

Fluorescent molecular tension sensors reveal the dynamics of force transmission at cellular adhesion complexes.
Abstract
Integrin-based adhesion complexes link the cytoskeleton to the extracellular matrix (ECM) and are central to the construction of multicellular animal tissues. How biological function emerges from the tens to thousands of proteins present within a single adhesion complex remains unclear. We used fluorescent molecular tension sensors to visualize force transmission by individual integrins in living cells. These measurements revealed an underlying functional modularity in which integrin class controlled adhesion size and ECM ligand specificity, while the number and type of connections between integrins and F-actin determined the force per individual integrin. In addition, we found that most integrins existed in a state of near-mechanical equilibrium, a result not predicted by existing models of cytoskeletal force transduction. A revised model that includes reversible cross-links within the F-actin network can account for this result and suggests one means by which cellular mechanical homeostasis can arise at the molecular level.
INTRODUCTION
Integrins are heterodimeric transmembrane proteins that form the core of micrometer-sized protein assemblies, here referred to generically as focal adhesions (FAs). These structures link the cytoskeleton to the extracellular matrix (ECM) and hence play a central role in the construction of multicellular tissues (1–3). Proteomics studies demonstrate that ~60 proteins constitute the core integrin adhesion machinery and that >2400 proteins are potential members of the integrin adhesome (4). Previous studies have uncovered a dense web of interactions between FA proteins (5), the complexity of which poses a challenge in understanding how FAs function as an integrated whole.
In this study, we sought to better understand how FA-mediated force transmission arises at the molecular level. The rationale in doing so is that the transmission of forces between the cytoskeleton and ECM constitutes a core function of FAs and is required both for tissue morphogenesis and many forms of cell migration. Force transmission is commonly described in terms of the molecular clutch model, in which continuous slippage between the rearward-flowing actin cytoskeleton and FA components mediates force transmission to the ECM (6–10). This model reproduces important biological observations, for example biphasic traction forces as a function of substrate stiffness (11–13). However, to our knowledge, the clutch model has not been directly tested by the observation of the dynamics of force transmission at the single-molecule level in living cells.
RESULTS
We used fluorescence resonance energy transfer (FRET)–based molecular tension sensors (MTSs) to measure the loads experienced by individual integrin heterodimers in human foreskin fibroblasts (HFFs) (Fig. 1, A to D). MTSlow and MTSFN9–10 report on loads between 2 and 7 pN and present either a linear arginine-glycine-aspartate (RGD) containing peptide or the fibronectin type III domains 9 and 10, respectively (14, 15). In addition, we developed a new sensor termed MTShigh that measures forces between 7 and 11 pN and contains the same RGD motif as MTSlow (fig. S1) (16). We found that HFFs had similar morphology, adhesion formation, and myosin activity when adhering to surfaces functionalized with either MTSlow or MTShigh (fig. S2).
Fig. 1. Single-molecule tension measurements in living cells reveal distinct subpopulations of load-bearing integrins.
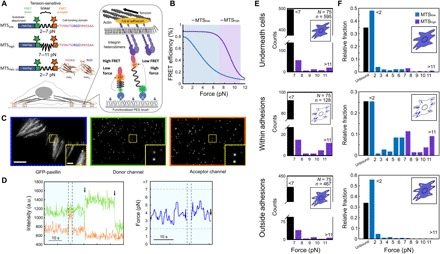
(A) FRET-based MTSs. MTSs are attached to the coverslip surface via the HaloTag domain. (B) FRET-force calibration curves for MTSlow (blue) and MTShigh (purple) (16, 51). (C) Representative images showing green fluorescent protein (eGFP) (left), FRET donor (middle), and acceptor channels (right) for HFFs adhering to a surface functionalized with MTSlow. Scale bar, 5 μm; inset scale bar, 2 μm. (D) Example intensity traces (left) for the FRET donor (green) and acceptor (orange). Vertical dashed lines delineate frames during which the acceptor dye was directly excited with 633-nm light; arrows mark acceptor or donor bleaching; horizontal gray dashed lines indicate upper and lower force measurement limits. Right: Corresponding load time series before acceptor photobleaching (light blue). Intensity, arbitrary units (a.u.). (E) Single-molecule load distributions for MTShigh underneath cells, within adhesions, and outside adhesions. N = number of cells, n = number of sensors. (F) Combined single-molecule load distributions for MTSlow and MTShigh sensors underneath cells, within adhesions, and outside adhesions for MTSlow [blue; data from (14)] and MTShigh (purple).
In previous studies, we found that most ligand-bound integrins exist in a minimally tensioned state (<2 pN) that does not depend on the actin cytoskeleton (14), which we confirm in this study (Fig. 1E). Measurements using MTShigh further revealed that the distribution of loads on individual integrins was highly asymmetric, with a small minority of integrins within the adhesions of HFFs bearing loads of ~6 pN and >11 (Fig. 1, E and F). The presence of the latter subpopulation is consistent with previous studies demonstrating that at least some integrins experience peak loads >50 pN (17–21).
How these different load subpopulations arise at the molecular level was unclear. A plausible explanation was that these subpopulations might correspond to ligation by different integrin heterodimers, a scenario supported by studies reporting distinct roles for α5β1- and αv-class integrins in adhesion and traction generation (22–25). To test this hypothesis, we made use of pan-integrin knockout (pKO) mouse kidney fibroblasts rescued with the integrin αv subunit (pKO-αv), which forms predominantly αvβ3 and αvβ5 heterodimers, the β1 subunit (pKO-β1), which forms only α5β1 integrin in these cells, or both subunits (pKO-αv/β1), which form all three integrin heterodimers (22). pKO-αv and pKO-αv/β1 cells spread normally on coverslips functionalized with either MTSlow or MTShigh and formed sizeable FAs (Fig. 2A, top and middle), while most of the pKO-β1 cells failed to spread on either sensor (see insets). In contrast, all three cell types spread on coverslips functionalized with MTSFN9–10. However, pKO-αv cells yielded lower integrated traction forces, without significant changes in adhesion size, compared to the other two cell types [Fig. 2, A (bottom), B, and D]. Thus, integrin usage and ligand identity strongly influenced adhesion and traction generation at the whole-cell level, an outcome consistent with previous observations (14, 24, 26, 27).
Fig. 2. Integrin class does not determine the force per integrin but affects ligand specificity.
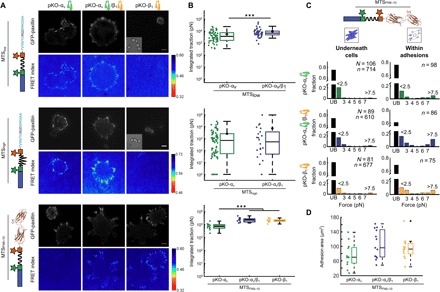
(A) Images of eGFP-paxillin and ensemble FRET maps for pKO-αv, pKO-αv/β1, and pKO-β1 cells adhering to MTSlow (top), MTShigh (middle), and MTSFN9–10 (bottom). Insets show corresponding bright-field images for pKO-β1 cells, which rarely spread on surfaces functionalized with MTSlow and MTShigh. Scale bars, 10 μm. (B) Ensemble quantification of pKO-αv, pKO-αv/β1, and pKO-β1 cells adhering to MTSlow, MTShigh, and MTSFN9–10. When adhering to MTSlow, pKO-αv/β1 cells exert more integrated traction compared to pKO-αv cells (pKO-αv: 67 cells, mean: 5.5 nN; pKO-αv/β1: 43 cells, mean: 9.8 nN) (***P = 3 × 10−4). When adhering to MTShigh, pKO-αv/β1 and pKO-αv cells produce comparable traction overall (pKO-αv: 77 cells, mean: 2.6 nN; pKO-αv/β1: 22 cells, mean: 7 nN). For MTSFN9–10, pKO-αv/β1 and pKO-β1 cells exert a higher integrated traction as compared to pKO-αv cells (pKO-αv: 13 cells, mean: 7.9 nN; pKO-αv/β1: 12 cells, mean: 26.9 nN; pKO-αv/β1: 12 cells, mean: 23.8 nN) (***P < 10 to 3). (C) Single-molecule load distributions for pKO cell lines adhering to MTSFN9–10. Black bars indicate unbound molecules. (D) Adhesion area measured for pKO-αv, pKO-αv/β1, and pKO-β1 cells adhering to MTSFN9–10. Areas were calculated from the thresholded eGFP-paxillin signal. Differences in adhesion area were not significant between the three cell types.
We next measured the distribution of loads experienced by individual integrins for pKO-αv, pKO-β1, and pKO-αv/β1 cells adhering to coverslips functionalized with MTSFN9–10. Contrary to expectation, the distributions of loads for integrins bearing >2 pN were notably similar across all three cell lines (Fig. 2B and fig. S3). However, the fraction of integrin-bound sensors underneath cells was significantly higher for pKO-αv/β1 and pKO-β1 cells as compared to pKO-αv cells (Fig. 2C and table S1), a factor that can largely account for the differences in traction generation at the whole-cell level. Thus, integrin usage indirectly influenced overall cellular traction by modulating the fraction of engaged integrins but did not influence the distribution of loads borne by individual, ligand-bound integrins.
An alternate hypothesis was that the load experienced by an integrin, regardless of class, is determined by the nature of its linkages to the actin cytoskeleton. The cytosolic protein vinculin reinforces the talin-actin linkage and may influence the load borne by individual integrins. We quantified traction generation at the whole-cell and single-integrin level for wild-type (WT) and vinculin-null (vin−/−) mouse embryonic fibroblasts (MEFs) adhering to MTSlow and MTShigh (Fig. 3, A and B) (28). WT but not vin−/− MEFs generated appreciable regions with low FRET when adhering to MTShigh (Fig. 3B), indicating that vinculin was required for the subpopulation of integrins transmitting loads >7 pN (Fig. 3C). This observation was likewise borne out by the distribution of loads on single integrins (Fig. 3D). Thus, linkages to F-actin, but not integrin heterodimer type, helped to determine the loads transmitted by individual integrins.
Fig. 3. Vinculin is required for higher forces per integrin.
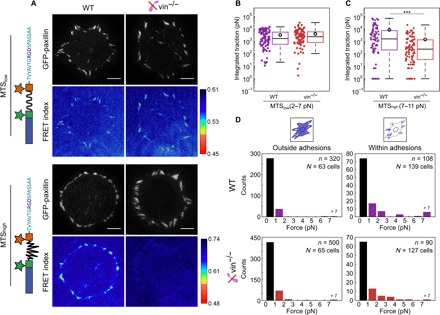
(A) Ensemble FRET maps for WT and vin−/− MEFs transfected with eGFP-paxillin and seeded on coverslips functionalized with MTSlow and MTShigh sensors. Scale bar, 10 μm. (B) Total integrated traction per cell for forces <7 pN measured with MTSlow. Open circles indicate the mean value. (WT: 96 cells, mean: 3.6 nN; vin−/−: 89 cells, mean: 4.4 nN.) (C) Total integrated traction per cell for forces between 7 and 11 pN measured with MTShigh. Open circles indicate the mean. (WT: 71 cells, mean: 8.7 nN; vin−/−: 99 cells, mean: 1.5 nN.) (D) Histograms of the single-molecule load measurements for WT and vin−/− MEFs measured for cells adhering to MTSlow for sensors outside adhesions (left) and within adhesions (right). ***P < 0.001 using two-sided Wilcoxon rank sum test.
We next examined the dynamics of force transmission at individual MTSs (Fig. 4). A large subpopulation of MTSlow and MTSFN9–10 yielded close-to-constant FRET levels corresponding to measurable loads <2 pN (Figs. 1D and 4). As noted above, we attribute this subpopulation to integrins that experience low loads that are independent of the cytoskeleton, for example, due to glycocalyx compression (29). In addition, we observed MTSlow, MTSFN9–10, and MTShigh sensor molecules that experienced loads consistent with cytoskeletally generated forces (>2 pN for MTSlow and MTSFN9–10; >7pN for MTShigh). Of these, most remained bound for up to tens of seconds at a close-to-constant force (Figs. 1D and 4B and table S2). This observation is in apparent contradiction with previous formulations of the clutch model, which predict continuous load-and-fail dynamics stemming from the progressive loading and failing of connections to F-actin (fig. S5). A minority of MTS FRET traces did exhibit anticorrelated changes in FRET donor and acceptor intensities indicative of dynamic changes in load (table S3 and figs. S6 to S8). We have classified these as either step (close to instantaneous at our time resolution of ~1 s) or more gradual ramp transitions (Fig. 4, A and C, and table S4). Although the clutch model predicts ramp increases and step decreases in load, it does not predict the step increases or ramp decreases in load that we observed (figs. S6 to S8). Although we cannot exclude the possibility that a subset of these events may be due to dye blinking, we rarely observe these events in our no-load control measurements (table S5). Measurements in U2OS osteosarcoma cells, which have been extensively used in studies of cell migration (30), did not show step or ramp transitions (fig. S9). Thus, dynamic changes in load, at least as assayed here, were evidently dispensable for cell adhesion.
Fig. 4. Dynamic transitions in load constitute a minority of sensor measurements.
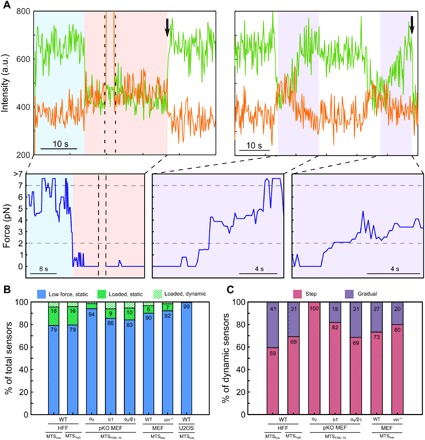
(A) Representative traces showing step (left) and gradual ramp (right) load transitions (FRET donor: green; FRET acceptor: orange; load: blue) for HFFs adhering MTSlow. Black arrows mark acceptor or donor bleaching; dashed black lines indicate direct excitation of the FRET acceptor. Horizontal gray dashed lines indicate upper and lower force measurement limits for MTSlow. (B) Percentage low force (defined as <2 for MTSlow or < 7 pN for MTShigh) (blue), higher force but static (green), and dynamic (hashed; subset of loaded integrins) sensors for a variety of cell types adhering to different MTSs. (C) Percent of dynamic sensors with step (magenta) and ramp (purple) transitions. U2OS cells had no observable dynamic events.
Our observations prompted us to explore extensions of the clutch model that incorporated known aspects of the architecture of FAs and the actin cytoskeleton. In established versions of the clutch model, all the F-actin filaments move with the same instantaneous velocity, which scales inversely with the total tension summed over all clutches (12). Individual clutches undergo repeated cycles of loading and failure as the monolithic F-actin moves rearward (fig. S5). We explored multiple extended models that included multiple clutch-actin connections, akin to multiple vinculins linking talin to actin, viscous relaxation of the clutch, catch bond behavior, or reversible actin cross-linkers (see Model Comparisons in the Supplementary Materials and Fig. 5). Among the models examined, only the addition of reversible cross-links between actin filaments (e.g., by α-actinin, filamin, nonmuscle myosin II, or other cross-linkers), yielded long periods of close-to-constant loads analogous to those observed in single-molecule measurements (Fig. 5, A to C). This result reflected the establishment of temporary mechanical equilibria between discrete clusters of motors (e.g., nonmuscle myosin II) and clutches (talin and vinculin). In addition, simulations recapitulated occasional step transitions, which reflected the disconnection or reattachment of an individual clutch to an actin filament, and ramp transitions, whose time scale reflected the equilibration of loads within the cross-linker network.
Fig. 5. A modified model of cytoskeletal force transduction yields mechanical equilibrium at individual integrins.
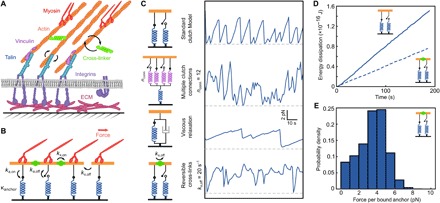
(A) Simplified cartoon of a FA: Nonmuscle myosin II pulls on reversibly cross-linked actin filaments, which are linked to integrins by vinculin and talin. (B) Cytoskeletal dynamics model: F-actin filaments bind to anchors (blue) and are linked by cross-linking proteins (green). (C) An example force trace of the standard clutch model and possible clutch model extensions that account for multivalent clutch connections, viscous relaxation, or reversible cross-links. Reversible cross-links allow for stable force plateaus as well as sporadic ramp and step events. The dashed gray lines indicate zero force. (D) Calculated energy dissipation from simulations with irreversible (top) and reversible (bottom) cross-links. (E) Force distribution for simulated anchors with reversible cross-linking (kx,off = 20 s−1).
Besides the addition of cross-linker binding and release rates [based on those of α-actinin (31)], the model required only minor tuning compared to a previously published clutch model (8). In contrast, irreversible cross-links between F-actin, as well as other clutch model extensions, resulted in load-and-fail dynamics, analogous to previous clutch models (Fig. 5C). These load-and-fail cycles are predicted to be costly in terms of energy dissipated in the repeated stretching of individual anchor linkages: In line with this understanding, models featuring reversible cross-linker dynamics predicted lower energy dissipation for similar overall force levels (Fig. 5D). Simulations performed with low substrate stiffnesses produced similar load dynamics (fig. S10). This result reflects the use of a clutch rupture force (Fb; table S6) that is larger than the typical load borne by an individual clutch linkage, a choice in parameterization that was necessary to reproduce the long-lived binding events that we observe (Figs. 1 and 4). It is possible that the dynamics and force sensitivity of the clutch connection to F-actin may differ in different cell types, and possibly in different compartments of the cell, for example, in nascent adhesions versus stable FAs.
The above model describes the loads borne by individual linkages to F-actin rather than by the integrins themselves. However, the anchor force distributions for simulations with reversible cross-linkers qualitatively match the observation of a peak in the measured load distributions for MTSlow and MTShigh (Figs. 5E and 1, E and F). Talin contains three actin binding sites and can recruit up to 11 vinculin molecules (32, 33). It is plausible that these connections to F-actin act in parallel, resulting in a broad range of loads transmitted by individual integrins. This possibility is supported by our observation of distinct subpopulations of integrins bearing ~6 and >11 pN within the FAs of HFFs (Fig. 1F), potentially reflecting multiple connections to F-actin. Single-pN loads are broadly consistent with a report that the average load experienced by talin was <6 pN (34). However, previous reports also describe >11-pN loads for a subset of talin molecules (35) and peak loads of >50 pN for a subset of integrins (21); these higher loads likely reflect additional, vinculin-mediated connections to F-actin (Fig. 3, A and C). In total, these observations are consistent with the hypothesis that the addition of multiple linkages to F-actin can result in a wide range of loads on individual integrins.
Actin retrograde flow rates provide an independent means of testing cytoskeletal clutch models (8, 12, 24). To examine how the simulated actin velocities compared with our system, we measured the velocity of F-actin filaments by treating living HFFs with 50 nM SiR-actin, a fluorogenic small-molecule probe that binds to F-actin. The mean speed for F-actin within both adhesions and linear F-actin–rich structures (e.g., stress fibers) was 7.9 nm/s (95% confidence interval: 7.6-8.1 nm/s; 9 cells, 2355 tracks), comparable to the mean velocity of 5 nm/s observed in reversible cross-linker simulations. These measured and simulated velocities are approximately one-half to one-third the magnitude of F-actin speeds measured in the lamellipodia of Xenopus XTC cells, respectively, differences that may reflect a decrease in F-actin velocities near adhesions (36).
DISCUSSION
Previously, we found that most of the integrins exist in a minimally tensioned state (14). Here, we extend this result and report that a small fraction of ligand-engaged integrins support loads >11 pN. The large majority of integrins thus experience loads substantially less than their maximum capacity. This mechanical reserve may allow cells to withstand external stresses that would threaten tissue integrity. Conversely, the ability to exert large, localized forces via a few integrins may be essential for cell migration and mechanosensing, for example, in fibrous ECM networks, where local effective stiffnesses can span several orders of magnitude (37, 38). Integrin complexes thus represent an interesting example of how a highly asymmetric distribution of activity at the molecular level (here, force transmission) can yield flexible and robust functionality at the cell and tissue levels.
Contrary to expectation, most of the integrins experienced close-to-constant loads within the resolution of our measurements (10). Although several nonexclusive factors, for example, domain unfolding in talin (33), may contribute to this observation, a model that incorporates reversible cross-links in the F-actin cytoskeleton is sufficient to account for our observations. This model is consistent with reports demonstrating that α-actinin cross-linking activity can change the mechanical properties of F-actin networks (39) and influence cell migration and traction force generation (40), although multiple actin cross-linkers are likely to contribute. Force transmission through a network of dynamic cross-linkers also reduced energy consumption compared to a system that underwent repeated load-and-fail cycles (Fig. 5D). We suggest that, despite the complexity of adhesion complexes, cellular mechanical homeostasis and efficient force transmission may arise from the core dynamical properties of the cytoskeleton. Additional tests in other model systems will, however, be required to establish the generality of this supposition.
Our data imply that the chain of molecular linkages between individual tension sensors and F-actin can remain stable for tens of seconds even under appreciable loads (Figs. 1 and 4 and table S2). This observation, in turn, suggests that the load on individual clutch linkages is, on average, appreciably less than their characteristic Fb (table S6). In the modified clutch model, this parameterization predicts adhesions whose stability is relatively insensitive to substrate stiffness (fig. S10). In contrast, adhesions with smaller Fb, or equivalently a higher average load per clutch, are predicted to yield load-and-fail dynamics at individual clutches and sensitivity to substrate stiffness as predicted in the original clutch model (8, 12). Determining whether the force sensitivity of adhesions differs as a function of cell type, matrix properties, and/or adhesion maturation provides an important target for future work. We speculate that modulation of key parameters such as the average load per clutch may provide a potent yet flexible method for cells to change mechanical states in response to external stimuli.
A core result of systems biology is that cellular subsystems, for example, signal transduction pathways, are often organized into semiautonomous functional modules, an outcome thought to enhance both robustness and evolvability (41, 42). Although previously proposed (5), whether a similar functional modularity might apply to complex structural assemblies such as FAs has been unclear. Our observations suggest that, despite a dense web of protein-protein interactions (43), FAs maintain modularity at a functional level. In the model systems studied here, the force-transducing machinery linking F-actin to adhesions resulted in per-integrin load distributions that were essentially identical regardless of integrin heterodimer usage (Fig. 2C). Integrin heterodimer usage in turn determined both ligand specificity and adhesion stability and, hence, influenced cellular adhesion and traction output. The flexibility afforded by this modular organization is likely to have greatly facilitated the evolution of the remarkable functional diversity of integrin-based adhesion complexes in metazoans.
Our findings complement work demonstrating that some proteins are recruited to FAs as part of preassembled complexes (44–46), suggestive of a hierarchical assembly process. These preassembled protein complexes are, however, not necessarily synonymous with single, defined functions; in other systems, evolutionary data demonstrate that biological function is often preserved even when the protein(s) fulfilling that function are not (47). Compositional and functional modularity may thus constitute distinct, and complementary, principles that govern the form and function of complex macromolecular assemblies.
MATERIALS AND METHODS
Sensor construct design
MTSlow and MTSFN9–10 were prepared as previously described (14, 15). The high-force MTS (MTShigh) was adapted from MTSlow by replacing the (GPGGA)8 module with another tension-sensitive domain, termed HPst (LSDED FKAVF GMTRS AFANL PLWKQ QALMK EKGLF), derived from the villin headpiece (16). The DNA encoding this construct was assembled by Epoch Life Sciences Inc. (Missouri City, TX) and was cloned into the pJ414 expression vector (DNA 2.0). We used Alexa 546 maleimide (Thermo Fisher Scientific, A10258) as the FRET donor and an Alexa 647 maleimide dye (Thermo Fisher Scientific, A20347) as the FRET acceptor. This modified MTS presents the identical RGD ligand derived from fibronectin as used in MTSlow. The entire MTShigh sequence is presented below:
M G S E I G T G F P F D P H Y V E V L G E R M H Y V D V G P R D G T P V L F L H G N P T S S Y V W R N I I P H V A P T H R S I A P D L I G M G K S D K P D L G Y F F D D H V R F M D A F I E A L G L E E V V L V I H D W G S A L G F H W A K R N P E R V K G I A F M E F I R P I P T W D E W P E F A R E T F Q A F R T T D V G R K L I I D Q N V F I E G T L P M G V V R P L T E V E M D H Y R E P F L N P V D R E P L W R F P N E L P I A G E P A N I V A L V E E Y M D W L H Q S P V P K L L F W G T P G V L I P P A E A A R L A K S L P N A K A V D I G P G L N L L Q E D N P D L I G S E I A R W L S T L E I S G G A G E F K C A G L S D E D F K A V F G M T R S A F A N L P L W K Q Q A L M K E K G L F G K C A G S E N L Y F Q G T V Y A V T G R G D S P A S S A A H H H H H H.
Expression and purification of MTS constructs
Sensors were expressed in BL21(DE3) competent Escherichia coli. Cultures (500 ml) were grown overnight at 30°C with ampicillin (100 μg/ml) and induced with 1 mM isopropyl-β-d-thiogalactopyranoside at an optical density of 0.6. The bacteria were then spun down at 6000g for 30 min and resuspended in 10 ml of lysis buffer [50 mM sodium phosphate, 300 mM NaCl, and 10 mM imidazole, (pH 8)] with a protease inhibitor cocktail (11873580001, Roche) and 10 μM lysozyme. The resuspended cells were rocked for 30 min at 4°C, lysed with a tip sonicator, and spun at 14,000g for 30 min. The supernatant was incubated with 2 ml of nickel–nitrilotriacetic acid HisPur Resin (Thermo Fisher Scientific) and rocked at 4°C for 2 hours. The solution was then packed into a gravity column, washed three times with 5 ml of wash buffer [50 mM sodium phosphate, 300 mM NaCl, and 20 mM imidazole (pH 7.4)], and incubated with 4 ml of elution buffer [50 mM sodium phosphate, 300 mM sodium chloride, and 250 mM imidazole (pH 7.4)] for 5 min. The eluate was collected and dialyzed overnight into storage buffer [1× phosphate-buffered saline (PBS), 1 mM EDTA, and 2 mM β-mercaptoethanol], flash-frozen, and stored at −80°C. Fractions were characterized by SDS–polyacrylamide gel electrophoresis (SDS-PAGE), and the concentration was determined by ultraviolet–visible (UV-Vis) spectroscopy (fig. S1).
Labeling of MTS constructs
Labeling of MTSs was performed through dual cysteine labeling and subsequent purification to separate the population of sensors with a single donor and acceptor dye. The cysteines were first reduced with 2 mM tris(2-carboxyethyl)phosphine for 30 min at room temperature and buffer exchanged into labeling buffer [50 mM phosphate buffer, 150 mM NaCl, and 1 mM EDTA (pH 7.4)] using three 7K Zeba desalting columns (89883, Thermo Fisher Scientific) in series. Alexa 546 and Alexa 647 maleimide dyes were added at a protein:donor:acceptor ratio of 1:1.5:2 for 1 hour at room temperature and overnight at 4°C. To help remove free dye and exchange the protein into fast protein liquid chromatography (FPLC) buffer A [50 mM tris buffer (pH 8) and 5 mM β-mercaptoethanol], the solution was passed through two PD MiniTrap desalting columns (45001529, GE Healthcare) in series. To separate out the sensors with a single donor and single acceptor, we used an AKTA Pure FPLC (GE Healthcare) with a MonoQ PC 1.6/5 (GE Healthcare) ion exchange column and a 10 mM/ml linear salt gradient with buffer B [50 mM tris (pH 8), 5 mM β-mercaptoethanol, and 2 M NaCl]. Fractions were characterized using SDS-PAGE, UV-Vis spectroscopy, and single-molecule imaging. The desired fractions were concentrated and exchanged into PBS using 3K centrifugal filters (Amicon) and stored at −80°C.
Preparation of functionalized Halo ligand coverslips
Coverslips were prepared as previously described (14). Briefly, 24 mm by 50 mm no. 1 coverslips (Fisherbrand) were sonicated in a bath sonicator (Kendall) for 20 min with isopropanol, Milli-Q water, and 5 M KOH, with Milli-Q water rinses between each step. The coverslips were then sonicated for 5 min in methanol and transferred to a solution of 2-ml N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (97%) (A0700, UCT Specialties), 10-ml glacial acetic acid, and 200-ml methanol. The coverslips were incubated in the silane mixture for 10 min, sonicated for 1 min, and then incubated for another 10 min. They were then rinsed with Milli-Q water and dried with nitrogen. To passivate the coverslips, 100 mg of maleimide polyethylene glycol (PEG) succinimidyl carboxymethyl ester (molecular weight, 5000; A5003-1, JenKem Technology) was dissolved in 1 ml of 100 mM phosphate buffer (pH 7.0). Two coverslips were sandwiched with 100 μl of the PEG solution in between for 1 hour at room temperature and protected from light. The coverslips were then washed with Milli-Q water and dried before being incubated overnight with 100 μl of 3 mM Halo ligand thiol (P6761 Promega or AcmeBiosciences Inc.) in 100 mM phosphate buffer (pH 7.0). Afterward, the coverslips were washed with Milli-Q water, dried, and stored in vacuum-sealed bags at −20°C.
Flow chamber preparation and imaging
Flow chambers were attached to PEGylated coverslips as previously described (15). For ensemble experiments, chambers were prepared with 100 nM double-labeled sensor and incubated at room temperature for 30 min. For the single-molecule assay, 100 nM unlabeled sensor with 100 pM labeled sensor was mixed in PBS and added to the flow cells for 30 min. The chambers were then washed with 200 μl of PBS and Pluronic F-127 (0.2% w/v) for ~1 min to prevent nonspecific cell attachment. The chambers were washed again with PBS to remove excess Pluronic. Cells were then added and incubated for at least 1 hour at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) high-glucose medium. FRET measurements were made within 3 hours of plating the cells and acquired with an objective heater (Bioptechs) set to 37°C. Images were prepared in Fiji (48) and analyzed using custom MATLAB scripts.
For immunofluorescence, cells were fixed with 4% paraformaldehyde for 15 min at 37°C and washed with PBS. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 5 min, washed with PBS, and then blocked with 5% bovine serum albumin (BSA) for 1 hour at room temperature. Antibodies for myosin IIa (Sigma-Aldrich, no. M8064; 1/100 dilution) and phosphorylated myosin light chain (Cell Signaling Technologies, no. 3675S; 1/200 dilution) were incubated with 5% BSA for 45 min at room temperature. Secondary antibodies (anti-rabbit 647, Cell Signaling Technologies, no. 4414S; and anti-mouse 555, Cell Signaling Technologies, no. 4409S; 1/200 dilution) were incubated with 5% BSA for 45 min at room temperature.
Total internal reflection fluorescence FRET imaging
Single-molecule and ensemble FRET fluorescence measurements were performed with objective-type total internal reflection fluorescence (TIRF) microscopy on an inverted microscope (Nikon TiE) with an Apo TIRF 100× oil objective lens, numerical aperture 1.49 (Nikon) as described previously (14) and controlled using Micromanager (49). Samples were excited with 473-nm OBIS laser (Coherent), 532-nm (Crystalaser), or 635-nm (Blue Sky Research) lasers. For single-molecule data, emission for the FRET donor and emission channels were separated as previously described and recorded on an electron-multiplying charge-coupled device camera (Andor iXon) (15). For collection of the green fluorescent protein (GFP) signal, we used an additional set of emission filters mounted on a motorized flip mount (Thorlabs Inc.) placed the donor fluorescence emission path. Filters used included a 593/40 nm filter (Semrock Inc.) for the collection of donor emission, a 675/30 nm filter for the collection of acceptor emission, and a 514/30 nm filter (Semrock Inc.) for GFP emission collection. For ensemble FRET maps taken for whole cells, emitted light passed through a quad-edge laser-flat dichroic with center/bandwidths of 405/60 nm, 488/100 nm, 532/100 nm, and 635/100 nm from Semrock Inc. (Di01-R405/488/532/635-25×36) and corresponding quad-pass filter with center/bandwidths of 446/37 nm, 510/20 nm, 581/70 nm, 703/88 nm band-pass filter (FF01-446/510/581/703-25). GFP, donor, and acceptor images were taken through separate additional cubes stacked into the light path (GFP: 470/40 nm, 495 nm long-pass, and 525/50 nm; donor: 550 nm long-pass; acceptor: 679/41 nm and 700/75 nm) and recorded on a Hamamatsu Orca Flash 4.0 camera.
Cell culture
HFF cells CCD-1070Sk (American Type Culture Collection CRL-2091) were cultured in DMEM high-glucose medium (Gibco, catalog no. 21063-029) in the absence of phenol red and supplemented with 10% fetal bovine serum (FBS; Axenia Biologix LLC ), sodium pyruvate (1 mM, Gibco), MEM nonessential amino acids (1×; Gibco), and penicillin/streptomycin (100 U/ml and 100 μg/ml; Gibco), herein referred to as normal culture medium. The cells were grown at 37°C with 5% CO2. Fibroblasts with stably expressing eGFP-paxillin (fused at the C terminus) were prepared as previously described (15).
pKO mouse kidney fibroblasts rescued with either αv, β1, or both αv and β1 integrin subunits were a gift from R. Fässler (Max Planck Institute Martinsried) (22). Cells were cultured on fibronectin-coated plastic (5 μg/ml; Corning, diluted in PBS and incubated at 37°C for 1 hour) in normal culture medium described above. pKO-β1 cells in particular were sensitive to the quality of fibronectin coating; thus, a minimum of 1 and 4 ml of the diluted fibronectin solution (5 μg/ml) were used per well for a six-well and 10-cm dish, respectively. Cells were grown at 37°C with 5% CO2.
WT and vin−/− MEFs were a gift from K. Rothenberg and B. Hoffman (Duke University) (28). Cells were cultured on tissue culture plastic in normal culture medium at 37°C and 5% CO2.
U2OS cells were a gift from M. Franklin and J. Liphardt (Stanford University). Cells were cultured on tissue culture plastic in normal culture medium at 37°C and 5% CO2.
Transfection
pKO-integrin cells and WT and vinculin KO MEF cells were transfected using a similar protocol to the one previously described for eGFP-paxillin human fibroblasts (15). Cells were trypsinized, pelleted, resuspended in medium lacking FBS and penicillin/streptomycin, and counted. pKO-integrin cells (2 × 106) and WT (5 × 105) and vinculin KO cells were repelleted at 800 rpm for 10 min. P4 Nucleofector solution (82 μl) was added to 18 μl of P4 supplement in a 1.5-ml Eppendorf tube and used to resuspend the cell pellet. DNA for C-terminal eGFP-paxillin (Addgene, no.15233) cloned into the DNA 2.0 PiggyBac vector (~4 μg) was added to the cells and gently flicked before transferring to a Lonza nucleofection cuvette. Cuvettes were placed in a Lonza 4D-Nucleofector system and program C2167 (for MEFs) was used. Warm medium (500 μl) was added to the cuvette, and cells were transferred to a six-well plate with medium equilibrated at 37°C with 5% CO2 using a pipette bulb without pipetting up and down. Cells were selected with puromycin (1 to 2.0 μg/ml) 24 hours after transfection for 4 to 6 days.
Calculating single-molecule FRET efficiency
Single-molecule data were acquired and analyzed as described previously (14). Briefly, data were acquired with excitation with a 532-nm laser at 5 frames/s for 300 or 600 frames and with direct acceptor excitation at 635 nm for approximately 10 frames at roughly frame 100. The direct excitation helped to distinguish between low-FRET sensors and sensors without an acceptor dye.
Traces were analyzed using a custom MATLAB code, and donor and acceptor channels were aligned using a single-molecule high-resolution colocalization map generated by scanning across a field of beads (50). The positions of individual sensors were then detected using a spot-finding algorithm (T. Ursell, Stanford University) and were determined to be colocalized if within two pixels. Intensities were calculated on the basis of an average of 7 × 7 pixels centered around the detected spot and corrected for spectral bleedthrough.
Intensities for each dye were averaged over manually identified FRETing, non-FRETing, and bleached regions. When the acceptor bleached before the donor, we used the following expression to calculate FRET efficiency
where Ia is the acceptor intensity during FRET, Ia,back is the acceptor background intensity, Id is the donor intensity during FRET, Id,0 is the donor intensity after acceptor photobleaching, Id,back is the donor background intensity, and γ is the correction factor accounting for relative dye quantum yields and instrument detection efficiencies.
When the donor fluorophore bleached first, the FRET efficiency was calculated as
Values for γ0 were 0.40 for MTSlow, 0.52 for MTSFN, and 0.52 for MTShigh.
Events were double-checked by generating a series of z projections for the donor and acceptor molecule during FRETing, non-FRETing, and bleached states. The autoGaussianSurf Matlab function (P. Mineault) was used to fit a two-dimensional Gaussian to the 7 ×7–pixel area to determine whether the spot represented a single emitting fluorophore. Low-FRET events were verified as having a functional acceptor by direct excitation with a 635-nm laser.
Combined force histograms
Combined single-molecule histograms for MTSlow and MTShigh were created by normalizing the proportion of molecules for the overlapping force bins. The proportion of molecules greater than 7 pN measured previously using MTSlow nearly matched the proportion of molecules bearing greater than 7 pN measured using MTShigh (14). The final histograms were created by scaling the force distribution measured by MTShigh by the proportion of molecules bearing greater than 7 pN measured using MTSlow for molecules within adhesions, underneath cells, and outside adhesions separately.
Obtaining theoretical FRET-force calibration curve
The FRET versus force response of the (GPGGA)8 linker used here was previously reported by Grashoff et al., and an updated calibration was recently reported by LaCroix et al. (51, 52). We used the updated MATLAB calibrations from LaCroix et al. to generate improved FRET versus force calibration curves. Using 43 amino acids (for the eight repeats of GPGGA plus the two cysteines and a single lysine), a fluorophore radius of 0.5 nm, a Forster radius of 6.95 nm, and persistence lengths from 0.87 to 0.98 nm, we constructed three FRET-force calibration curves to account for the slightly different resting FRET efficiencies determined experimentally (fig. S11).
Ensemble FRET analysis
Ensemble measurements were performed as previously described (14). In summary, images of eGFP-paxillin marked cells were acquired using a Hamamatsu Orca Flash 4.0 camera and were subsequently corrected for illumination spatial inhomogeneities, background-subtracted, and intensity-normalized. The GFP image was then boxcar-averaged (“moving average v3.1” from MATLAB Central File Exchange) at 10 different rotations of the original image at 20° intervals, thresholded, and segmented using a watershed algorithm. The segmented image was then corrected to combine adjacent islands representing a single adhesion and filtered to exclude islands below a lower limit (0.5 μm2). The segmented GFP image was then used to mask the corresponding FRET signal.
FRET images were converted to FRET index values by dividing the acceptor intensity over the sum of the donor and the acceptor signal. Then, the FRET images were converted to FRET efficiency after correcting to dye labeling efficiency, bleedthrough, the measured no-load FRET efficiency, and the FRET-index measured outside the cell (14). The total integrated traction of a cell was calculated by summing the force contributions (defined as the average pixel value times number of pixels) for pixels within adhesions. For MTSlow or MTSFN9–10, forces corresponding to >7pN were set to 7.1 pN. For MTShigh measurements, calculated forces corresponding to <7 pN were set to 0 pN.
Dynamics analysis
Traces with potential dynamic behavior, identified by having distinct anticorrelated signals, were marked and analyzed individually. Step events were classified by having large anticorrelated changes in the donor and acceptor signal within the period of one to three frames (0.2 to 0.6 s) and were manually annotated. Force traces were smoothed using a median filter over five frames. Ramp events were classified by having more gradual anticorrelated changes and were manually marked. Ramp traces were then converted to the force domain, and only changes in load between 2 and 7 pN were fit. Dynamic events were only accepted if the acceptor was confirmed to be active and could not be accounted for by either donor or acceptor photobleaching. Very few dynamic-like sensors were observed under no-load conditions (table S5).
Actin tracking imaging
Halo-PEG coverslips were incubated with 100 nM unlabeled RGD sensor at room temperature for 30 min, and cells were seeded and allowed to spread for at least ~1 hour. After a 9-min incubation with 50 nM SiR-actin (Cytoskeleton Inc., no. CY-SC001), the sample was incubated with Prolong Live Antifade Reagent (Invitrogen, P36975) for 1 hour. The low dilution of SiR-actin allowed for individual molecules to be tracked, and the addition of Prolong reduced photobleaching. For each cell, a 100-ms exposure of the GFP-paxillin channel (for masking) was first acquired, followed by a 60-frame sequence in the far red channel (SiR-actin) with 300-ms exposures taken every 2 s.
For fixed cell control data, cells were allowed to spread on functionalized coverslips and then fixed in 4% paraformaldehyde for 15 min at room temperature. After rinsing, the cells were treated with SiR-actin and Prolong reagent as above.
F-actin tracking analysis
Speckles and tracks were identified using quantitative fluorescent speckle microscopy software made available by the Danuser laboratory (53). The localizations, which are given with pixel precision, are fit to subpixel positions by Gaussian fitting.
Every frame was used to track speckles, but velocities were calculated from speckle displacements over five frames (10 s), giving velocities on the time scale of our FRET measurements. Adhesions were masked by an Otsu threshold of the eGFP-paxillin image after background subtraction to remove the diffuse cytoplasmic signal. F-actin stress fibers were masked as the brightest 3% of pixels of a time-series projection of the F-actin tracks. We measure the actin velocities of tracks that originate both over stress fibers and adhesions.
From our mean velocity measurement, <d>, we calculate a corrected velocity, s, using the following relation (50)
where σ is the localization error (SD) of single-molecule localizations (estimated to be 47 nm here).
Reversible cross-linker model
In the reversible cross-linker model, F-actin was treated as a network of filaments connected by noncompliant dynamic cross-linkers (Fig. 4). These cross-linkers bind and unbind at rates kx,on and kx,off. Each clutch is bound to an individual filament, and the number of motors per filament is dictated by simulation parameters (table S6). Force traces were averaged over 1-s time steps to reflect the time scale of the processed force traces from the single-molecule measurement. Individual clutches experience load routed from different combinations of motors at any given instant, where individual loads are dictated by both the force-velocity relationships of individual motors and the loading history of the clutches within the cluster. The forces on individual clutches build until the F-actin retrograde flow rate is close to 0 and the motor stall force and clutch force are nearly equal. When a cross-linker binds or unbinds, the forces on the associated clutches are no longer balanced, and the F-actin velocities adjust to reestablish mechanical equilibrium.
In the resulting simulations, dynamic clusters of clutches continuously stretch and relax, oscillating around force plateaus for periods of 10 to 60 s. Ramp and step transitions are observed throughout these simulations, in a manner consistent with our experimental observation: A step transition occurs when a binding clutch quickly builds force or when an unbinding clutch instantaneously returns to 0 force. More gradual ramp transitions occur in neighboring clutches as the associated loads readjust to achieve a force balance within the cluster.
Although a variety of models were explored, the dynamic F-actin network best captured the behavior observed experimentally. In contrast to the other models, the force plateaus persisted the longest with minimal fluctuations, and the single-clutch dynamics were consistent across simulation parameters (i.e., relatively insensitive to small parameter changes). Although further testing of the dynamic F-actin network model is still needed and alternate models have not been definitively ruled out, the dynamic F-actin model best captures the experimentally observed behavior of the individual sensor force distribution and force dynamics.
Energy dissipation was calculated as the sum of stored energy in anchors upon unbinding. When an unbinding event occurred, the stored energy in the anchor was calculated using the spring constant of the anchor and its displacement
The running sum of this value was recorded for the duration of the experiment. This does not account for energy dissipation within the motor-actin system.
Supplementary Material
Acknowledgments
We thank K. Rothenberg of the Hoffman laboratory (Duke University) for the vin−/− MEFs, M. Franklin from the Liphardt laboratory (Stanford University) for the U2OS cells, and R. Fässler (Max Planck Institute, Martinsried) for the pKO cells. We thank the Khosla laboratory for their protein purification expertise and access to their equipment. We would also like to thank A. LaCroix from the Hoffman laboratory (Duke University) for useful discussions on FRET-force calibrations and B. Zhong, E. Korkmazhan, C. Garzon-Coral, W. Weis, and O. Chaudhuri for useful discussions and feedback. The data reported in this paper are further detailed in the Supplementary Materials. Funding: Research reported in this publication was supported by grants R01-CA172986 and U54-CA210190 to D.J.O. and R01-GM112998-01 and R35-GM130332 to A.R.D. from the National Institutes of Health (NIH). The research of A.R.D. was supported, in part, by a Faculty Scholar from the Howard Hughes Medical Institute. S.J.T. was supported by the John Stauffer Stanford Graduate Fellowship from Stanford, and A.C.C., C.M.M., and L.S.P. were supported by Graduate Research Fellowships from the National Science Foundation (00039202). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Author contributions: S.J.T., A.C.C., and C.M.M. performed experiments and analyzed data. S.M.A., L.S.P., and A.R.D. created and ran clutch model simulations. All authors contributed to the writing and editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaax0317/DC1
REFERENCES AND NOTES
- 1.Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Margulies S. S., Dembo M., Boettiger D., Hammer D. A., Weaver V. M., Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Mekhdjian A. H., Kai F., Rubashkin M. G., Prahl L. S., Przybyla L. M., McGregor A. L., Bell E. S., Barnes J. M., DuFort C. C., Ou G., Chang A. C., Cassereau L., Tan S. J., Pickup M. W., Lakins J. N., Ye X., Davidson M. W., Lammerding J., Odde D. J., Dunn A. R., Weaver V. M., Integrin-mediated traction force enhances paxillin molecular associations and adhesion dynamics that increase the invasiveness of tumor cells into a three-dimensional extracellular matrix. Mol. Biol. Cell 28, 1467–1488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bökel C., Brown N. H., Integrins in development: Moving on, responding to, and sticking to the extracellular matrix. Dev. Cell 3, 311–321 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Horton E. R., Byron A., Askari J. A., Ng D. H. J., Millon-Frémillon A., Robertson J., Koper E. J., Paul N. R., Warwood S., Knight D., Humphries J. D., Humphries M. J., Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 17, 1577–1587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaidel-Bar R., Itzkovitz S., Ma’ayan A., Iyengar R., Geiger B., Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–867 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward M. D., Hammer D. A., Focal contact assembly through cytoskeletal polymerization: Steady state analysis. J. Math. Biol. 32, 677–704 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Hu K., Ji L., Applegate K. T., Danuser G., Waterman-Storer C. M., Differential transmission of actin. Science 315, 111–115 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Chan C. E., Odde D. J., Traction dynamics of filopodia on compliant substrates. Science 322, 1687–1691 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Case L. B., Waterman C. M., Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 17, 955–963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown C. M., Hebert B., Kolin D. L., Zareno J., Whitmore L., Horwitz A. R., Wiseman P. W., Probing the integrin-actin linkage using high-resolution protein velocity mapping. J. Cell Sci. 119, 5204–5214 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Ghibaudo M., Saez A., Trichet L., Xayaphoummine A., Browaeys J., Silberzan P., Buguin A., Ladoux B., Traction forces and rigidity sensing regulate cell functions. Soft Matter 4, 1836–1843 (2008). [Google Scholar]
- 12.Bangasser B. L., Rosenfeld S. S., Odde D. J., Determinants of maximal force transmission in a motor-clutch model of cell traction in a compliant microenvironment. Biophys. J. 105, 581–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elosegui-Artola A., Oria R., Chen Y., Kosmalska A., Pérez-González C., Castro N., Zhu C., Trepat X., Roca-Cusachs P., Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 18, 540–548 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Chang A. C., Mekhdjian A. H., Morimatsu M., Denisin A. K., Pruitt B. L., Dunn A. R., Single molecule force measurements in living cells reveal a minimally tensioned integrin state. ACS Nano 10, 10745–10752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morimatsu M., Mekhdjian A. H., Chang A. C., Tan S. J., Dunn A. R., Visualizing the interior architecture of focal adhesions with high-resolution traction maps. Nano Lett. 15, 2220–2228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Austen K., Ringer P., Mehlich A., Chrostek-Grashoff A., Kluger C., Klingner C., Sabass B., Zent R., Rief M., Grashoff C., Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nat. Cell Biol. 17, 1597–1606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurchenko C., Chang Y., Narui Y., Zhang Y., Salaita K. S., Integrin-generated forces lead to streptavidin-biotin unbinding in cellular adhesions. Biophys. J. 106, 1436–1446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galior K., Liu Y., Yehl K., Vivek S., Salaita K., Titin-based nanoparticle tension sensors map high-magnitude integrin forces within focal adhesions. Nano Lett. 16, 341–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Blanchfield L., Ma V. P.-Y., Andargachew R., Galior K., Liu Z., Evavold B., Salaita K., DNA-based nanoparticle tension sensors reveal that T-cell receptors transmit defined pN forces to their antigens for enhanced fidelity. Proc. Natl. Acad. Sci. U.S.A. 113, 5610–5615 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roein-Peikar M., Xu Q., Wang X., Ha T., Ultrasensitivity of cell adhesion to the presence of mechanically strong ligands. Phys. Rev. X. 6, 011001 (2016). [Google Scholar]
- 21.Chowdhury F., Li I. T. S., Leslie B. J., Doğanay S., Singh R., Wang X., Seong J., Lee S.-H., Park S., Wang N., Ha T., Single molecular force across single integrins dictates cell spreading. Integr. Biol. 7, 1265–1271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiller H. B., Hermann M.-R., Polleux J., Vignaud T., Zanivan S., Friedel C. C., Sun Z., Raducanu A., Gottschalk K.-E., Théry M., Mann M., Fässler R., β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 15, 625–636 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Bharadwaj M., Strohmeyer N., Colo G. P., Helenius J., Beerenwinkel N., Schiller H. B., Fässler R., Müller D. J., αV-class integrins exert dual roles on α5β1 integrins to strengthen adhesion to fibronectin. Nat. Commun. 8, 14348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elosegui-Artola A., Bazellières E., Allen M. D., Andreu I., Oria R., Sunyer R., Gomm J. J., Marshall J. F., Jones J. L., Trepat X., Roca-Cusachs P., Rigidity sensing and adaptation through regulation of integrin types. Nat. Mater. 13, 631–637 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Mets R., Wang I., Balland M., Oddou C., Moreau P., Fourcade B., Albiges-Rizo C., Delon A., Destaing O., Cellular tension encodes local Src-dependent differential β1 and β3 integrin mobility. Mol. Biol. Cell 30, 181–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benito-Jardón M., Klapproth S., Gimeno-LLuch I., Petzold T., Bharadwaj M., Müller D. J., Zuchtriegel G., Reichel C. A., Costell M., The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. eLife 6, e22264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García A. J., Schwarzbauer J. E., Boettiger D., Distinct activation states of α5β1 integrin show differential binding to RGD and synergy domains of fibronectin. Biochemistry 41, 9063–9069 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Mierke C. T., Kollmannsberger P., Zitterbart D. P., Diez G., Koch T. M., Marg S., Ziegler W. H., Goldmann W. H., Fabry B., Vinculin facilitates cell invasion into three-dimensional collagen matrices. J. Biol. Chem. 285, 13121–13130 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paszek M. J., DuFort C. C., Rossier O., Bainer R., Mouw J. K., Godula K., Hudak J. E., Lakins J. N., Wijekoon A. C., Cassereau L., Rubashkin M. G., Magbanua M. J., Thorn K. S., Davidson M. W., Rugo H. S., Park J. W., Hammer D. A., Giannone G., Bertozzi C. R., Weaver V. M., The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardel M. L., Schneider I. C., Aratyn-Schaus Y., Waterman C. M., Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 26, 315–333 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J., Wirtz D., Pollard T. D., Dynamic cross-linking by α-actinin determines the mechanical properties of actin filament networks. J. Biol. Chem. 273, 9570–9576 (1998). [DOI] [PubMed] [Google Scholar]
- 32.Yao M., Goult B. T., Chen H., Cong P., Sheetz M. P., Yan J., Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep. 4, 4610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao M., Goult B. T., Klapholz B., Hu X., Toseland C. P., Guo Y., Cong P., Sheetz M. P., Yan J., The mechanical response of talin. Nat. Commun. 7, 11966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A., Ouyang M., Van den Dries K., McGhee E. J., Tanaka K., Anderson M. D., Groisman A., Goult B. T., Anderson K. I., Schwartz M. A., Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol. 213, 371–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ringer P., Weißl A., Cost A.-L., Freikamp A., Sabass B., Mehlich A., Tramier M., Rief M., Grashoff C., Multiplexing molecular tension sensors reveals piconewton force gradient across talin-1. Nat. Methods 14, 1090–1096 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Yamashiro S., Tanaka S., McMillen L. M., Taniguchi D., Vavylonis D., Watanabe N., Myosin-dependent actin stabilization as revealed by single-molecule imaging of actin turnover. Mol. Biol. Cell 29, 1941–1947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beroz F., Jawerth L. M., Münster S., Weitz D. A., Broedersz C. P., Wingreen N. S., Physical limits to biomechanical sensing in disordered fibre networks. Nat. Commun. 8, 16096 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Owen L. M., Adhikari A. S., Patel M., Grimmer P., Leijnse N., Kim M. C., Notbohm J., Franck C., Dunn A. R., A cytoskeletal clutch mediates cellular force transmission in a soft, three-dimensional extracellular matrix. Mol. Biol. Cell 28, 1959–1974 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J., Tseng Y., Wirtz D., Strain hardening of actin filament networks. Regulation by the dynamic cross-linking protein α-actinin. J. Biol. Chem. 275, 35886–35892 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Ehrlicher A. J., Krishnan R., Guo M., Bidan C. M., Weitz D. A., Pollak M. R., Alpha-actinin binding kinetics modulate cellular dynamics and force generation. Proc. Natl. Acad. Sci. U.S.A. 112, 6619–6624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitano H., Biological robustness. Nat. Rev. Genet. 5, 826–837 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Atay O., Skotheim J. M., Modularity and predictability in cell signaling and decision making. Mol. Biol. Cell 25, 3445–3450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geiger T., Zaidel-Bar R., Opening the floodgates: Proteomics and the integrin adhesome. Curr. Opin. Cell Biol. 24, 562–568 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Ye F., Lagarrigue F., Ginsberg M. H., SnapShot: Talin and the modular nature of the integrin adhesome. Cell 156, 1340–1340.e1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffmann J.-E., Fermin Y., Stricker R. L., Ickstadt K., Zamir E., Symmetric exchange of multi-protein building blocks between stationary focal adhesions and the cytosol. eLife 3, e02257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toplak T., Pandzic E., Chen L., Vicente-Manzanares M., Horwitz A. R., Wiseman P. W., STICCS reveals matrix-dependent adhesion slipping and gripping in migrating cells. Biophys. J. 103, 1672–1682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebé-Pedrós A., Roger A. J., Lang F. B., King N., Ruiz-Trillo I., Ancient origin of the integrin-mediated adhesion and signaling machinery. Proc. Natl. Acad. Sci. U.S.A. 107, 10142–10147 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Edelstein A. D., Tsuchida M. A., Amodaj N., Pinkard H., Vale R. D., Stuurman N., Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Churchman L. S., Ökten Z., Rock R. S., Dawson J. F., Spudich J. A., Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc. Natl. Acad. Sci. U.S.A. 102, 1419–1423 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grashoff C., Hoffman B. D., Brenner M. D., Zhou R., Parsons M., Yang M. T., McLean M. A., Sligar S. G., Chen C. S., Ha T., Schwartz M. A., Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466, 263–266 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaCroix A. S., Lynch A. D., Berginski M. E., Hoffman B. D., Tunable molecular tension sensors reveal extension-based control of vinculin loading. eLife 7, e33927 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mendoza M. C., Besson S., Danuser G., Quantitative fluorescent speckle microscopy (QFSM) to measure actin dynamics. Curr. Protoc. Cytom. 62, 2.18.1–2.18.26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang D. L., Bax N. A., Buckley C. D., Weis W. I., Dunn A. R., Vinculin forms a directionally asymmetric catch bond with F-actin. Science 357, 703–706 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaax0317/DC1