

Abstract
Loss of function of voltage-gated potassium (Kv) channels is linked to a range of lethal or debilitating channelopathies. New pharmacological approaches are warranted to isoform-selectively activate specific Kv channels. One example is KCNA1 Potassium Voltage-Gated Channel Subfamily A Member 1 (KCNA1) (Kv1.1), an archetypal Shaker-type Kv channel, in which loss-of-function mutations cause episodic ataxia type 1 (EA1). EA1 causes constant myokomia and episodic bouts of ataxia and may associate with epilepsy and other disorders. We previously found that the inhibitory neurotransmitter γ-aminobutyric acid and modified versions of glycine directly activate Kv channels within the KCNQ subfamily, a characteristic favored by strong negative electrostatic surface potential near the neurotransmitter carbonyl group. Here, we report that adjusting the number and positioning of fluorine atoms within the fluorophenyl ring of glycine derivatives produces isoform-selective KCNA1 channel openers that are inactive against KCNQ2/3 channels, or even KCNA2, the closest relative of KCNA1. The findings refine our understanding of the molecular basis for KCNQ versus KCNA1 activation and isoform selectivity and constitute, to our knowledge, the first reported isoform-selective KCNA1 opener.
SIGNIFICANCE STATEMENT
Inherited loss-of-function gene sequence variants in KCNA1, which encodes the KCNA1 (Kv1.1) voltage-gated potassium channel, cause episodic ataxia type 1 (EA1), a movement disorder also linked to epilepsy and developmental delay. We have discovered several isoform-specific KCNA1-activating small molecules, addressing a notable gap in the field and providing possible lead compounds and a novel chemical space for the development of potential future therapeutic drugs for EA1.
Introduction
Currents generated by voltage-gated potassium (Kv) channels are essential for repolarizing excitable cells to end each action potential and, depending on the cell type, are also instrumental in modulating resting membrane potential, fluid and ion homeostasis, and direct regulation of sodium-coupled solute transporters. Native Kv channels are composed of complexes of pore-forming α subunits and a variety of regulatory subunits. Kv-α subunits are encoded by genes within a family comprising 40 members in the human genome, distributed between 12 subfamilies (Jan and Jan, 2012; Abbott, 2014). Typically, little functional redundancy is observed even among closely related Kv-α subunits (or, indeed, among β subunits) (Roepke and Abbott, 2006; Abbott, 2014; Crump and Abbott, 2014), explaining the expanding list of channelopathies linked to pathologic Kv-α subunit gene sequence variants and highlighting the need for development of isoform-selective Kv channel therapeutic small molecules.
Development of small molecules that isoform-selectively modulate Kv channels has lagged behind the discovery of Kv channelopathies, one reason being that Kv-α subunits within a subfamily often share very high sequence homology, especially among the pore and voltage sensor domains (VSDs) that are the most viable targets for pharmacological modulation. In addition, many Kv channelopathies involve loss of function (e.g., Long QT syndrome and most cases of inherited Kv channel epilepsy syndromes), requiring small molecules that activate rather than inhibit the channel, which is technically more challenging.
One example of a Kv channel that to date lacked an isoform-selective activator is KCNA1, also termed Kv1.1. KCNA1 is the first-cloned human Kv channel α subunit and is closely related to the first cloned, and extensively studied, Drosophila melanogaster Kv channel named Shaker (Jan and Jan, 2012). More than 40 years ago, a clinical entity now named episodic ataxia type 1 (EA1) was defined, involving bouts of constant myokomia, loss of balance, and motor coordination; EA1 sufferers are also predisposed to epilepsy (Spauschus et al., 1999; Eunson et al., 2000; Chen et al., 2007; Demos et al., 2009; Ishida et al., 2012). EA1 is caused by loss-of-function mutations in KCNA1 and has a prevalence of around 1 in 500,000 (Browne et al., 1994; Ptácek and Fu, 2002). The disease linkage was discovered >25 years ago, but in that time, no KCNA1-selective openers had been reported despite the acknowledged need for this as a therapeutic strategy (D’Adamo et al., 2015b). Very recently, complete loss of KCNA1 in a patient with a homozygous variant (p.Val368Leu) in the pore region that eliminated channel function was found to cause recessive neonatal epileptic encephalopathy and dyskinesia (Verdura et al., 2020). In addition, a de novo dominant-negative p.Leu328Val variant was recently found in a patient with tetany and hypomagnesemia (van der Wijst et al., 2018), following from previous work showing autosomal dominant linkage of human KCNA1 variants with hypomagnesemia (Glaudemans et al., 2009; van der Wijst et al., 2010). KCNA1 mutations are also thought to cause migraine, hyperthermia (D’Adamo et al., 2015a), motor developmental delay, and skeletal muscle deformities (Kinali et al., 2004). Inherited loss-of-function (typically dominant-negative) gene variants in related potassium channel–encoding gene KCNA2 are also associated with neurologic diseases, including episodic ataxia (Corbett et al., 2016), epileptic encephalopathies (Syrbe et al., 2015), and hereditary spastic paraplegia (Helbig et al., 2016). As KCNA2-specific small molecule activators are also to our knowledge lacking, new approaches are warranted for pharmacological targeting of KCNA family potassium channels. Here, we focus on KCNA1.
We previously made the surprising discovery that the primary inhibitory neurotransmitter γ-amino-butyric acid (GABA) directly activates neuronal Kv channels from the KCNQ (Kv7) family (KCNQ3 and KCNQ5) by binding in a pocket between the pore module and VSD (Manville et al., 2018). More recently, we discovered that glycine, another prominent inhibitory neurotransmitter, does not activate KCNQ channels, but we were able to re-engineer it to do so by introducing electrostatic properties predicted to be important in KCNQ channel activation (Manville and Abbott, 2019b). Here, we report that by testing a series of related glycine derivatives and introducing chemical properties predicted to disfavor targeting of KCNQ channels, we have discovered, to our knowledge, the first-reported isoform-selective activator of KCNA1.
Materials and Methods
Channel Subunit cRNA Preparation and Xenopus laevis Oocyte Injection.
We generated complementary RNA (cRNA) transcripts encoding human KCNA1, KCNA2, KCNQ2, KCNQ3, and Glycine Receptor Alpha 1 (GLRA1) using in vitro transcription with the mMessage mMachine kits (Thermo Fisher Scientific) following vector linearization. We injected defolliculated stage V and VI X. laevis oocytes (Xenocyte, Dexter, MI) with α subunit cRNAs (0.1–10 ng). We incubated the oocytes at 16°C in standard ND96 storage solution containing penicillin and streptomycin and washed them daily for 2 to 3 days prior to two-electrode voltage-clamp recording.
Two-Electrode Voltage Clamp.
We performed two-electrode voltage-clamp recording at room temperature with an OC-725C amplifier (Warner Instruments, Hamden, CT) and pClamp10 software (Molecular Devices, Sunnyvale, CA) 2–5 days after cRNA injection as described above. The oocytes were placed in a small-volume oocyte bath (Warner) and viewed with a dissection microscope. Unless otherwise stated, chemicals were from Sigma (St. Louis, MO). Bath solution was (in millimolars) as follows: 96 NaCl, 4 KCl, 1 MgCl2, 1 CaCl2, and 10 HEPES (pH 7.6). Chemicals were sourced from Sigma Aldrich, Matrix Scientific (Columbia, SC), and Santa Cruz Biotechnology (Dallas, TX). Glycine, (2-fluorophenyl) glycine (2FPG), N-(3-fluorophenyl)-N-(methylsulfonyl) glycine (3FMSG), 2-(Trifluoromethyl)-DL-phenylglycine (2TFMPG), and Z-D-Cyclohexylglycine were each solubilized in bath solution at a stock concentration of 10 mM; 4-(Fluorophenyl)glycine (4FPG) and 4-(trifluoromethyl)-L-phenylglycine (4TFMPG) were each solubilized in 1 M hydrochloric acid at a stock concentration of 10 mM. All stock solutions were diluted in bath solution on the day of experiments. Channel activation was screened for using either 30 or 100 µM concentrations of each of the compounds, and then dose responses were conducted as appropriate. We introduced the drugs into the oocyte recording bath by gravity perfusion at a constant flow of 1 ml per minute for 3 minutes prior to recording. Pipettes were of 1 to 2 MΩ resistance when filled with 3 M KCl.
Currents were recorded in response to pulses between −80 and +40 mV at 10 or 20 mV intervals, followed by a single tail pulse to −30 mV (to quantify voltage dependence of activation at a constant electrochemical driving force) from a holding potential of −80 mV. This yielded current-voltage relationships (mean and normalized) and current magnitude at baseline and in the presence of the compounds described. For GLRA1 studies, oocytes were clamped at -50 mV. Data were analyzed using Clampfit (Molecular Devices) and Graphpad Prism software (GraphPad, San Diego, CA); values are stated as mean ± S.E.M. Raw or normalized tail currents were plotted versus prepulse voltage and fitted with a single Boltzmann function:
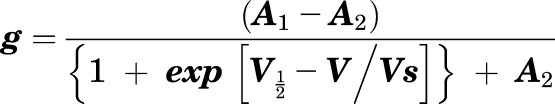 |
(1) |
where g is the normalized tail conductance, A1 is the initial value at −∞, A2 is the final value at +∞, V1/2 is the half-maximal voltage of activation, and Vs the slope factor.
Chemical Structures.
We plotted and viewed chemical structures and electrostatic surface potentials using Jmol, an open-source Java viewer for chemical structures in three dimensions (http://jmol.org/).
Statistical Analysis.
All values are expressed as mean ± S.E.M. One-way ANOVA was applied for all other tests; if multiple comparisons were performed, a post hoc Tukey’s Honestly Significant Difference (HSD) test was performed following ANOVA. All P values were two-sided. Statistical significance was defined as P < 0.05.
Results
Glycine-Derived 2FPG Activates KCNA1 but Not KCNA2.
We previously found that the inhibitory neurotransmitter GABA is able to directly activate KCNQ2/3, KCNQ3, and KCNQ5 channels, facilitated by negative electrostatic surface potential centered on the GABA carbonyl group (Manville et al., 2018), a property found to enable synthetic anticonvulsants such as retigabine (ezogabine) to also activate KCNQ channels that possess a specific tryptophan residue (e.g., W265 in human KCNQ3) on the S5 segment (Kim et al., 2015) (Fig. 1A). In contrast, we discovered that glycine, another prominent inhibitory neurotransmitter, lacks both the strong negative electrostatic surface potential at its carbonyl group (Fig. 1B) and the ability to activate KCNQ2/3 channels (Manville and Abbott, 2019b). However, we found that by chemically modifying glycine to introduce a carbonyl-centered negative electrostatic surface potential, we produced three novel glycine-based KCNQ activators: 2FPG, 4FPG, and 3FMSG (Fig. 1C) (Manville and Abbott, 2019b). Docking results suggested the importance for interaction with these compounds of an arginine at the junction between S4 and the S4-S5 linker (Manville and Abbott, 2019b), a residue that, unlike the KCNQ3-W265 residue, is conserved in all the KCNQs and other Kv channels such as KCNA1 and KCNA2 (Fig. 1D).
Fig. 1.

Lead compounds and sequence alignment. (A) GABA structure, electrostatic surface potentials (red, electron-dense; blue, electron-poor; green, neutral), and an overlay of the two, all calculated and plotted using Jmol. Arrow, carbonyl group. (B) Glycine, parameters as in (A). (C) Structures and surface potential plots, as in (A), for three glycine derivatives bearing a phenyl ring; arrows, carbonyl group. (D) Sequence alignment including the S4 and S5 segments (orange) of human KCNA1, KCNA2, and KCNQ1-5; amino acid numbering shown on left. Yellow, conserved arginine (e.g., KCNQ3-R242) positioned at the junction between S4 and the S4-5 linker. Cyan, S5 tryptophan (e.g., KCNQ3-W265) conserved in KCNQ2-5.
Here, we therefore tested whether KCNA1 and KCNA2 are also sensitive to 2FPG, 4FPG, and 3FMSG, using heterologous expression in Xenopus laevis oocytes and two electrode voltage-clamp electrophysiology. KCNA1 and KCNA2 each lack the tryptophan analogous to KCNQ3-W265, but each possess an ortholog of KCNQ3-R242 (Fig. 1D). Strikingly, while 4FPG showed little to no activity (Fig. 2, A–C), 2FPG was highly effective at activating KCNA1 (Fig. 2, D–F); 3FMSG was active but less effective than 2FPG (Fig. 2, G–I) (each at 100 µM) (Fig. 2, A and B). These effects resulted in the ability of 2FPG and 3FMSG, but not 4FPG, to hyperpolarize the membrane potential (EM) of oocytes expressing KCNA1 (Fig. 2, C, F, and I). In contrast, 2FPG, 3FMSG, and 4FPG (at 100 µM) neither activated KCNA2 (Fig. 2, J and K) nor hyperpolarized the EM of oocytes expressing KCNA2 (Fig. 2L).
Fig. 2.

2FPG activates KCNA1 but not KCNA2. All error bars indicate S.E.M. Dashed lines indicate zero current level here and in all other figures. (A) Mean traces showing effects of 4FPG on KCNA1; n = 8. Lower right inset: voltage protocol used for all Kv channel recordings in this study. Arrow shows time point at which tail currents are quantified. (B) Mean and normalized (G/Gmax) KCNA1 tail current vs. prepulse voltage relationships calculated from traces as in (A); n = 8. (C) Effects of 4FPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1; n = 8. (D) Mean traces showing effects of 2FPG (100 µM) on KCNA1; n = 5. Lower insets: expanded view of tail currents; arrows show time point at which tail currents are quantified. (E) Mean and normalized (G/Gmax) KCNA1 tail currents vs. prepulse voltage relationships calculated from traces as in (D); n = 5. (F) Effects of 2FPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1; n = 5. (G) Mean traces showing effects of 3FMSG (100 µM) on KCNA1; n = 5. (H) Mean and normalized (G/Gmax) KCNA1 tail current vs. prepulse voltage relationships calculated from traces as in (G); n = 5. (I) Effects of 3FMSG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1; n = 5. (J) Mean traces showing effects of 4FPG, 2FPG, and 3FMSG (100 µM) on KCNA2; n = 5. (K) Mean and normalized (G/Gmax) KCNA2 tail current vs. prepulse voltage relationships calculated from traces as in (A); n = 5. (L) Effects of 4FPG, 2FPG, and 3FMSG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA2; n = 5.
Z-D-Cyclohexylglycine is a KCNA1-Selective Opener.
Despite displaying isoform selectivity for KCNA1 versus the closely related KCNA2 (Fig. 2), 2FPG also activates KCNQ2/3 (Manville and Abbott, 2019). We therefore next pursued a more selective KCNA1 opener. The KCNQ2/3 openers we derived from glycine share two prominent features with KCNQ2/3 opener retigabine, i.e., strong negative electrostatic surface potential centered on a carbonyl oxygen, as well as one or more fluorine atoms on a phenyl ring. We therefore tested whether removing one of these properties would maintain KCNA1 activity but improve selectivity.
Based on their chemical structures and electrostatic surface potential maps, we selected three compounds, the first of which was Z-D-Cyclohexylglycine, which lacks halides but bears negative electrostatic surface potential foci centered on two carbonyl groups (Fig. 3A). This modification achieved the desired result; i.e., Z-D-Cyclohexylglycine (100 µM) activated KCNA1 but not KCNA2 or KCNQ2/3 (Fig. 3B), inducing a negative shift in the voltage dependence of activation (V0.5act) only in KCNA1 (Fig. 3, C and D). Accordingly, Z-D-Cyclohexylglycine hyperpolarized the membrane potential of oocytes expressing KCNA1 but not of oocytes expressing KCNA2 or KCNQ2/3 (Fig. 3E).
Fig. 3.

A fluorine-free glycine derivative that lacks KCNQ2/3 activity but activates KCNA1. All error bars indicate S.E.M. (A) Chemical properties of Z-D-Cyclohexylglycine: structure, electrostatic surface potentials (red, electron-dense; blue, electron-poor; green, neutral), and an overlay of the two, all calculated and plotted using Jmol. Arrows, carbonyl groups. (B) Mean traces showing effects of Z-D-Cyclohexylglycine (100 µM) on KCNA1, KCNA2, and KCNQ2/3; n = 4 to 5. (C) Effects of Z-D-Cyclohexylglycine (100 µM) on raw tail current vs. prepulse voltage relationships, calculated from traces as in (B); n = 4 to 5. (D) Effects of Z-D-Cyclohexylglycine (100 µM) on normalized (G/Gmax) tail current vs. prepulse voltage relationships, calculated from traces as in (B); n = 4 to 5. (E) Effects of Z-D-Cyclohexylglycine (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1, KCNA2, or KCNQ2/3; n = 4 to 5.
Trifluoromethylphenyl Ring Glycine Derivatives Isoform-Selectively Activate KCNA1.
2TFMPG, which bears three fluorine atoms on a methyl group on its phenyl ring but has negligible negative electrostatic potential, including at its glycine carbonyl (Fig. 4A), also activated KCNA1 (at 100 µM) but not KCNA2 or KCNQ2/3 (Fig. 4B), again by negative shifting the V0.5act (Fig. 4C). This again resulted in membrane potential hyperpolarization only for oocytes expressing KCNA1 (Fig. 4D). As the activation by 2TFMPG was relatively weak (a −6.9 mV shift in V0.5act at 100 µM), we next tested closely related 4TFMPG. By shifting the trifluoromethyl group 2 positions along the phenyl ring, the predicted resultant negative electrostatic potential in 4TFMPG was weak, but unlike in 2TFMPG, it was predicted to be centered on the carbonyl group (Fig. 5A). This combination proved optimal, producing a robust activation (at 100 µM 4TFMPG) of KCNA1 (Fig. 5, B–D), especially at negative voltages (Fig. 5E), and no effects on KCNA2 or KCNQ2/3 (Fig. 5, B–E). Again, 4TFMPG only hyperpolarized the oocyte membrane potential when KCNA1 was expressed (Fig. 5F).
Fig. 4.

Additional fluorine atoms remove KCNQ2/3 activity of 2FPG but retain KCNA1 activity. All error bars indicate S.E.M. (A) Chemical properties of 2FPG vs. 2TFMPG: structure, electrostatic surface potentials (red, electron-dense; blue, electron-poor; green, neutral), and an overlay of the two, all calculated and plotted using Jmol. Arrows, carbonyl groups. Fluorine atoms are circled. (B) Mean traces showing effects of 2TFMPG (100 µM) on KCNA1, KCNA2, and KCNQ2/3; n = 5 to 6. (C) Effects of 2TFMPG (100 µM) on raw tail current vs. prepulse voltage relationships, calculated from traces as in (B); n = 5 to 6. (D) Effects of 2TFMPG (100 µM) on normalized (G/Gmax) tail current vs. prepulse voltage relationships, calculated from traces as in (B); n = 5 to 6. (E) Effects of 2TFMPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1, KCNA2, or KCNQ2/3; n = 5 to 6.
Fig. 5.

Repositioning the trifluoromethyl group on 2TFMPG increases efficacy for KCNA1 and maintains isoform selectivity. All error bars indicate S.E.M. (A) Chemical properties of 2TFMPG vs. 4TFMPG: structure, electrostatic surface potentials (red, electron-dense; blue, electron-poor; green, neutral), and an overlay of the two, all calculated and plotted using Jmol. Large arrows, carbonyl groups. Beige line, repositioning of trifluoromethyl group on 4TFMPG. The trifluoromethyl group is circled on 2TFMPG. (B) Mean traces showing effects of 4TFMPG (100 µM) on KCNA1 and KCNA2; n = 4 to 5. (C) Effects of 4TFMPG (100 µM) on KCNA1, KCNA2, and KCNQ2/3 raw tail current vs. prepulse voltage relationships; n = 4 to 5. (D) Effects of 4TFMPG (100 µM) on KCNA1, KCNA2, and KCNQ2/3 normalized (G/Gmax) tail current vs. prepulse voltage relationships; n = 4 to 5. (E) Effects of 4TFMPG (100 µM) on KCNA1, KCNA2, and KCNQ2/3 raw tail current quantified as current fold-change vs. voltage; n = 4 to 5. (F) Effects of 4TFMPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1, KCNA2, or KCNQ2/3; n = 4 to 5.
As an additional control, we tested the ability of the glycine derivatives to activate a canonical glycine receptor, GLRA1. While GLRA1 was robustly activated by glycine, none of the derivatives tested activated the channel (each at 100 µM) (Fig. 6).
Fig. 6.

KCNA1-activating glycine derivatives do not activate canonical glycine receptor GLRA1. All error bars indicate S.E.M. (A) Exemplar trace showing lack of GLRA1 activation by Z-D-Cyclohexylglycine (ZDG), 2TFMPG, or 4TFMPG compared with robust activation by glycine alone (all compounds applied at 100 µM); application indicated by colored bars at top. Gray, application of bath solution alone. (B) Mean data from traces as in (A) (n = 3 oocytes, performed three times for each oocyte and then averaged).
Having completed the initial screens at 100 µM (summarized in Fig. 7A by comparing current fold increase in KCNA1 vs. KCNA2), we conducted dose responses for the KCNA1-selective compounds Z-D-Cyclohexylglycine and 4TFMPG, comparing them to 2FPG. As quantified by current fold-increase at −60 mV, 4TFMPG exhibited similar potency to that of the less selective 2FPG (136 vs. 116 nM EC50, respectively) but ∼twofold-lower efficacy (17.4-fold vs. 31-fold maximal current increase). Z-D-Cyclohexylglycine was less potent with respect to KCNA1 activation (EC50 = 2.1 ± 0.6 µM) and less efficacious (3.9-fold maximal current increase) than either (Fig. 7B). Comparing next by ΔV0.5act, Z-D-Cyclohexylglycine exhibited similar maximal effect to 2FPG (−12.2 vs. −15.3 mV shifts, respectively) but lower potency (972 vs. 64 nM, respectively). 4TFMPG (25 nM EC50) exhibited higher potency than 2FPG but half the maximal effect (−7.6 vs. −15.3 mV shifts, respectively) (Fig. 7C).
Fig. 7.

Potency and efficacy of isoform-selective and -nonselective KCNA1 openers derived from glycine. All error bars indicate S.E.M. (A) Effects of glycine derivatives indicated (100 µM) on KCNA1 (left) and KCNA2 (right) activity quantified as tail current fold-change vs. prepulse voltage; n = 4–6. (B) Concentration/response relationships for glycine derivatives indicated on KCNA1 activity quantified as tail current fold-change at −60 mV prepulse voltage vs. concentration; n = 4–6. (C) Concentration/response relationships for glycine derivatives as in (B) on KCNA1 activity quantified as shift in midpoint voltage dependence of activation (ΔV0.5activation) vs. concentration; n = 4–6.
We next tested the efficacy of 4TFMPG in rescue of EA1 mutants E283K (Imbrici et al., 2017), V408A (Browne et al., 1994), G311D (Karalok et al., 2018), and hypomagnesemia/tetany mutant L328V (van der Wijst et al., 2018) KCNA1 channels. We tested homomeric and heteromeric channels, mimicking possible homozygous and heterozygous disease states. All four mutants were previously shown to reduce KCNA1 current in a dominant-negative manner. Accordingly, we observed peak currents at +40 mV ranging from 0 to 2 µA for the mutant channels (Fig. 8) compared with up to 15 µA for wild-type KCNA1 channels (Figs. 2–5). As we had observed for wild-type channels (Figs. 2–5), 4TFMPG was ineffective at increasing peak prepulse or tail currents at positive voltages in homomeric KCNA1-E283K channels (Fig. 8, A and B). In contrast, 4TFMPG shifted homomeric KCNA1-E283K voltage dependence of activation by > −5 mV (from −19.1 ± 0.8 mV to −24.9 ± 1.2 mV) (Fig. 8B). This was associated with a 4TFMPG-induced −8 mV shift in EM of cells expressing KCNA1-E283K (Fig. 8C). The shifts were not as apparent in heteromeric KCNA1/KCNA1-E283K tail currents (data not shown), but the prepulse current/voltage (I/V) relationship for the heteromeric channel revealed a negative shift in the −30 to 0 mV range (Fig. 8, D and E) as well as a negative shift in EM of cells expressing KCNA1/KCNA1-E283K that shifted this value more negative than that of wild-type KCNA1-expressing cells (Fig. 8F).
Fig. 8.

Effects of 4TFMPG on EA1-associated KCNA1 mutant channels All error bars indicate S.E.M. (A) Effects of 4TFMPG (100 µM) on KCNA1-E238K; n = 6. Left, mean traces; right, peak prepulse current vs. voltage. (B) Mean (left) and normalized (G/Gmax) (right) KCNA1-E283K tail current vs. prepulse voltage relationships calculated from traces as in (A); n = 6. Red dashed line, superimposed wild-type KCNA1 G/V curve (from Fig. 5D), baseline-adjusted to match that of the mutant channels. (C) Effects of 4TFMPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1-E283K; n = 6. Red dashed line, baseline EM of wild-type KCNA1 for comparison (from Fig. 5F). (D) Mean traces showing effects of 4TFMPG (100 µM) on heteromeric KCNA1/KCNA1-E238K; n = 6. (E) Peak prepulse current vs. voltage for cells as in (D) (n = 6). (F) Effects of 4TFMPG (100 µM) on resting membrane potential (EM) of unclamped X. laevis oocytes expressing KCNA1/KCNA1-E283K; n = 6. Red dashed line, baseline EM of wild-type KCNA1 for comparison (from Fig. 5F). (G) Mean traces showing effects of 4TFMPG (100 µM) on KCNA1-V408A; n = 6. (H) Left, mean KCNA1-V408A tail currents vs. prepulse voltage relationships calculated from traces as in (G); n = 6. Red dashed line, superimposed normalized wild-type KCNA1 G/V curve (from Fig. 5D). Right, effects of 4TFMPG (100 µM) on EM of unclamped X. laevis oocytes expressing KCNA1-V408A; n = 6. Red dashed line, baseline EM of wild-type KCNA1 for comparison (from Fig. 5F). (I) Effects of 4TFMPG (100 µM) on homomeric KCNA1-G311D; left, mean traces; right, EM; n = 6. Red dashed line, baseline EM of wild-type KCNA1 for comparison (from Fig. 5F). (J) Effects of 4TFMPG (100 µM) on heteromeric KCNA1/KCNA1-G311D; left, mean traces; right, EM; n = 6. Red dashed line, baseline EM of wild-type KCNA1 for comparison (from Fig. 5F). (K) Effects of 4TFMPG (100 µM) on homomeric KCNA1/KCNA1-L328V; left, mean traces; right, EM; n = 5. (L) Effects of 4TFMPG (100 µM) on heteromeric KCNA1/KCNA1-L328V; left, mean traces; right, EM; n = 5.
KCNA1-V408A channels exhibit accelerated C-type inactivation (D’Adamo et al., 1998), and 4TFMPG did not alleviate this or otherwise alter prepulse currents (only homomeric data shown) (Fig. 8G). However, 4TFMPG appeared to augment recovery from inactivation during the tail pulse, as it increased by ≥twofold tail current after prepulses between +10 and +40 mV, without enhancing currents after prepulses from −80 to 0 mV. Accordingly, 4TFMPG did not alter EM of cells expressing this mutant (Fig. 8H). 4TFMPG was ineffective at rescuing homomeric KCNA1-G311D current or EM (Fig. 8I) but was able to hyperpolarize EM of cells expressing heteromeric KCNA1/KCNA1-G311D beyond that of baseline wild-type (Fig. 8J). Finally, 4TFMPG was ineffective at rescuing the strongly dominant-negative mutant, KCNA1-L328V, in both homomeric (Fig. 8K) and heteromeric (Fig. 8L) forms.
Discussion
The findings herein are mechanistically unexpected and also of potential translational significance. To our knowledge, there is only one previously reported small-molecule opener of KCNA1, pimaric acid, which is KCNA isoform-nonselective and also activates Kv channels outside the KCNA subfamily (Sakamoto et al., 2017). As KCNA1 loss of function causes type 1 episodic ataxia (EA1) (Browne et al., 1994), selective openers of this channel are sought after for their possible therapeutic benefits.
Previous studies showed that negative electrostatic surface potential centered on a carbonyl group enabled synthetic anticonvulsants such as retigabine (ezogabine) to activate KCNQ channels that possess a specific tryptophan residue (W265 in human KCNQ3) on the S5 segment (Kim et al., 2015); we previously found that the inhibitory neurotransmitter GABA shares this chemical property and also activates KCNQ2/3, KCNQ3, and KCNQ5 (Manville et al., 2018), whereas glycine does not. Retigabine activates neuronal KCNQ2, 3, 4, and 5, which all possess the KCNQ3-W265 equivalent. However, like GABA and the drug gabapentin (each of which activates KCNQ3 and KCNQ5) (Manville and Abbott, 2018; Manville et al., 2018), retigabine does not activate KCNQ1, which lacks the requisite S5 tryptophan (Wuttke et al., 2005; Lange et al., 2009).
Unexpectedly, we previously found that 4FPG and 3FMSG (but not 2FPG) each activated KCNQ1 despite the lack of a KCNQ3-W265 equivalent and despite showing other isoform selectivity within the KCNQ subfamily (Manville and Abbott, 2019). In addition, we found that the selectivity was not primarily endowed by binding selectivity, which we quantified directly using competition with [3H]-GABA. These and other prior data suggest that small molecules bearing resemblance to GABA and glycine can bind to various KCNQ channels without necessarily activating them, although we also previously confirmed that glycine, unlike GABA, does not bind to KCNQ2/3 channels (Manville et al., 2018, 2019; Manville and Abbott, 2019b).
The prior findings also suggested that the requirement for the KCNQ3-W265 equivalent is context-dependent; e.g., mutating it to leucine prevents activation of KCNQ3 by 3FMSG, yet 3FMSG activates wild-type KCNQ1, which inherently lacks the W265 equivalent. Additionally, we previously found that an arginine juxtaposed between the S4 and S4-5 linker predicted to be located near the intracellular face of the cell membrane is also influential in both activation by, and binding to, KCNQ channels by glycine derivatives, GABA, and various compounds from medicinal plants (De Silva et al., 2018; Manville et al., 2018, 2019; Manville and Abbott, 2019a,b). The arginine, KCNQ3-R242 and its equivalents, is conserved between KCNQs and, for example, KCNA1 and 2 (Fig. 1D). All the above suggested the possibility that glycine derivatives, despite demonstrating some KCNQ isoform selectivity, might also activate non-KCNQ channels, which we tested herein.
While 4TFMPG successfully and selectively hyperpolarized the voltage dependence of activation of wild-type KCNA1, it was in general less effective at rescuing EA1-mutant KCNA1, either in homomeric or heteromeric (with wild-type) channels (Fig. 8). The most positive aspect of mutant rescue was that 4TFMPG was still able to hyperpolarize cells expressing E283K or G311D mutants while also effecting hyperpolarizations in the voltage dependence of activation of E283K and V408A tail currents. The mutants we examined covered sites across several important functional domains of KCNA1; E283K is in the S3-4 extracellular linker in the KCNA1 VSD, G311D is in the S4-5 linker that connects the VSD to the pore, L328V is at the intracellular side of S5, and V408A is located at the C-terminal end of S6. 4TFMPG was probably least effective at ameliorating effects of G311D and L328V, the two mutants closest to the binding pocket occupied by glycine derivatives in KCNQ channels (Manville and Abbott, 2019). It is possible that the G311D and L328V substitutions directly disrupt functional effects or binding of 4TFMPG, or the greater disruptive effects of these two mutants compared with E328K and V408A make the former particularly recalcitrant to rescue. 4TFMPG was ineffective at recovering, in either homomeric or heteromeric mutant channels, the large currents seen with wild-type; this was consistent with the inability to increase peak wild-type KCNA currents at positive potentials. The findings suggest that future therapeutic use of 4TFMPG or optimized derivatives would likely only be effective if used in conjunction with a gene therapy, such as an allele-specific knockdown approach designed to diminish expression of the mutant allele, relieving the dominant-negative effect so that the small molecule could then hyperpolarize the voltage dependence of the residual wild-type current, augmenting activity at negative voltages. This approach would not work for patients with homozygous mutations but has shown promise, for example, in proof-of-principle in vitro studies and in a mouse model humanized in one allele to introduce a hypertrophic cardiomyopathy myosin heavy chain 7 dominant-negative mutant (Anderson et al., 2020).
There is much still to learn about the mechanisms underlying activation of Kv channels by neurotransmitter derivatives and other channel openers, especially with respect to the molecular basis for selectivity of activation. For example, we do not yet know why small molecules such as 2FPG can activate both KCNQ2/3 and KCNA1 but not KCNA2, which is more closely related to KCNA1. Answering this question will require not only complete mapping of the binding sites but also an appreciation of the residues within the activation machinery that permit a specific small molecule to isoform-selectively activate certain KCNQ or KCNA isoforms despite binding to several more without activating them.
The ultimate therapeutic success of Kv channel openers relies on their safety as well as efficacy; factors that depend on criteria not tested here, including specificity for other channels and nonchannel proteins outside the KCNA and KCNQ families; and also pharmacokinetics and pharmacodynamics of the molecule in vivo.
Abbreviations
- 2FPG
(2-fluorophenyl) glycine
- 2TFMPG
2-(Trifluoromethyl)-DL-phenylglycine
- 3FMSG
N-(3-fluorophenyl)-N-(methylsulfonyl) glycinea
- 4FPG
4-(Fluorophenyl)glycine
- 4TFMPG
4-(Trifluoromethyl)-phenylglycine
- EA1
episodic ataxia 1
- Kv
voltage-gated potassium
- V0.5act
voltage dependence of activation
- VSD
voltage sensor domain
Authorship Contributions
Participated in research design: Manville, Abbott.
Conducted experiments: Manville.
Performed data analysis: Manville, Abbott.
Wrote or contributed to the writing of the manuscript: Manville, Abbott.
Footnotes
This study was supported by the National Institutes of Health, National Institute of General Medical Sciences [Grant R35 GM130377] and National Institute of Neurologic Disorders and Stroke [Grant R01 NS107671] to G.W.A.
References
- Abbott GW. (2014) Biology of the KCNQ1 potassium channel. New J Sci 2014:26. [Google Scholar]
- Anderson BR, Jensen ML, Hagedorn PH, Little SC, Olson RE, Ammar R, Kienzle B, Thompson J, McDonald I, Mercer S, et al. (2020) Allele-selective knockdown of MYH7 using antisense oligonucleotides. Mol Ther Nucleic Acids 19:1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8:136–140. [DOI] [PubMed] [Google Scholar]
- Chen H, von Hehn C, Kaczmarek LK, Ment LR, Pober BR, Hisama FM. (2007) Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia. Neurogenetics 8:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett MA, Bellows ST, Li M, Carroll R, Micallef S, Carvill GL, Myers CT, Howell KB, Maljevic S, Lerche H, et al. (2016) Dominant KCNA2 mutation causes episodic ataxia and pharmacoresponsive epilepsy. Neurology 87:1975–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump SM, Abbott GW. (2014) Arrhythmogenic KCNE gene variants: current knowledge and future challenges. Front Genet 5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamo MC, Gallenmüller C, Servettini I, Hartl E, Tucker SJ, Arning L, Biskup S, Grottesi A, Guglielmi L, Imbrici P, et al. (2015a) Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol 5:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamo MC, Hasan S, Guglielmi L, Servettini I, Cenciarini M, Catacuzzeno L, Franciolini F. (2015b) New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci 9:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamo MC, Liu Z, Adelman JP, Maylie J, Pessia M. (1998) Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J 17:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demos MK, Macri V, Farrell K, Nelson TN, Chapman K, Accili E, Armstrong L. (2009) A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord 24:778–782. [DOI] [PubMed] [Google Scholar]
- De Silva AM, Manville RW, Abbott GW. (2018) Deconstruction of an African folk medicine uncovers a novel molecular strategy for therapeutic potassium channel activation. Sci Adv 4:eaav0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JB, Hanna MG, et al. (2000) Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 48:647–656. [PubMed] [Google Scholar]
- Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, Knoers NV, Hoenderop JG, Bindels RJ. (2009) A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest 119:936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig KL, Hedrich UB, Shinde DN, Krey I, Teichmann AC, Hentschel J, Schubert J, Chamberlin AC, Huether R, Lu HM, et al. (2016) A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann Neurol 80:638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrici P, Altamura C, Gualandi F, Mangiatordi GF, Neri M, De Maria G, Ferlini A, Padovani A, D’Adamo MC, Nicolotti O, et al. (2017) A novel KCNA1 mutation in a patient with paroxysmal ataxia, myokymia, painful contractures and metabolic dysfunctions. Mol Cell Neurosci 83:6–12. [DOI] [PubMed] [Google Scholar]
- Ishida S, Sakamoto Y, Nishio T, Baulac S, Kuwamura M, Ohno Y, Takizawa A, Kaneko S, Serikawa T, Mashimo T. (2012) Kcna1-mutant rats dominantly display myokymia, neuromyotonia and spontaneous epileptic seizures. Brain Res 1435:154–166. [DOI] [PubMed] [Google Scholar]
- Jan LY, Jan YN. (2012) Voltage-gated potassium channels and the diversity of electrical signalling. J Physiol 590:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karalok ZS, Megaro A, Cenciarini M, Guven A, Hasan SM, Taskin BD, Imbrici P, Ceylaner S, Pessia M, D’Adamo MC. (2018) Identification of a new de novo mutation underlying regressive episodic ataxia type I. Front Neurol 9:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RY, Yau MC, Galpin JD, Seebohm G, Ahern CA, Pless SA, Kurata HT. (2015) Atomic basis for therapeutic activation of neuronal potassium channels. Nat Commun 6:8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinali M, Jungbluth H, Eunson LH, Sewry CA, Manzur AY, Mercuri E, Hanna MG, Muntoni F. (2004) Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul Disord 14:689–693. [DOI] [PubMed] [Google Scholar]
- Lange W, Geissendörfer J, Schenzer A, Grötzinger J, Seebohm G, Friedrich T, Schwake M. (2009) Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol Pharmacol 75:272–280. [DOI] [PubMed] [Google Scholar]
- Manville RW, Abbott GW. (2018) Gabapentin is a potent activator of KCNQ3 and KCNQ5 potassium channels. Mol Pharmacol 94:1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Abbott GW. (2019a) Cilantro leaf harbors a potent potassium channel-activating anticonvulsant. FASEB J 33:11349–11363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Abbott GW. (2019b) In silico re-engineering of a neurotransmitter to activate KCNQ potassium channels in an isoform-specific manner. Commun Biol 2:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, Papanikolaou M, Abbott GW. (2018) Direct neurotransmitter activation of voltage-gated potassium channels. Nat Commun 9:1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville RW, van der Horst J, Redford KE, Katz BB, Jepps TA, Abbott GW. (2019) KCNQ5 activation is a unifying molecular mechanism shared by genetically and culturally diverse botanical hypotensive folk medicines. Proc Natl Acad Sci USA 116:21236–21245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptácek LJ, Fu YH. (2002) Molecular biology of episodic movement disorders. Adv Neurol 89:453–458. [PubMed] [Google Scholar]
- Roepke TK, Abbott GW. (2006) Pharmacogenetics and cardiac ion channels. Vascul Pharmacol 44:90–106. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Suzuki Y, Yamamura H, Ohya S, Muraki K, Imaizumi Y. (2017) Molecular mechanisms underlying pimaric acid-induced modulation of voltage-gated K+ channels. J Pharmacol Sci 133:223–231. [DOI] [PubMed] [Google Scholar]
- Spauschus A, Eunson L, Hanna MG, Kullmann DM. (1999) Functional characterization of a novel mutation in KCNA1 in episodic ataxia type 1 associated with epilepsy. Ann N Y Acad Sci 868:442–446. [DOI] [PubMed] [Google Scholar]
- Syrbe S, Hedrich UBS, Riesch E, Djémié T, Müller S, Møller RS, Maher B, Hernandez-Hernandez L, Synofzik M, Caglayan HS, et al. EuroEPINOMICS RES consortium (2015) De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat Genet 47:393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wijst J, Glaudemans B, Venselaar H, Nair AV, Forst AL, Hoenderop JG, Bindels RJ. (2010) Functional analysis of the Kv1.1 N255D mutation associated with autosomal dominant hypomagnesemia. J Biol Chem 285:171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wijst J, Konrad M, Verkaart SAJ, Tkaczyk M, Latta F, Altmüller J, Thiele H, Beck B, Schlingmann KP, de Baaij JHF. (2018) A de novo KCNA1 mutation in a patient with tetany and hypomagnesemia. Nephron 139:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdura E, Fons C, Schlüter A, Ruiz M, Fourcade S, Casasnovas C, Castellano A, Pujol A. (2020) Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J Med Genet 57:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. (2005) The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol 67:1009–1017. [DOI] [PubMed] [Google Scholar]