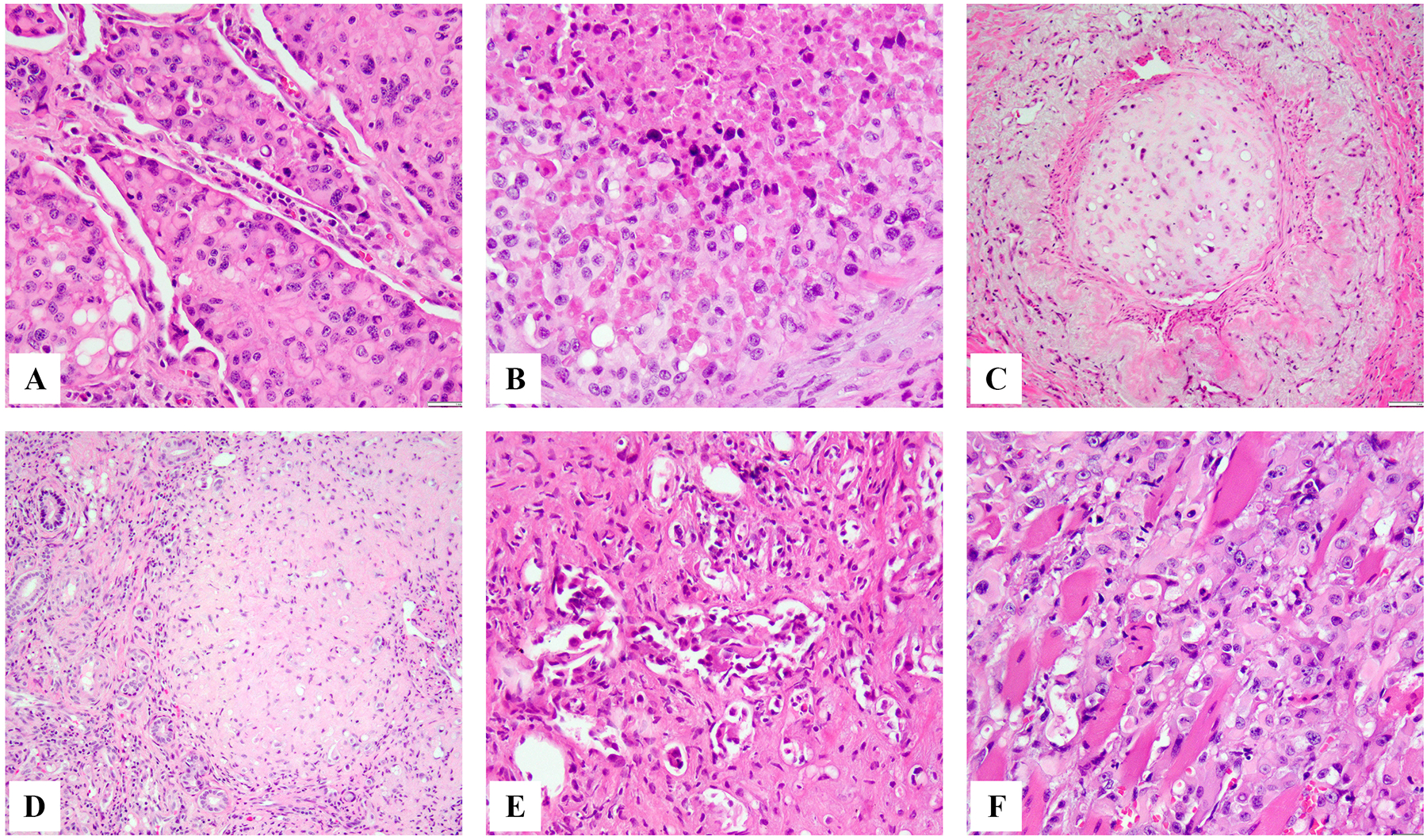
Microscopic spectrum of epithelioid hemangioendothelioma with WWTR1-CAMTA1 fusion in different clinical presentations. Pulmonary involvement showing solid nodular growth of epithelioid hemangioendothelioma present as tumor plugs within still visible alveolar spaces (A) or completely effacing the alveolar architecture and showing confluent growth with central necrosis (B). Intravascular growth within variable caliber vessels is a common finding in different clinical presentations of epithelioid hemangioendothelioma (C). Hepatic epithelioid hemangioendothelioma often shows a sclerotic, scar-like appearance with lesional cells barely visible (D) or frequent intravascular growth within sinusoids and small vessels (E). Soft tissue epithelioid hemangioendothelioma showing infiltrative growth within skeletal muscle (F).
Figure 4.
Radiographic and pathologic appearance of pleural involvement of epithelioid hemangioendothelioma. MRI and PET scan showing complete encasement of the left lung by a circumferential pleural involvment (A,B), which required a total pneumonectomy (C), confirming the pleural-confined disease, without parenchymal involvement. Microscopic appearance showing variable degrees of pleural involvement, ranging from more confluent, expansile areas (D) to linear, narrow thickening (E); other cases showed extension of disease from the pleural surface along the fissures, which appeared thickened, often lacking evidence of intra-alveolar spread (E,F). High magnification of pleural disease often shows a more spindle/sarcomatoid phenotype, with a variable number of interspersed epithelioid cells (H,I), highlighted by ERG immunostaining (J).
Liver epithelioid hemangioendothelioma. A common denominator for hepatic epithelioid hemangioendothelioma was the fibrotic stromal background, most lesions appeared hypocellular and deceptively bland. Diagnosis was especially challenging in small core biopsies, where some of these features could easily be overlooked, in the absence of supporting immunohistochemistry for endothelial markers. A consistent feature was the presence of intravascular disease, either within the hepatic sinusoids or within small and medium sized vessels (Fig 3).
Soft Tissue. Epithelioid hemangioendothelioma arising in soft tissues often displayed the classic morphologic features, including epithelioid endothelial cells arranged in cords, or nests, lacking vasoformative features (Fig 3). The lesional cells were often embedded within the distinctive chondromyxoid or hyaline stromal component.
Histologic grading: Among the WWTR1-CAMTA1 fusion positive tumors, 40 cases had a grade I, defined as mild nuclear pleomorphism, <2 mitotic figures/10 high power fields and lack of necrosis. 43 tumors were classified as malignant or grade II, if they had at least 2 of the following criteria: moderate to marked nuclear pleomorphism, increased mitotic activity >2 mitotic figures/10 high power fields and necrosis (Figs 2–5).
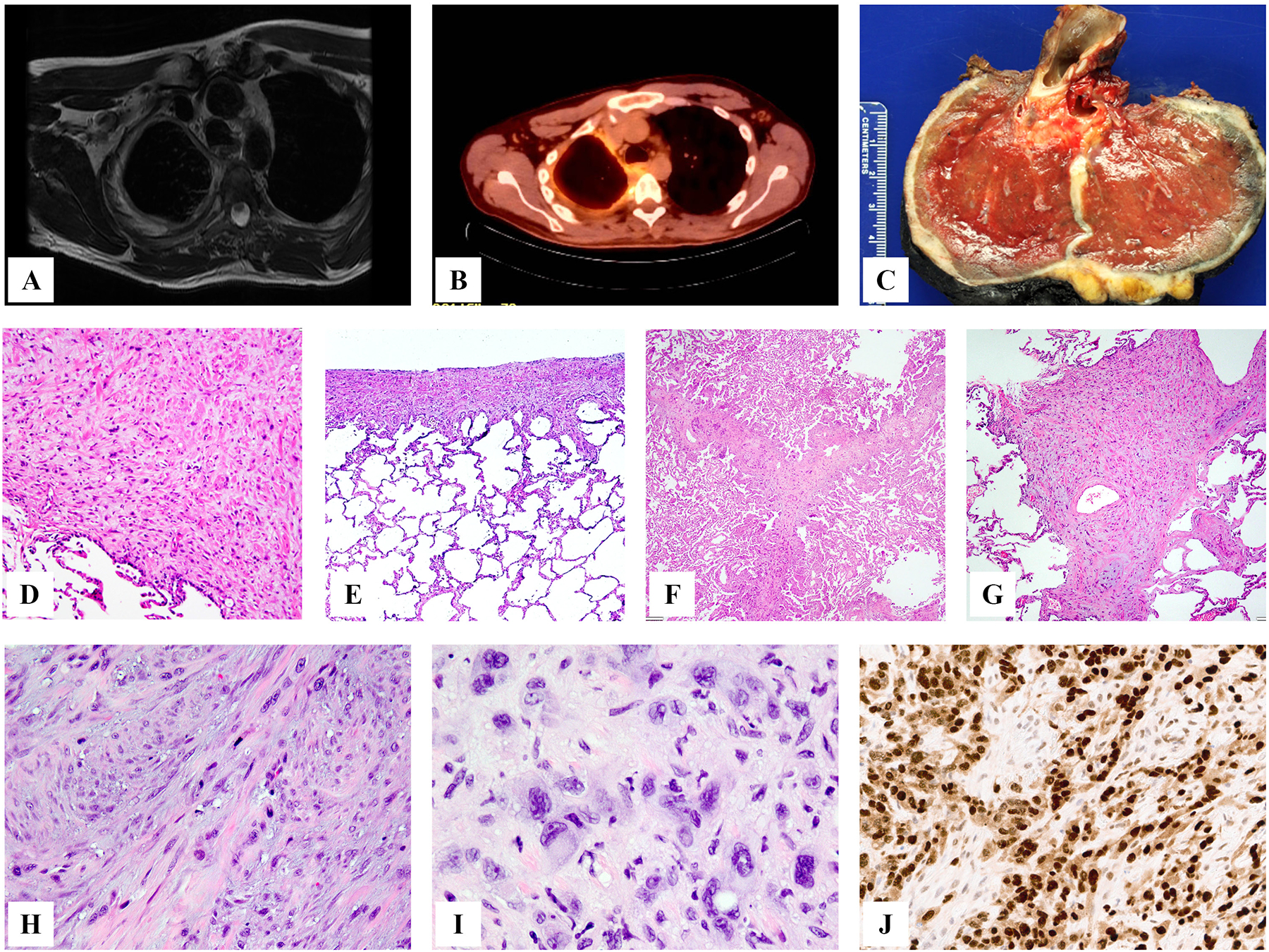
Figure 5.
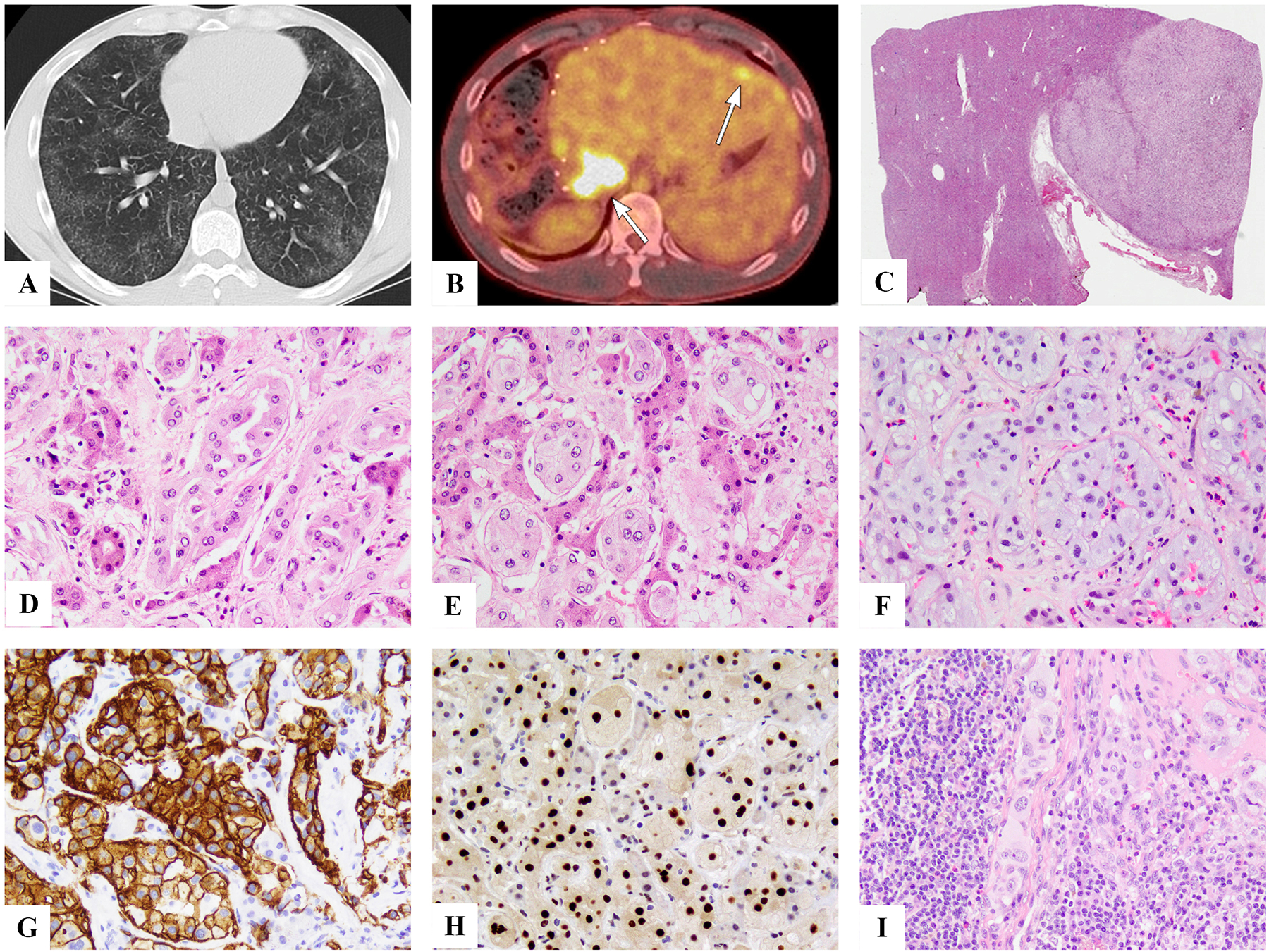
Clinical and pathologic features of YAP1-TFE3-positive epithelioid hemangioendothelioma. This patient presented with milliary lung involvement (A) and few liver nodules which appeared well-defined (B, C). Microscopic appearance reveals the distinct morphologic appearance, with well-formed vascular channels, lined by epithelioid cells with abundant eosinophilic to amphophilic cytoplasm, infiltrating in between hepatocytes (D,E) or completely obliterating alveolar architecture (F). Immunostains showed positiveity for CD34 (H) and TFE3 (I). Lymph node spread was also documented (J).
Survival Analyses
In the WWTR1-CAMTA1 fusion-positive cohort, the median follow-up time for survivors was 24 months (range: 0 – 289 months). Overall survival at 3 and 5 years for the entire cohort was 70% and 59%, respectively. Among patients with lung primary tumors, 3 and 5 year overall survival was 64% and 49%; among liver primary tumors, 77% and 67%, and among soft tissue primaries, all patients were alive at 3 and 5 years. Patients with tumors of the soft tissue had a significantly longer overall survival than patients with lung, liver, or bone primaries (p=0.004), while those with lung primaries had a significantly shorter overall survival (p=0.01) (Fig 6).
Figure 6.
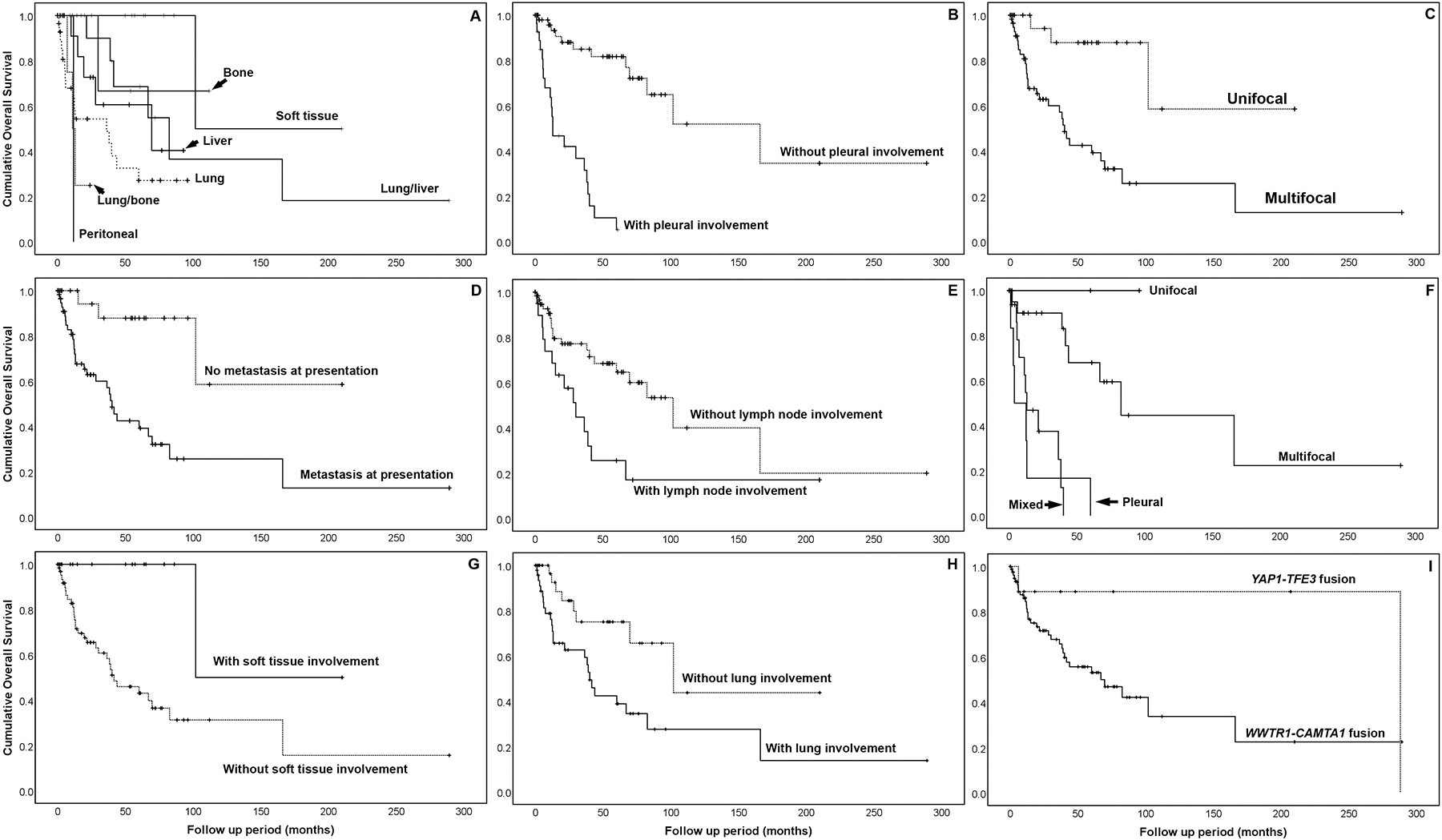
Kaplan-Meier survival curves showing overall survival for (A) site of primary tumor; (B) pleural involvement; (C) focality; (D) metastasis; (E) lymph node involvement; (F) type of lung involvement; (G) soft tissue; (H) lung involvement in WWTR1-CAMTA1 fusion-positive epithelioid hemangioendothelioma; and (I) molecular subtype.
To stratify patients into prognostically favorable or unfavorable subgroups, we perfomed univariate analyses of overall survival by the following clinicopathologic factors: sex, age, primary site of disease, pattern of pulmonary spread (if lung primary), multifocality at presentation, presence of metastases, lymph node metastases, pleural involvement, or histologic grade. Primary disease site (p=0.001), pattern of pulmonary spread (p<0.001), tumor multifocality (p=0.001), presence of metastases (p=0.001), lymph node involvement (p=0.006), pleural involvement (p<0.001), and histologic grade (p=0.049) were significant predictors of shorter overall survival.
As there were 4 distinct subgroups within the ‘pattern of spread’ category, multivariable analysis could not be performed using this classification given the low patient number in each group. On multivariate analysis, multifocal tumors (hazard ratio [HR] 68.1 95% confidence interval [CI] 3.3 – 1425; p=0.006), presence of metastases (HR 19.8 95% CI 1.4 – 281; p=0.027), lymph node involvement (HR 2.7 95% CI 1.2 – 6.2; p=0.014), and lung primary tumors (HR 4.2 95% CI 1.2 – 14.8; p=0.026) were factors associated with a shorter overall survival. Pleural involvement did not reach statistical significance, but was also associated with a shorter overall survival (HR 4.6 95% CI 0.99 – 21.4; p = 0.053).
The median follow-up time of survivors in the YAP1-TFE3 cohort was 17.9 months. The 3 and 5 year survival of YAP1-TFE3 fusion-positive patients was 88% and 86%, respectively. Although the disease specific survival rate at 5 years was 86% in TFE3-rearranged patients compared to 58% in CAMTA1-rearranged patients, the difference between groups did not reach statistical signficance (p = 0.095).
Initial Treatment Modality
Treatment records were available for 55 WWTR1-CAMTA1 and 7 YAP1-TFE3 fusion-positive patients; the remaining cases were pathology consultations from outside institutions or were lost to follow up shortly after presentation.
One-third of these patients (n=18) were managed initially with expectant observation. Half of those observed had lung disease only and half had lung plus liver involvement. Eight of these patients (44%) had no sign of disease progression at the date of last follow-up, 7 (39%) were started on systemic treatment after disease progression, and 3 (17%) died without initiating systemic therapy (1 was lost to follow up and 2 were referred for local therapies for symptom control and likely succumbed to rapid disease progression). Of the patients who progressed after initial observation, 7 had lung plus liver primary disease, 3 developed pleural metastases, and 4 had lymph node metastases.
Twenty-four patients (44% of patients with treatment records) underwent upfront surgical resection as initial management, 75% of whom had no evidence of disease recurrence at the time of last follow-up. Among those who underwent surgery, 71% had unifocal disease. The patients with multifocal disease that underwent upfront surgery had only one involved organ (lung or liver) in 6 of 7 cases. Patients who ultimately had curative resection (n=18) had primary epithelioid hemangioendothelioma of the soft tissue (67%), liver (17%), bone (11%) or lung (6%). One patient with clinical follow-up available underwent primary liver transplant for multifocal liver epithelioid hemangioendothelioma, developed recurrent liver disease less than two years after transplant, and ultimately distant metastases requiring systemic treatment.
Thirteen patients (24% of patients with treatment records) received upfront systemic therapy, all of whom had multifocal disease at the time of diagnosis. Five patients had lung involvement only, 3 patients each had lung plus liver or lung plus bone involvement, and one patient each with liver or peritoneal involvement at diagnosis. The majority (58%) of patients with pleural disease at diagnosis received upfront systemic treatment and none were taken to surgery.
Among the 7 patients with the YAP1-TFE3 rearrangement with clinical follow-up, 3 underwent initial surgery and 2 were managed with either observation or systemic treatment, respectively. Two of the three patients who underwent surgery remained without evidence of disease, one of whom required resection of a local recurrence. One of two patients who were observed initially remained on expectant observation, while one progressed and initiated systemic therapy. Both patients treated with initial systemic therapy had multifocal disease at diagnosis, one of which involved the pleura.
Systemic Therapy
Systemic treatment regimens varied widely between patients and included single-agent cytotoxic chemotherapy, multi-agent cytotoxic chemotherapy, multitargeted tyrosine kinase inhibitors, monoclonal antibodies against VEGF and PDGFRA, and anti-PD1-based immunotherapy, among other targeted agents (e.g. mTOR and MEK inhibition). In addition, as demonstrated in the Swimmer’s plot (Supplem Fig 1), local treatment modalities, such as surgery, embolization, or radiation, were frequently utilized in conjunction with systemic therapy and often contributed to prolonged disease control. Patients who had systemic treatment had a median of 2 lines of treatment (range: 1–5). The median time on treatment per line of therapy was 73 days (range: 1–767 days). The most common regimens were liposomal doxorubicin, sorafenib, pazopanib, and gemcitabine-based therapy (Supplem Table 1). Patients on anti-PD-1 based immunotherapy had the longest median duration of treatment (210 days) when compared to other systemic treatment categories (Supplem Fig 2), although only 4 patients were treated with checkpoint blockade.
Next Generation Sequencing
Fifteen patients with WWTR1-CAMTA1 and 3 patients with YAP1-TFE3 fusion-positive epithelioid hemangioendothelioma underwent IMPACT testing. Two patients underwent germline genetic testing. The WWTR1-CAMTA1 fusion was confirmed by Archer in 5 cases and 3 YAP1-TFE3 fusions were identified on IMPACT. All 15 WWTR1-CAMTA1 tumor samples had matched normal tissue comparators. Eight patients had no detectable somatic alterations. Seven had additional mutations beyond the canonical disease-defining fusion and one patient had multiple copy number alterations (Supplem Fig 3). Three of fifteen (20%) patients had at least one likely oncogenic alteration. Two patients had loss-of-function mutations in ATRX (E2262 and K1317 nonsense mutations), one of whom also had an oncogenic FAT1-intragenic fusion. Other oncogenic alterations included a frameshift deletion in XRCC2 (K182Rfs*33), a splice site alteration in KMT2C (X130_splice), and deep deletions in CDKN2A and CDKN2B. No likely oncogenic somatic alterations were detected in any of the patients with YAP1-TFE3 fusion. On germline genetic testing, one patient with a YAP1-TFE3 tumor had a loss-of-function missense mutation in APC (I1307K). Overall, 78% of samples had at least one genetic alteration on next generation sequencing.
DISCUSSION
Weiss and Enzinger coined the term epithelioid hemangioendothelioma to describe an epithelioid and angiocentric soft tissue tumor with a clinical course less aggressive than conventional angiosarcoma, but more aggressive than benign hemangiomas. Nearly one-fifth of patients in their first reported case series developed distant metastases to the lung, liver, bone or lymph nodes, indicating that a subset of patients have an aggressive clinical course [1]. The potentially malignant nature of epithelioid hemangioendothelioma was confirmed in subsequent series by Mentzel et al [6] and then by Dail and colleagues [17], in which greater than 20% of patients developed distant metastases, one-quarter developed clinical lymphadenopathy, and a subset had pleural involvement. These patients were noted to have worse outcomes and chemotherapy had no appreciable clinical effect, findings which were later confirmed by other groups [17, 18]. Furthermore, in a prior molecular study we have shown that multifocal liver epithelioid hemangioendothelioma represents a clonal process, demonstrating that separate lesions harbor fusions with an identical genomic breakpoint, in keeping with a single clone, i.e. metastatic dissemination confined to one organ [19].
The present study represents the most comprehensive clinicopathologic and molecular investigation of epithelioid hemangioendothelioma to date, including 93 patients with translocation-confirmed tumors. Although other large series have evaluated the clinicopathologic features of epithelioid hemangioendothelioma [5, 6, 20–22], our study is unique by including only patients with molecularly confirmed tumors and the incorporation of next generation sequencing in select cases. As detailed in the pathologic description of various anatomic sites, epithelioid hemangioendothelioma spans a wide morphologic spectrum, and the differential diagnosis includes not only other vascular tumors, but also metastatic carcinoma or mesothelioma. Thus, it is quite possible that prior studies did not include tumors deviating from the classic morphology, especially lesions with an unusual spindle/sarcomatoid phenotype or showing significant nuclear pleomorphism, which would have been regarded as angiosarcomas or other neoplasms.
Furthermore, this is the first study investigating the two distinct molecular subsets of epithelioid hemangioendothelioma side-by-side, in an attempt to compare their clinical features and biologic behavior. While our results confirm many of the key early findings, it underscores the remarkable dichotomy in the clinical behavior of this disease, despite a homogeneous initiating genetic event. At one end of the spectrum, patients with either solitary soft tissue or multifocal lung plus liver disease follow a relatively indolent course in keeping with a low grade process and for which surgical resection appears at least beneficial, if not curative in localized lesions. Expectant observation is a reasonable approach for such tumors that are unresectable or those that may be highly morbid if resected. At the other end of the spectrum, patients with either pleural or lymph node involvement, regardless of their primary site or pathologic grade, follow a highly aggressive clinical course akin to a high grade sarcoma, despite multimodality therapy. Based on these clinical and pathologic findings, a risk assesment can be performed to stratify patients based on the presence of adverse factors and select patients with high risk disease who may benefit from aggressive systemic therapy in conjunction with appropriate local therapies to achieve disease control.
The most common sites of disease among epithelioid hemangioendothelioma with WWTR1-CAMTA1 fusion were the lung, soft tissue, and liver, with approximately 70% of patients having multifocal disease at presentation. Distant metastatic disease was common, manifesting in 51% of cases at diagnosis. Pleural and lymph node metastases represent poor prognostic signs and were significantly associated with a worse overall survival. Only 22% and 30% of patients with pleural or lymph node metastases, respectively, were alive at 5 years. This is compared to a greater than 70% survival rate at 5 years in patients lacking these 2 adverse factors. Only one patient in our cohort presented with diffuse peritoneal involvement, including omental caking and ‘serosal carcinomatosis’, and showed a fulminant clinical progression, likely representing similar disease biology to those with pleural involvement.
Ten patients with the YAP1-TFE3 fusion were included in this series. Similar to WWTR1-CAMTA1 positive cases, soft tissue, liver, and lung were common sites of primary disease and distant metastatic spread occurred in 7 of 10 patients at diagnosis, as well as locoregional lymph node involvement in the majority of patients. Patients with YAP1-TFE3-fusion positive tumors tended to be younger at diagnosis, have less frequent pleural disease, and trended towards an improved overall survival, potentially representing a more indolent subtype of epithelioid hemangioendothelioma. These findings support our previous studies [10] that found a distinct morphology and clinical behavior among the YAP1-TFE3 positive epithelioid hemangioendothelioma patients, potentially reflecting a distinct pathogenesis of the underlying fusion.
In keeping with the heterogeneity of the clinical presentations, the treatment modalities applied varied depending on the primary tumor site, disease focality, and presence of symptoms. Most patients with liver and/or lung disease were initially observed, with roughly half of them remaining stable without need for systemic therapy. Patients with unifocal disease, especially in the soft tissue or liver, did well with surgical resection as the primary treatment modality, with 75% demonstrating no evidence of recurrence at last follow-up. Systemic therapy was reserved for those with symptomatic multifocal or metastatic disease or those who failed a trial of observation or those who recurred after surgical resection.
As demonstrated previously, epithelioid hemangioendothelioma patients can benefit from systemic therapy, although mainly through disease stabilization rather than tumor shrinkage [23]. As objective responses are uncommon, it is often difficult to determine whether clinical benefit is derived from the treatment or due to underlying indolent disease. Based on the poor survival outcomes of patients with pleural involvement and lymph node metastases, initiating systemic therapy early in the treatment course of these patients may be beneficial. Anti-angiogenic agents were among the most commonly used systemic treatment in this cohort, likely reflecting the early-phase prospective data demonstrating favorable responses with agents such as bevacizumab and sorafenib [24, 25]. Both single and multiagent cytotoxic regmens were also commonly used, either doxorubicin-, gemcitabine-, or taxane-based. Interestingly, four patients were treated with anti-PD-1-based therapy and had favorable disease stabilization rates in comparison with other treatment regimens used in our cohort. This is merely a signal of efficacy, though, as the number of patients treated were small and the indolent nature of select epithelioid hemangioendothelioma cases may be confounding.
Our next generation sequencing findings suggest that a personalized approach to epithelioid hemangioendothelioma treatment may be promising. More than half of sequenced patients had a genetic alteration beyond the disease-defining gene fusion, while 22% had putatively oncogenic alterations. These loss-of-function alterations involved the DNA damage response, cell cycle, and epigenetic pathways. XRCC2 is a member of the RAD51 complex essential in the repair of double-stranded breaks via homologous recombination [26]. Pre-clinical data suggests that XRCC2 mutant tumors may be susceptible to treatment with poly(ADP-ribose) polymerase 1 (PARP) inhibitors [27]. Interestingly, a recently published case report noted other DNA damage response alterations, ERCC1 and XRCC1 mutations, in an epithelioid hemangioendothelioma patient [28]. CDKN2A and CDKN2B are tumor suppressors essential in the cell cycle regulation that inhibit CDK4 and CDK6 and activate RB1, leading to cell cycle arrest [29, 30]. Tumors with CDKN2A alterations have demonstrated sensitivity to CDK4/6 inhibitors [28]. ATRX is essential to chromatin remodeling and histone deposition in areas of repetitive DNA repeats, including telomeres [31]. Loss of ATRX impairs homologous recombination repair after DNA double stranded breaks and leads to an alternative lengthening of telomere phenotype [32]. Therapeutic approaches to target ATRX-altered tumors are actively being studied [31, 33].
In summary, this study highlights the relatively high 5-year overall survival rate among patients with epithelioid hemangioendothelioma, indicating that this disease is often indolent. Yet, a subset of patients with pleural and lymph node involvement have significantly shorter survival and a much more aggressive clinical course, likely reflecting inherent differences in underlying tumor biology. Currently, there is no standard of care for treating this disease and multiple systemic therapies offer long term benefit, primarily in the form of disease stabilization. Immune checkpoint blockade with anti-PD1 antibodies demonstrated early signs of clinical efficacy in this study and may represent a novel approach to treating epithelioid hemangioendothelioma. In addition to identifying the gene-defining fusions, our next generation tumor sequencing platform detected potentially targetable oncogenic alterations. Larger studies are needed to further explore the genomic landscape of this rare sarcoma.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
Supported in part by: P50 CA140146-01 (SS, CRA); P50 CA217694 (SS, CRA, NPA); P30-CA008748 (CRA, NPA); Kristen Ann Carr Foundation (CRA); Cycle for Survival (CRA), Epithelioid Hemangioendothelioma Foundation (CRA, WDT)
Footnotes
CONFLICTS OF INTEREST
The authors delare that they have no conflicts of interest
Supplementary information is available at Modern Pathology’s website
REFERENCES
- 1.Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer. 1982;50:970–81. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher CDM, World Health Organization., International Agency for Research on Cancer. WHO classification of tumours of soft tissue and bone. 4th ed IARC Press: Lyon, 2013. 468 p. pp. [Google Scholar]
- 3.Antonescu C. Malignant vascular tumors--an update. Mod Pathol. 2014;27 Suppl 1:S30–8. [DOI] [PubMed] [Google Scholar]
- 4.Lau K, Massad M, Pollak C, Rubin C, Yeh J, Wang J, et al. Clinical patterns and outcome in epithelioid hemangioendothelioma with or without pulmonary involvement: insights from an internet registry in the study of a rare cancer. Chest. 2011;140:1312–8. [DOI] [PubMed] [Google Scholar]
- 5.Shiba S, Imaoka H, Shioji K, Suzuki E, Horiguchi S, Terashima T, et al. Clinical characteristics of Japanese patients with epithelioid hemangioendothelioma: a multicenter retrospective study. BMC Cancer. 2018;18:993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mentzel T, Beham A, Calonje E, Katenkamp D, Fletcher CD. Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol. 1997;21:363–74. [DOI] [PubMed] [Google Scholar]
- 7.Deyrup AT, Tighiouart M, Montag AG, Weiss SW. Epithelioid hemangioendothelioma of soft tissue: a proposal for risk stratification based on 49 cases. Am J Surg Pathol. 2008;32:924–7. [DOI] [PubMed] [Google Scholar]
- 8.Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer. 2011;50:644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Science translational medicine. 2011;3:98ra82. [DOI] [PubMed] [Google Scholar]
- 10.Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52:775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanas MR, Ma S, Jadaan FO, Ng CK, Weigelt B, Reis-Filho JS, et al. Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene. 2016;35:929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD. 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature medicine. 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 14.Chakravarty D, Gao J, Phillips S, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. 2017:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dail DH, Liebow AA, Gmelich JT, Friedman PJ, Miyai K, Myer W, et al. Intravascular, bronchiolar, and alveolar tumor of the lung (IVBAT). An analysis of twenty cases of a peculiar sclerosing endothelial tumor. Cancer. 1983;51:452–64. [DOI] [PubMed] [Google Scholar]
- 18.Kitaichi M, Nagai S, Nishimura K, Itoh H, Asamoto H, Izumi T, et al. Pulmonary epithelioid haemangioendothelioma in 21 patients, including three with partial spontaneous regression. Eur Respir J. 1998;12:89–96. [DOI] [PubMed] [Google Scholar]
- 19.Errani C, Sung YS, Zhang L, Healey JH, Antonescu CR. Monoclonality of multifocal epithelioid hemangioendothelioma of the liver by analysis of WWTR1-CAMTA1 breakpoints. Cancer Genetics. 2012;205:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flucke U, Vogels RJ, de Saint Aubain Somerhausen N, Creytens DH, Riedl RG, van Gorp JM, et al. Epithelioid Hemangioendothelioma: clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol. 2014;9:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson T, Zhang L, Hameed M, Rusch V, Travis WD, Antonescu CR. Thoracic epithelioid malignant vascular tumors: a clinicopathologic study of 52 cases with emphasis on pathologic grading and molecular studies of WWTR1-CAMTA1 fusions. Am J Surg Pathol. 2015;39:132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelini A, Mavrogenis AF, Gambarotti M, Merlino B, Picci P, Ruggieri P. Surgical treatment and results of 62 patients with epithelioid hemangioendothelioma of bone. J Surg Oncol. 2014;109:791–7. [DOI] [PubMed] [Google Scholar]
- 23.Cioffi A, Italiano A, Penel N, Berge Y, Toulmonde M, Salas S, et al. Metastatic epitheloid hemangioendothelioma (EHE): Role of systemic therapy and survival. 2011;29:10079-. [Google Scholar]
- 24.Agulnik M, Yarber JL, Okuno SH, von Mehren M, Jovanovic BD, Brockstein BE, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257–63. [DOI] [PubMed] [Google Scholar]
- 25.Chevreau C, Le Cesne A, Ray-Coquard I, Italiano A, Cioffi A, Isambert N, et al. Sorafenib in patients with progressive epithelioid hemangioendothelioma: a phase 2 study by the French Sarcoma Group (GSF/GETO). Cancer. 2013;119:2639–44. [DOI] [PubMed] [Google Scholar]
- 26.Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature. 1999;401:397–9. [DOI] [PubMed] [Google Scholar]
- 27.Xu K, Song X, Chen Z, Qin C, He Y. XRCC2 rs3218536 polymorphism decreases the sensitivity of colorectal cancer cells to poly(ADP-ribose) polymerase 1 inhibitor. Oncol Lett. 2014;8:1222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell. 2017;32:761–76 e6. [DOI] [PubMed] [Google Scholar]
- 29.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. [DOI] [PubMed] [Google Scholar]
- 30.Siebert R, Willers CP, Opalka B. Role of the cyclin-dependent kinase 4 and 6 inhibitor gene family p15, p16, p18 and p19 in leukemia and lymphoma. Leuk Lymphoma. 1996;23:505–20. [DOI] [PubMed] [Google Scholar]
- 31.Udugama M, Sanij E, Voon HPJ, Son J, Hii L, Henson JD, et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. 2018;115:4737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juhasz S, Elbakry A, Mathes A, Lobrich M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol Cell. 2018;71:11–24 e7. [DOI] [PubMed] [Google Scholar]
- 33.Han M, Napier CE, Frolich S, Teber E, Wong T, Noble JR, et al. Synthetic lethality of cytolytic HSV-1 in cancer cells with ATRX and PML deficiency. J Cell Sci. 2019; 132: jcs222349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.