

Nano-mediated tumor tissue–cell–mitochondria–targeted multistage delivery and controlled release of CO augment therapy outcomes.
Abstract
CO gas molecule not only could selectively kill cancer cells but also exhibits limited anticancer efficacy because of the lack of active tumor-targeted accumulation capability. In this work, a multistage assembly/disassembly strategy is developed to construct a new intelligent nanomedicine by encapsulating a mitochondria-targeted and intramitochondrial microenvironment–responsive prodrug (FeCO-TPP) within mesoporous silica nanoparticle that is further coated with hyaluronic acid by step-by-step electrostatic assembly, realizing tumor tissue–cell–mitochondria–targeted multistage delivery and controlled release of CO in a step-by-step disassembly way. Multistage targeted delivery and controlled release of CO involve (i) the passive tumor tissue–targeted nanomedicine delivery, (ii) the active tumor cell–targeted nanomedicine delivery, (iii) the acid-responsive prodrug release, (iv) the mitochondria-targeted prodrug delivery, and (v) the ROS-responsive CO release. The developed nanomedicine has effectively augmented the efficacy and safety of CO therapy of cancer both in vitro and in vivo. The proposed multistage assembly/disassembly strategy opens a new window for targeted CO therapy.
INTRODUCTION
Chemotherapy and radiotherapy are being widely used as first-line treatment methods against cancers. However, high nonspecific toxicity of most existing chemo/radiotherapeutic agents to normal tissues and blood frequently leads to a low quality of life. The development of low-toxicity and high-efficacy anticancer drugs and new therapy strategies is highly desired (1–3). Fortunately, it was found that some gaseous signaling molecules such as CO and NO can selectively induce the apoptosis of cancer cells and also protect normal cells in the appropriate concentration range (4–8). Such an anticancer selectivity is clearly superior to traditional chemo/radiotherapeutic drugs. Therefore, gas therapy is emerging as a promising therapy strategy (9–14).
CO has high but aimless transmembrane diffusibility owing to small molecular size. However, high blood concentration and low intratumoral accumulation could cause CO poisoning risk and limited CO therapy efficacy, respectively. In the current clinical practice regarding CO therapy, two main CO administration routes are direct inhalation of CO gas and uptake of CO prodrugs. It is hard for direct inhalation of CO gas to control the dose of CO intake, while existing CO prodrugs lack tumor-targeting capabilities. Both CO administration routes bring a high risk of CO poisoning. Therefore, the targeted delivery and controlled release of CO is vitally important to enhance the efficacy and biosafety of CO therapy (2, 9, 15). By virtue of multifunctional nanomaterials, the tumor-targeted delivery of CO prodrug [or CO-releasing molecules (CORMs)] and intratumoral controlled release of CO by intelligent nanomedicines is an important strategy (2, 16–20). Some nanomedicines have been developed for tumor-targeted delivery and controlled release of CO, but they can only deliver CO into lysosome or cytoplasm rather than mitochondria. It is well known that mitochondria are the targets for CO to exert its physiological functions (2, 21, 22), where the administration of CO can inhibit mitochondrial respiration by intensively competing with oxygen for binding to cytochrome c oxidase to impair mitochondrial bioenergetics of tumor cells, leading to the apoptosis of tumor cells (2, 4–6). Therefore, we here hypothesize that the sequentially targeted delivery of CO prodrug to tumor tissue to cell to mitochondria could possibly achieve ideal CO therapy efficacy. Furthermore, we also hypothesize that the tumor mitochondrial microenvironment–triggered release of CO from prodrug would further enhance the efficacy of targeted therapy (23).
In this work, we propose a multistage targeted delivery and controlled release strategy and construct a versatile intelligent nanomedicine (FeCO-TPP@MSN@HA) to orderly realize the tumor tissue–targeted delivery of the nanomedicine (first-step targeting), the tumor cell–targeted delivery of the nanomedicine (second-step targeting), the acid-responsive release of a new CO prodrug from the nanomedicine (first-step controlled release), the mitochondria-targeted delivery of the prodrug (third-step targeting), and the intramitochondrial reactive oxygen species (ROS)–responsive release of CO (second-step controlled release), as illustrated in Fig. 1 (A and B). The core of this work is the design and synthesis of the mitochondria-targeted and intramitochondrial microenvironment–responsive prodrug [FeCO–triphenylphosphonium (TPP)] by a coordination linkage reaction between mercapto propyltriphenylphosphonium (TPP-SH) and Fe3CO12 (FeCO). Negatively charged mesoporous silica nanoparticle (MSN) is selected as a biocompatible carrier to efficiently load the positively charged FeCO-TPP prodrug by electrostatic assembly, realizing the acid-responsive disassembly and release of the FeCO-TPP prodrug. Further coating of hyaluronic acid (HA) on the surface of small-sized MSN (FeCO-TPP@MSN@HA) by electrostatic assembly achieves both passive tumor targeting by the enhanced permeability and retention (EPR) effect and active targeting to CD44-overexpressed tumor cells (24–26). The innovation and clinical benefits of this work involves the development of multistage targeted delivery and controlled release by the FeCO-TPP@MSN@HA nanomedicine in a step-by-step disassembly way to realize high-efficacy and safe mitochondria-targeted CO therapy of cancer in vitro and in vivo. In comparison with other common nanomedicines for tumor-targeted CO delivery, the developed FeCO-TPP@MSN@HA nanomedicine can particularly deliver CO to the mitochondria of cancer cells, exhibiting maximal outcome of cancer therapy.
Fig. 1. Construction of the FeCO-TPP@MSN@HA nanomedicine by the multistage assembly/disassembly strategy for tumor-targeted CO delivery.
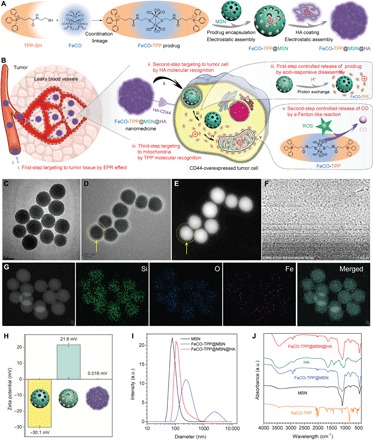
(A) Schematic illustration of multistage assembly method for construction of the FeCO-TPP@MSN@HA nanomedicine. (B) Schematic illustration of multistage disassembly strategy and mechanisms for tumor tissue–cell–mitochondria–targeted delivery and controlled release of CO by the FeCO-TPP@MSN@HA nanomedicine. The transmission electron microscopy (TEM) image of the MSN carrier (C); the TEM (D), high angle annular dark field (HADDF) (E), scanning electron microscopy (SEM) (F), and element mapping (G) images of the FeCO-TPP@MSN@HA nanomedicine; the zeta potentials (H), dynamic light scattering (DLS) particle size distributions (I), and FTIR spectra (J) of MSN, FeCO-TPP@MSN, and FeCO-TPP@MSN@HA are shown. a.u., arbitrary units.
RESULTS AND DISCUSSION
Synthesis and characterization of the FeCO-TPP prodrug and the FeCO-TPP@MSN@HA nanomedicine
The key of the work is to synthesize the new prodrug that is able to both accumulate into mitochondria in a targeted way and release CO in mitochondria in a responsive way. Positively charged TPP ion has been well verified to be an excellent mitochondria-targeting moiety owing to its electrostatic and lipophilic attractions with mitochondrial membrane (2, 27–29). Moreover, we have confirmed that metal carbonyl compounds are able to ROS-responsively decompose into CO by the Fenton-like reaction. Therefore, we conjugated a TPP derivate [3-carboxypropyl triphenylphosphonium bromide (TPP-COOH)] with a metal carbonyl compound (FeCO) to synthesize the above-designed prodrug (FeCO-TPP) in this work, as shown in Fig. 1A. First, TPP-SH was synthesized by an esterification reaction between TPP-COOH and H2N-EtSH (H2N-C2H4-SH, cysteamine) and was then conjugated with FeCO to form FeCO-TPP by the ligand-exchange reaction (30). The structures of the reagents, intermediates, and final product FeCO-TPP were confirmed by proton nuclear magnetic resonance (1H NMR) and mass spectrometry (MS) spectra characterization (fig. S1, A to F). Furthermore, from Fourier transform infrared (FTIR) spectra (fig. S1G), FeCO-TPP inherited the characteristic bands of TPP and carbonyl, as indicated by the cyan and yellow zones, respectively, further verifying the structure of FeCO-TPP. In addition, the ultraviolet (UV) adsorption bands of FeCO shifted to longer wavelength (fig. S1H) owing to the coordination between TPP-SH and FeCO.
To protect the FeCO-TPP prodrug and to deliver it into tumor cells for subsequent mitochondrial targeting, we used the negatively charged MSN to encapsulate the positively charged FeCO-TPP for tumor-targeted delivery by the EPR effect and then coated a layer of HA (a receptor of CD44) by electrostatic assembly to recognize tumor cells that overexpressed CD44 protein (Fig. 1B). The structure, morphology, composition, and potential of the synthesized FeCO-TPP@MSN@HA nanomedicine were characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), dynamic light scattering (DLS), and FTIR techniques (Fig. 1, C to J). From panels C and I of Fig. 1, the synthesized MSN exhibited uniform particle size (about 70 nm), good dispersion, and highly negative surface potential in favor of prodrug loading and HA coating. From panels D and E of Fig. 1, it could be found that a thin layer of HA (about 15 nm in thickness) with low contrast was coated on the surface of MSN, as directed by yellow arrows. The synthesized FeCO-TPP@MSN@HA nanomedicine maintained good dispersion and had a slightly increased hydrated diameter owing to HA coating [3.9 weight % (wt %) HA coating capacity], as indicated by SEM image (Fig. 1F) and DLS data (Fig. 1I). The elementary mapping results (Fig. 1G) clearly indicated that the FeCO-TPP prodrug was well encapsulated into MSN with a loading capacity of 353.9 mg of FeCO-TPP per gram of MSN (47.4 mg of CO per gram of MSN), which caused the surface potential reversion after prodrug loading (Fig. 1H) as expected. The highly positive potential of FeCO-TPP@MSN made it easy to be coated with HA by electrostatic assembly, leading to approximately neutral surface potential of the synthesized FeCO-TPP@MSN@HA nanomedicine and preventing the leakage of the FeCO-TPP prodrug and the HA coating with high stability in aqueous solution (fig. S2A). Moreover, FTIR results further indicated the successful FeCO-TPP loading and HA coating onto MSN (Fig. 1J). In addition, it can clearly be found from Brunauer-Emmett-Teller (BET) data (fig. S2, B and C) that the porous channels of MSN were filled with the prodrug.
Behavior and mechanism of the release of prodrug and CO
We hypothesized that the FeCO-TPP@MSN@HA nanomedicine could recognize/adhere onto tumor cells after reaching the tumor site and then take off HA during endocytosis into lysosome, so we checked the sequential process step by step, involving the tumor tissue–targeted delivery of the nanomedicine, the tumor cell–targeted delivery of the nanomedicine, the acid-responsive release of prodrug, the mitochondria-targeted delivery of prodrug, and the ROS-responsive release of CO. The sequentially acid-/ROS-responsive release profiles of the FeCO-TPP@MSN nanomedicine were checked first under in vitro simulated conditions. From Fig. 2A, FeCO-TPP@MSN was considerably stable in the phosphate-buffered saline (PBS) solution (pH 7.4), but the sharp release of the FeCO-TPP prodrug occurred in the slightly acid solution with pH 6.8 and higher acidity resulted in quicker release of prodrug. Such an acid-responsive behavior should derive from the proton exchange with the positively charged FeCO-TPP prodrug (Fig. 2A), which was just an opposite process of prodrug loading (Fig. 1A). Many silanol groups (Si─OH) on the surface of MSN could be ionized into silicate (Si─O−) to adsorb FeCO-TPP+ and could also be protonated under the acidic conditions to desorb/release FeCO-TPP+ (31). Furthermore, the ROS-responsive CO release behavior of the released FeCO-TPP prodrug was further investigated at different concentrations of ·OH, which were used to simulate the ROS-enriching microenvironment of mitochondria. The released CO concentration was monitored by the UV method using hemoglobin as probe (16). From Fig. 2B, FeCO-TPP decomposed into CO in the presence of ·OH owing to strong oxidation of ·OH, which was similar to the property of metal carbonyls (a kind of Fenton-like agents), as reported previously (17). Moreover, a higher concentration of ·OH caused faster CO release, meaning more intensive influence on more malignant tumor cells, as they have higher energy levels according to the Warburg effect of tumor (32).
Fig. 2. Controlled release profiles of FeCO-TPP@MSN.
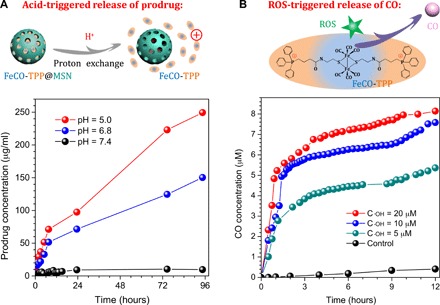
Acid-responsive prodrug release profiles of the FeCO-TPP@MSN nanomedicine under in vitro simulated conditions (A) and intramitochondrial ROS-responsive CO release profiles of the FeCO-TPP prodrug at different ·OH concentrations (B) are shown.
Targeted delivery and controlled release profiles in vitro
The in vitro sequential multistage targeted delivery and controlled release process in cells was further evaluated step by step through confocal laser scanning microscopic imaging. First, the tumor cell–targeted delivery profiles of the FeCO-TPP@MSN@HA nanomedicine (step i in Fig. 1B) were investigated using various cancer and normal cells. The MSN carrier and the FeCO-TPP prodrug were labeled with red rhodamine B isothiocyanate (RITC) and blue 8-mercaptoquinoline (QL), respectively, and then used to construct the FeCO-TPP@MSN@HA nanomedicine for fluorescence tracing. From Fig. 3A, HeLa and 4T1 cells can more effectively endocytose the FeCO-TPP@MSN@HA nanomedicine under the same conditions compared to the other cancer cells owing to remarkably higher overexpression of CD44 on them (fig. S3). Meanwhile, the nanomedicine was rarely endocytosed by normal MCF-10A (Michigan Cancer Foundation-10A) and human embryonic kidney (HEK) 293T cells with normal low expression of CD44. These results clearly indicated that the nanomedicine could specially target the CD44-overexpressed cancer cells for targeted drug delivery. HeLa cells with highest recognizability were therefore chosen for further investigation of the following targeted delivery and controlled release stages.
Fig. 3. Targeted delivery and controlled release profiles in vitro.
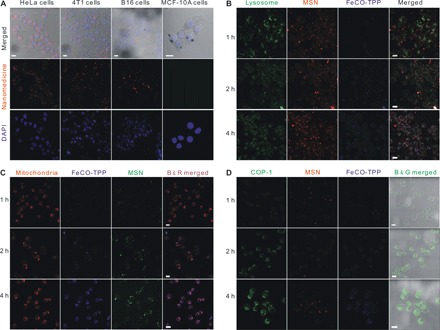
(A) CD44-dependent targeted endocytosis behaviors of the FeCO-TPP@MSN@HA nanomedicine shown by representative confocal fluorescence microscopy images of HeLa, 4T1, B16, and MCF-10A cells incubated with the FeCO-TPP@MSN-RITC@HA for 4 hours and stained for cellular nuclei with 4,6-diamino-2-phenylindole (DAPI). Therein, the MSN carrier was labeled with red RITC and then used to construct the FeCO-TPP@MSN-RITC@HA nanomedicine. (B) Intralysosome acid-responsive prodrug release behaviors of the FeCO-TPP@MSN@HA nanomedicine shown by the fluorescence images of HeLa cells treated with QL-FeCO-TPP@MSN-RITC@HA for the different time periods and stained with LysoTracker (green). Therein, the FeCO-TPP prodrug was labeled with blue QL (8-mercaptoquinoline). The increases in yellow and blue represent the lysosomal uptake of the nanomedicine and the FeCO-TPP prodrug release from lysosome, respectively. (C) Mitochondria-targeted prodrug delivery behaviors of the FeCO-TPP@MSN@HA nanomedicine shown by the fluorescence images of HeLa cells treated with QL-FeCO-TPP@MSN@HA for the different time periods and stained with MitoTracker (red). The increase in purple represents the mitochondrial uptake of the released FeCO-TPP prodrug. (D) Intramitochondrial CO release behaviors of the FeCO-TPP drug released from the FeCO-TPP@MSN@HA nanomedicine shown by the fluorescence images of HeLa cells treated with QL-FeCO-TPP@MSN-RITC@HA for the different time periods and stained with the COP-1 probe for detection of CO (green). The increase in green represents the release of CO. Scale bars, 20 μm.
During the endocytosis of the FeCO-TPP@MSN@HA nanomedicine by HeLa cells, it was found from Fig. 3B that the nanomedicine is mainly located in lysosomes (step ii in Fig. 1B) after entering into cells in a typical nanoparticle-endocytosis way, as reflected by the merging of green and red into yellow. Moreover, the blue gradually appeared, suggesting the sustained release of the FeCO-TPP prodrug from the nanomedicine. The prodrug release in lysosome should be attributed to the acid-responsive disassembly/release capability as mentioned above (Fig. 2A).
To track the intramitochondrial targeting of prodrug, we stained the mitochondria of HeLa cells with green MitoTracker dye after coincubation with the nanomedicine. The MSN carrier and the FeCO-TPP prodrug were labeled with red RITC and blue QL, respectively, and then used to construct the FeCO-TPP@MSN@HA nanomedicine for fluorescence tracing. To facilely distinguish the colors for monitoring of drug release, we set mitochondria and MSN channels in confocal images as red and green, respectively. The emerging of the red and blue yielded the purple, representing the intramitochondrial accumulation of the prodrug. From Fig. 3C, the increase in purple represented the liver (fig. S7, A to C) uptake of the released FeCO-TPP prodrug (step iii in Fig. 1B). With the release of the prodrug from the nanomedicine in lysosome, the prodrug was gradually accumulating into the mitochondria by TPP molecular recognition, exhibiting the mitochondria-targeted prodrug delivery stage. The mitochondria-targeting capability of the FeCO-TPP prodrug should be owed to its electrostatic and lipophilic attractions with mitochondrial membrane and also provided an opportunity for further mitochondrial-responsive CO release.
The fluorescent COP-1 (CO Probe-1) probe was used to detect the intracellular CO release (16). From Fig. 3D, the enhancement of the green fluorescence intensity with the increase of incubation time reflected the increasing release of CO in HeLa cells. It could be found that the intramitochondrial ROS triggered the release of CO from the FeCO-TPP prodrug when it reached the mitochondria of HeLa cells (step iv in Fig. 1B), owing to its ROS-responsive behavior (Fig. 2B). The above-mentioned four aspects of results accordantly confirmed the hypothesis of multistage sequential targeted delivery and controlled release process, as shown in Fig. 1B.
In vitro CO therapy efficacies and mechanisms
In vitro anticancer performances of the FeCO-TPP@MSN@HA nanomedicine were investigated with multiple cell lines: two cancer ones (HeLa and 4T1 cells) and two normal ones (HEK-293T and MCF-10A cells). From Fig. 4 (A to D), FeCO-TPP, FeCO-TPP@MSN, and FeCO-TPP@MSN@HA all exhibited a dosage- and time-dependent cytotoxicity behavior against investigated two types of cancer cells but insignificant toxicity to normal cells in the concentration range of 0 to 100 μg/ml despite incubation time (fig. S4), which could be attributed to cancer selectivity of CO and good biocompatibility of the MSN carrier. In comparison with FeCO-TPP and FeCO-TPP@MSN, the FeCO-TPP@MSN@HA nanomedicine exhibited largest damage to cancer cells at the same incubation concentration and time, possibly owing to targeted drug delivery. From Fig. 4 (A and B), the median effective doses (ED50) of FeCO-TPP@MSN@HA against HeLa and 4T1 cells were calculated to be 82.6 and 41.5 μg/ml, respectively (equal to 3.9 and 2.0 μg of CO per milliliter, according to the CO-loading capacity). In addition, we also used CORM-3 as CO donor (for sustained CO release) to measure the ED50 values of CO on HeLa and 4T1 cells to be 147.3 and 110.9 μM (equal to 4.1 and 3.1 μg of CO per milliliter), respectively (fig. S5). Therefore, despite sustained CO release, it was still easy for FeCO-TPP@MSN@HA to quickly achieve enough high CO concentration for killing tumor cells. Moreover, the cytotoxicity of the FeCO-TPP@MSN@HA nanomedicine remarkably increased with the increase of incubation time (Fig. 4, A and B, and fig. S4, A, B, E, and F) and was even higher than that of the free prodrug FeCO-TPP (fig. S4, E and F). Such a toxicity amplification effect should result from sustained releases of FeCO-TPP and then CO (Fig. 2). In addition, the free prodrug FeCO-TPP brought slight cytotoxicity to normal cells but could be shielded effectively by the formulation of FeCO-TPP@MSN@HA (fig. S4, G and H), which could be attributed to the inhibition of the HA-mediated targeting to the uptake of the FeCO-TPP@MSN@HA nanomedicine by normal cells and/or the protection effect of CO.
Fig. 4. In vitro selective anticancer effects and mechanisms of the FeCO-TPP@MSN@HA nanomedicine.
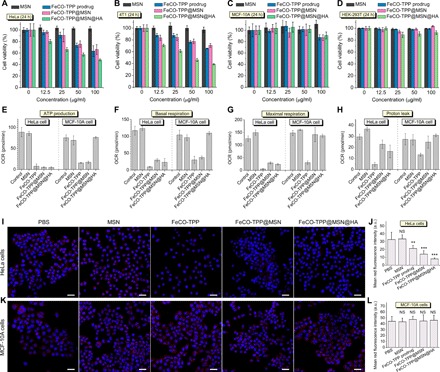
(A to D) Cytotoxicity of the FeCO-TPP@MSN@HA nanomedicine to cancer (HeLa and 4T1 cells) and normal (MCF-10A and HEK-293T cells) model cell lines. (E to H) Cell energy metabolism evaluation by ATP production (E), basal respiration (F), maximal respiration (G), and proton leak (H). (I and K) Confocal images of treated HeLa and MCF-10A cells where nuclei and mitochondria were stained into blue and red, respectively. (J) and (L) were the quantification of red fluorescence intensity in (I) and (K), respectively. Mean value and error bar were represented as means ± SD. P values were calculated by the two-tailed Student’s t test (**P < 0.01 and ***P < 0.001; NS, no significant difference). Scale bars (I and K), 20 μm. OCR, oxygen consumption rate.
Furthermore, the mechanism of cancer selectivity of the nanomedicine was investigated from the aspect of cell energy metabolism. From Fig. 4 (E to H), the MSN carrier almost had no influence on adenosine 5′-triphosphate (ATP) levels of HeLa and MCF-10A cells, while FeCO-TPP, FeCO-TPP@MSN, and FeCO-TPP@MSN@HA all remarkably inhibited the ATP level of HeLa cells in accordance with the reduction of mitochondrial amount (Fig. 4, I and J), causing the anticancer outcome. Furthermore, the intracellular level of lactate was remarkably enhanced after FeCO-TPP@MSN@HA treatment and also decreased gradually over treatment time (fig. S6), indicating that the aerobic respiration of HeLa cells was inhibited by CO during the glycolysis while anaerobic respiration and lactate consumption were activated (33), which led to the decrease in the ATP level of HeLa cells. Although FeCO-TPP and FeCO-TPP@MSN also inhibited the ATP level of MCF-10A cells (Fig. 4E), the mitochondrial amount had not decreased (Fig. 4, K and L), possibly because the positively charged FeCO-TPP and FeCO-TPP@MSN and/or CO that leaked from them were easily endocytosed to affect the mitochondrial functions to a certain extent while the neutral FeCO-TPP@MSN@HA without CO leaking did not. In particular, FeCO-TPP@MSN@HA demonstrated no impact on both the ATP level of MCF-10A cells and the mitochondrial amount (Fig. 4, E, K, and L) because of its CD44-recognizing ability, reflecting low cytotoxicity to normal cells. Moreover, the case of basal respiration was similar to that of ATP production (Fig. 4F), but the maximal respiration and proton leak of MCF-10A cells treated with FeCO-TPP@MSN and FeCO-TPP@MSN@HA were still maintained (Fig. 4G), further indicating the protection effect. Briefly, the FeCO-TPP@MSN@HA nanomedicine did damage to mitochondrial functions of cancer cells but protected the mitochondria of normal cells for maintaining normal energy metabolism.
In vivo therapeutic efficacies
Before the intravenous injection of the FeCO-TPP@MSN@HA nanomedicine for tumor therapy, its blood safety was first checked after intravenous injection of FeCO-TPP@MSN@HA (2.79 mg ml−1, 100 μl) into healthy mice (about 20 g) in comparison with PBS control (100 μl), MSN carrier (2 mg ml−1, 100 μl), FeCO-TPP prodrug (0.71 mg ml−1, 100 μl), and FeCO-TPP@MSN (2.71 mg ml−1, 100 μl), which contained equal amounts of MSN and FeCO-TPP. Blood samples at different time points (1, 7, and 20 days after injection) were collected for measurement. Compared with PBS control, MSN carrier, FeCO-TPP prodrug, and FeCO-TPP@MSN, FeCO-TPP@MSN@HA at the same dosage had not caused substantial damage to liver (fig. S7, A to C) and kidney functions (fig. S7, D and E) within 20 days (the same to treatment time) after injection. Moreover, there was no obvious difference in the levels of standard hematology markers including white blood cells, red blood cells, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, red blood cell volume distribution width, and lymphocytes percentage between PBS control and FeCO-TPP@MSN@HA (fig. S8), indicating high blood compatibility of FeCO-TPP@MSN@HA.
In vivo treatments with the FeCO-TPP@MSN@HA nanomedicine were further executed using the models of mice bearing B16 and 4T1 tumors, respectively. First, the tumor-targeted delivery behavior was investigated. From in vivo fluorescence imaging results in Fig. 5A, it can be found that free FeCO-TPP was quickly excreted with very little intratumoral accumulation, while both MSN and FeCO-TPP@MSN@HA could effectively accumulate into tumor. Furthermore, from CD31-stained (green vessel) tumor slice imaging, the FeCO-TPP@MSN@HA nanomedicine extravasated (Fig. 5B) and penetrated into tumor cells with the encapsulated FeCO-TPP prodrug together (Fig. 5C), indicating the EPR effect of investigated tumor and the effective tumor-targeted delivery of the nanomedicine (34–36). As to therapy experiments, B16 tumor–bearing mice were randomly divided into five groups (n = 5) to intravenously inject PBS as control (100 μl), MSN carrier (2 mg ml−1, 100 μl), FeCO-TPP prodrug (0.71 mg ml−1, 100 μl), FeCO-TPP@MSN (2.71 mg ml−1, 100 μl), and FeCO-TPP@MSN@HA (2.79 mg ml−1, 100 μl), respectively, at the equal molar concentration of MSN or FeCO-TPP. From therapy outcomes in Fig. 5 (D to F), the MSN carrier did not affect the tumor growth of B16 tumor–bearing mice. By comparison, FeCO-TPP inhibited tumor growth slightly after 20-day treatment despite having no tumor-targeting capability, but FeCO-TPP@MSN enhanced tumor therapy efficacy to a certain extent owing to its passive targeting behavior. Moreover, FeCO-TPP@MSN@HA can further augment tumor therapy efficacy owing to its passive and active targeting profiles (Fig. 5, D to F). The hematoxylin and eosin (H&E) staining of tumor slices further confirmed that the FeCO-TPP@MSN@HA nanomedicine can eliminate tumor cells most efficiently compared to FeCO-TPP and FeCO-TPP@MSN (Fig. 5G). In addition, all treatment groups did not bring obvious damage to main organs including heart, liver, spleen, lung, and kidney (fig. S9), suggesting good biocompatibility at the treated dose. No visible weight loss of mice treated with the FeCO-TPP@MSN@HA nanomedicine reflected good health status.
Fig. 5. In vivo tumor-targeted therapy efficacy of the FeCO-TPP@MSN@HA nanomedicine.
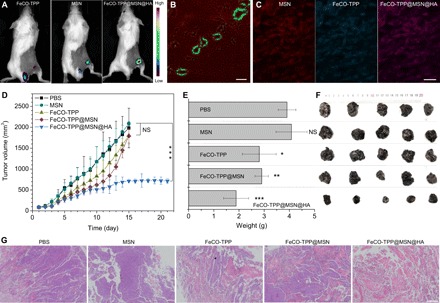
The tumor-targeted delivery behaviors testified by fluorescence images of the tumor-bearing mice intravenously injected with the FeCO-TPP prodrug, the MSN carrier, and the FeCO-TPP@MSN@HA nanomedicine, respectively (A), and the CD31-stained slice of FeCO-TPP@MSN@HA–treated tumor (B and C) where green, red, and blue represent vessel, MSN, and FeCO-TPP, respectively. The outcomes of B16 tumor therapy with the nanomedicine: The tumor volume change with time (D), the tumor weight comparison after 20 days of treatment (E), the mouse weight change with time (F), and the histological analysis of treated tumors by the H&E staining (G) are shown. Mean value and error bar were represented as means ± SD. P values were calculated by one-way analysis of variance (ANOVA) with Tukey post hoc testing to correct for multiple comparisons (*P < 0.05, **P < 0.005, and ***P < 0.0005). Scale bars, 200 (B), 20 (C), and 500 μm (G).
Furthermore, the tumor lung metastasis model was built with 4T1 breast cancer cells and labeled with fluorescence to evaluate the antimetastasis efficacy of the FeCO-TPP@MSN@HA nanomedicine. From Fig. 6A, the treatment outcomes of primary 4T1 tumors were similar to the case of B16 tumors as mentioned above. Neither MSN nor FeCO-TPP almost affected the growth of primary 4T1 tumors; FeCO-TPP@MSN inhibited their growth to a certain extent, and the primary tumor inhibition effect of FeCO-TPP@MSN@HA was most obvious and sustained (more than 46 days), indicating the contributions of passive and active targeting functions. Correspondingly, the survival rate of 4T1 tumor–bearing mice was remarkably enhanced by FeCO-TPP@MSN and FeCO-TPP@MSN@HA treatments (Fig. 6B). In particular, the FeCO-TPP@MSN@HA treatment increased the life time of 4T1 tumor–bearing mice by about 60% compared with the control group. Moreover, in vivo real-time fluorescence tracking results indicated that with the increasing growth of primary tumors, mice without any treatment suffered the metastasis in lung after about 25 days and quickly died after 29 days (Fig. 6C). Neither MSN nor FeCO-TPP significantly affected the evolution of primary tumor growth and lung metastasis. By comparison, the FeCO-TPP@MSN treatment distinctly reduced the probability of lung metastasis on day 25, and after the FeCO-TPP@MSN@HA treatment, it was until day 41 that lung metastasis occurred. The antimetastasis effect of the FeCO-TPP@MSN@HA nanomedicine might be one of main reasons of increasing the lifetime of mice. Furthermore, the histological analysis of lung and primary tumors of dead mice was executed with H&E staining. It was found that the number and size of lung metastasis tumors were remarkably reduced by the FeCO-TPP@MSN and FeCO-TPP@MSN@HA treatments (Fig. 6, D and E), further confirming the antimetastasis effect. In addition, the tumor cells were killed by the FeCO-TPP@MSN@HA nanomedicine most efficiently (Fig. 6F), further suggesting the antitumor effect in vivo. In addition, all treatment groups did not bring visible damage to heart, liver, spleen, and kidney (fig. S10), and there was no obvious weight loss of mice treated with the FeCO-TPP@MSN@HA nanomedicine.
Fig. 6. Inhibition of the growth and lung metastasis of primary breast tumor by the FeCO-TPP@MSN@HA nanomedicine.
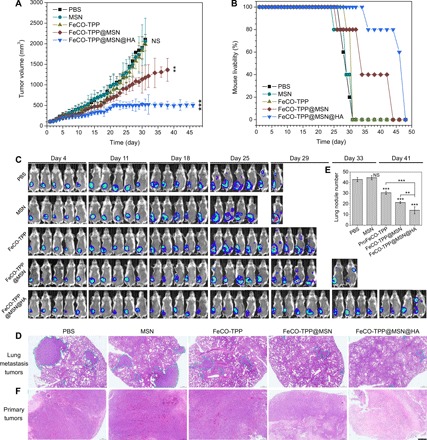
The volume change of primary 4T1 tumor with time (A), the survival rate of 4T1 tumor–bearing mice with and without drug treatment (B), the fluorescence tracking of 4T1 tumor growth and metastasis (C), the histological analysis of lung metastasis tumors (D) and primary tumors (F) by the H&E staining (scale bar. 500 μm), and the statics of the lung metastasis tumor number from the histological analysis (E) are shown. Lung metastasis tumors in (D) were indicated by blue dashed lines. Mean value and error bar were represented as means ± SD. P values were calculated by one-way ANOVA with Tukey post hoc testing to correct for multiple comparisons (**P < 0.005 and ***P < 0.0005).
CONCLUSION
In summary, an intelligent tumor tissue–cell–mitochondria–targeted delivery platform had been developed by the incorporation of FeCO-TPP prodrug into monodispersive MSN for augmented cancer therapy. The FeCO-TPP prodrug was prepared by coordinating FeCO with TPP-SH by the ligand-exchange reaction, which maintained the ROS-triggered CO release activity and could also effectively target the mitochondria for CO therapy. The encapsulation of the FeCO-TPP prodrug into MSN significantly enhanced its biocompatibility and their passive targeting to tumor tissue by the EPR effect. Moreover, the constructed FeCO-TPP@MSN@HA nanomedicine was designed to achieve the tumor cell targeting by the CD44 recognition of HA. These tumor tissue–cell–mitochondria–targeted delivery and subsequent triggered release of prodrug/CO had been verified by fluorescence colocalization in vitro. The cell energy metabolism studies revealed that the nanomedicine enhanced damage to mitochondrial functions of cancer cells but protect the mitochondrial functions of normal cells for maintaining normal energy metabolism. In vivo studies indicated that the FeCO-TPP@MSN@HA nanomedicine had augmented CO therapy, demonstrating effective inhibition to tumor growth and metastasis. Overall, these results suggest that the proposed multistage assembly/disassembly strategy provided a new route to efficient CO delivery for cancer treatment.
METHODS
Chemicals
TPP-COOH, cysteamine (H2N-EtSH), FeCO, triethylamine (TEA), dichloromethane (DCM), tetrahydrofuran (THF), 2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), CORM-3, tetraethyl orthosilicate (TEOS), cetyltrimethylammonium chloride [CTAC; 25 wt % in H2O], triethanolamine (TEAH), HA, d-luciferin, RITC, and Alcian Blue 8GX were purchased from Sigma-Aldrich. 8-Mercaptoquinoline hydrochloride was purchased from Meryer. The human and mouse CD44 enzyme-linked immunosorbent assay (ELISA) kits were purchased from Shanghai Yueyan Biological Technology. The Agilent Seahorse XFe24 FluxPak Assay Kit was purchased from Agilent Technologies Inc. The Cell-Counting Kit-8 (CCK-8) was purchased from Beyotime Biotechnology Co. Ltd. All other used agents were of the highest commercial grade available.
Synthesis and characterization of the prodrug FeCO-TPP
First, cysteamine (H2N-EtSH) was oxidized into H2N-EtS-SEt-NH2 by H2O2 in a solution of methanol (MeOH), and then, H2N-EtS-SEt-NH2 (1 mmol) was added into the 100-ml DCM solution of TPP-COOH (2.2 mmol) and TEA (6 mmol) together with HATU (2.2 mmol) under stirring. After 24 hours, the resultant (TPP-S-S-TPP) was washed with HCl (5%, 50 ml × 3), H2O (50 ml × 3), NaHCO3 (50 ml × 3), and H2O (50 ml × 3), in turn, and then dried with Na2SO4 and purified with column (SiO2, DCM:MeOH=20:1). Then, TPP-S-S-TPP was reduced into TPP-SH by NaBH4. FeCO (0.3 mmol), and TPP-SH (1 mmol) were dissolved together in 50 ml of THF, and the mixture was stirred under reflux. It could be observed that the green solution gradually turned into deep red, reflecting the occurrence of coordination reaction. After 1 hour, the mixture was cooled down and filtered. The solvent was evaporated, and then, the obtained red solid FeCO-TPP was purified with column. It was noted that the reaction and preservation were light-sealed. The chemical structure of FeCO-TPP was analyzed by matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF)/mass spectrometry on positive ion reflection mode, NMR (VNMRS 400 MHz WB), and FTIR [Thermo Nicolet Nexus 670 attenuated total reflectance (ATR)–IR spectrometer].
Synthesis and characterization of FeCO-TPP@MSN@HA
First, MSN was synthesized by a surfactant-directed self-assembly method. In a typical procedure, 2 g of CTAC (25 wt % in water) and 0.02 g of TEAH were dissolved into 20 ml of water under stirring, and the mixed solution was placed in 80°C water bath to stabilize for more than 30 min. TEOS (1.5 ml) was subsequently injected at a rate of 3 μl/min, and the mixture was stirred for another 1 hour. The precipitate was collected by centrifugation and then washed with ethanol (EtOH)/HCl solution to remove the surfactant.
The loading of the prodrug FeCO-TPP into the carrier MSN was executed using a nanocasting method. FeCO-TPP (2 mg) was completely dissolved into the EtOH solution of MSN (1 mg) to get a clear pale yellow solution (note that there is no intensive ultrasound). The mixed solution was light-sealed and degassed under vacuum until the volume decreased to ~200 μl. Nanoparticles were collected by centrifugation and washed with EtOH, and the supernatant/washing solutions were collected (metered to 500 μl) for the measurement of drug loading capacity. Several different concentrations of EtOH solution of FeCO-TPP were first prepared and detected by UV spectrophotometer to make a standard curve. The absorbances of the above-mentioned supernatant clear solutions of FeCO-TPP before and after loading with MSN at 329 nm were measured and used to calculate the FeCO-TPP loading capacity with the standard curve according to the Beer-Lambert law. Furthermore, collected FeCO-TPP was redispersed into water under shaking and gradually added into the PBS solution of HA. The mixture was stabilized for 2 hours under silent and cold surrounding, and then, FeCO-TPP@MSN@HA was collected by centrifugation and washed with PBS twice.
The morphology and size of MSN and FeCO-TPP@MSN@HA were measured by TEM (JEM-2100F). Hydrodynamic size and zeta potential were measured on a Malvern Zetasizer Nano ZS90. The composition of FeCO-TPP@MSN@HA was tested by ATR-FTIR spectroscopy on a Thermo Nicolet Nexus 670 ATR-IR spectrometer. The UV absorption spectra (200 to 800 nm) were recorded at room temperature on a GENESYS 10S UV-Vis spectrophotometer (Thermo Fisher Scientific). As to the stability of structure, the as-prepared FeCO-TPP@MSN@HA nanomedicine was dispersed in PBS (pH 7.4), and some solutions were taken at different time points (24 and 48 hours) to check the size distribution by DLS.
The quantitative spectrophotometric determination of the content of HA was done by the formation of soluble complexes between Alcian Blue dye (ABD) and HA. ABD (1.0 mg/ml) was dissolved in the aqueous solution of sodium acetate and then filtered using a 0.45-μm syringe filter. Fresh dye solution (2.4 ml), 1.2 ml of distilled water, and 0.4 ml of different concentrations of standard HA solutions (100 to 1000 ng/ml) were thoroughly mixed. The absorbance was measured after stabilizing for 10 min at 25°C. The linear standard curve of HA concentration versus the difference of absorbance between ABD and HA-ABD complex at 480 nm was fitted. HA was stripped off by decomposing MSN with sodium hydroxide solution and removing FeCO-TPP by dialysis [molecular weight cutoff (MWCO), 3500 Da] and then was mixed with ABD to form the HA-ABD complex for detection of the HA content in FeCO-TPP@MSN@HA. According to the standard curve, the HA content in FeCO-TPP@MSN@HA was calculated to be 3.9 wt %.
Measurement of acid-responsive prodrug release behaviors of FeCO-TPP@MSN
To facilitate the real-time monitoring of drug release from nanomedicine, we designed a specific cuvette, which consisted of two half general cuvettes connected with a piece of dialysis membrane (MWCO, 3000 Da) and appropriate AB glue. Two sides of the cuvette were filled with fresh PBS (bottom) and with the PBS solution of FeCO-TPP@MSN (top), respectively. Immediately, the cuvette was put on a UV spectrophotometer (keep the laser to only contact fresh PBS at the bottom) to collect UV spectra for monitoring prodrug release in real time. Meanwhile, a mini stirrer was preplaced in the cuvette for continuous stirring during prodrug release measurement to ensure the uniformity of solution. The absorbance at 273 nm was used to calculate prodrug release amount according to the Beer-Lambert law and the standard curve of FeCO-TPP solution.
Measurement of ROS-responsive CO release profiles of the FeCO-TPP prodrug
CO concentration in PBS was measured by the hemoglobin method according to the Beer-Lambert law. The released CO in PBS was detected spectrophotometrically by measuring the conversion of hemoglobin to carboxyhemoglobin (HbCO). First, hemoglobin from bovine erythrocytes (MP Biomedicals; final concentration, 5 μM) was dissolved completely in the PBS (pH 6.8) and then was reduced by adding sodium dithionite (0.52 mg) under a nitrogen atmosphere. Then, the FeCO-TPP prodrug and the fresh mixture of ferrous sulfate (FeSO4) and hydrogen peroxide (H2O2) were added into the above hemoglobin solution, in turn, to generate ·OH by the Fenton reaction and trigger CO release from the prodrug. Different amounts of FeSO4 and H2O2 were used to produce different concentrations of hydroxyl radicals. The UV adsorption spectra of the mixed solution (350 to 600 nm) were collected in real time on a GENESYS 10S UV-Vis spectrophotometer (Thermo Fisher Scientific). The Beer-Lambert law was used to calculate CO concentration with the following function
where CCO and CHb express the released CO concentration and the initial hemoglobin concentration, respectively, while I410nm and I430nm express the intensities of collected spectrum at 410 and 430 nm, respectively.
Synthesis of QL-FeCO-TPP
The FeCO-TPP prodrug was labeled in situ with fluorescent QL (8-mercaptoquinoline hydrochloride) by the ligand-exchange reaction. FeCO (32.82 mg), 8-mercaptoquinoline hydrochloride (12.92 mg), and TPP-SH (133 mg) in dimethyl sulfoxide (4 ml) were heated at 70°C for 2 hours under the N2 atmosphere. The green solution gradually turned deep red. Last, the red resultant was purified by the above-mentioned method.
Synthesis of MSN-RITC
RITC–3-aminopropyltriethoxysilane (APTES) was first prepared by an addition reaction. RITC (38.29 mg) was completely dissolved into 10 ml of MeOH; then, APTES (50 μl) was added dropwise, and the mixed solution was stirred for 2 days under the light-sealed condition to synthesize RITC-APTES. CTAC (0.5 g, 25 wt % in water) and TEAH (0.005 g) were dissolved into 6 ml of water in 80°C water bath. Then, the mixture of TEOS (0.5 ml) and RITC-APTES (250 μl) was injected at a rate of 3 μl/min and stirred for 1 hour. The removal of residual reactants and CTAC followed the above-mentioned method similar to the case of MSN.
Assessment of targeted endocytosis behaviors
HeLa, 4T1, B16, HEK-293T, and MCF-10A cells were cultured in a 37°C and 5% CO2 incubator. All cell culture–related reagents were purchased from Beyotime Biotechnology Co. Ltd. Confocal fluorescence imaging of cells was performed using a Leica TCS SP5 II laser scanning confocal microscope. For confocal imaging, cells were seeded on coverslips in a 12-well plate and incubated together with fluorescently labeled FeCO-TPP@MSN@HA (100 μg/ml) for 4 hours. After removing free nanoparticles, the cells were fixed with 4% formaldehyde, stained with 4,6-diamino-2-phenylindole (DAPI), and then imaged by a confocal fluorescence microscope.
Assessment of lysosomal uptake and mitochondrial uptake
The MSN carrier and the FeCO-TPP prodrug were labeled with red RITC and blue QL, respectively. HeLa cells were incubated with FeCO-TPP@MSN@HA (100 μg/ml) for 1, 2, 3, and 4 hours. Free nanoparticles were removed, and then, intracellular lysosome, mitochondria, and CO were detected with LysoTracker, MitoTracker, and COP-1, respectively. The cells were fixed with 4% formaldehyde. Then, the cell creepers were back-buckled on the slide. Last, a Leica TCS SP5 II confocal fluorescence microscope was used to image those cell creepers.
Assessment of in vitro cytotoxicity test
HeLa, 4T1, B16, HEK-293T, and MCF-10A cells at a density of 1 × 104 cells per well were seeded into 96-well cell culture plates in quintuplicate. After incubation for 24 hours, the culture medium was replaced with a fresh one containing various concentrations of drug (MSN, free FeCO-TPP prodrug, FeCO-TPP@MSN, and FeCO-TPP@MSN@HA) and incubated further for 24, 48, or 72 hours at 37°C in a humidified 5% CO2 atmosphere. Then, a standard CCK-8 essay was carried out to determine cell viability. The absorbance at 450 nm was measured using a microplate reader (Synergy H1, BioTek). In addition, the cytotoxicity of CO was also measured using CORM-3 as a CO donor.
Assessment of mitochondrial membrane impairment
HeLa and MCF-10A cells were seeded in a 24-well plate (1 × 104) 24 hours before treated with FeCO-TPP@MSN@HA (100 μg ml−1). After incubation for 24 hours, free nanoparticles were removed by washing, and MitoTracker Red and DAPI were added to stain mitochondria and nuclei. Cells were fixed with 4% formaldehyde and observed on the confocal fluorescence microscope.
Assessment of cellular energy metabolism
HeLa and MCF-10A cells were planted in a specific 24-well plate and incubated for 4 hours. After 6 hours of incubation with drug or nanomedicine, free drug/nanoparticles were removed by washing with the XF Base Medium (pH 7.4). With further incubation for 1 hour in a CO2-free incubator, the plate was detected using the Extracellular Flux Analyzer (XFe24). In addition, the level of lactic acid (LA) in cancer cells treated with FeCO-TPP@MSN@HA was also detected by LA assay kit (Solarbio, BC2235).
Assessment of the determination of CD44 levels
CD44 levels of the HEK-293T, MCF-10A, 4T1, HeLa, and B16 cells were investigated. Cells (1 million per ml) were lysed, and then, the supernatant was collected by centrifugation (20 min, 2000 revolutions per second) and subsequently analyzed using CD44 ELISA Kit.
In vivo tumor-targeted delivery study
BALB/c mice (~17 g) were purchased from Guangdong Medical Laboratory Animal Center and used under protocols approved by the Animal Ethical and Welfare Committee of Shenzhen University. The 4T1 tumor model was built by subcutaneous injection of 1 × 107 4T1 cells into the hindlimb. After mean volume of tumors reached about 60 mm3, three groups of 4T1 tumor–bearing mice were intravenously injected with QL–labeled FeCO-TPP (7.5 mg/kg), IR780-labeled MSN (7.5 mg/kg), and FeCO-TPP-QL@MSN-IR780@HA (15 mg/kg), respectively. After 4 hours, in vivo fluorescence imaging was conducted on an IVIS Spectrum (PerkinElmer). Then, tumors were extracted, sliced, and stained with green anti-CD31 antibody to check the tumor targeting of nanomedicine.
In vivo tumor therapy
For the metastatic 4T1 orthotopic murine breast cancer model therapy, 5 × 106 fLuc-4T1 cells were injected into the breast of each BALB/c mouse. 4T1 orthotopic tumor–bearing mice were randomized into five groups, with n = 5 tumors per group, when tumor volume reached approximately 100 mm3. Five groups were intravenously injected three times with PBS (100 μl), MSN (2 mg ml−1, 100 μl), free FeCO-TPP (0.71 mg ml−1, 100 μl), FeCO-TPP@MSN, and FeCO-TPP@MSN@HA (2.79 mg ml−1, 100 μl), respectively, once a week. At fixed time points, the metastasis process was monitored by bioluminescence imaging on an IVIS spectrum system after mice were intraperitoneally injected with d-luciferin. Tumor size was measured with a digital caliper, and tumor volumes were calculated according to the following formula: volume = width2 × length/2. At the end of treatment (mice naturally died or were humanely sacrificed when primary tumor volume of mice reached about 2000 mm3, which were statistically regarded as imminent deaths), primary and metastatic tumors and main organs (heart, liver, spleen, lung, and kidney) were extracted for the histological analysis. All the sample tissues were dissected to make paraffin section for H&E staining and histological observation under an optical microscope (ZEISS BX51). The diameter of lung metastasis nodules was measured under the microscope and divided into four grades (grade I < 0.5 mm, 0.5 mm ≤ grade II < 1 mm, 1 mm ≤ grade III < 2 mm, and grade IV ≥ 2 mm), and then, the number of metastases was normalized according to the following formula: I × 1 + II × 2 + III × 3 + IV × 4, where I, II, III, and IV represent the numbers of four grade metastases, respectively. As to B16 orthotopic murine skin cancer model therapy, the B16 tumor model was built by subcutaneous injection of 1 × 107 B16 cells into the hind limb. After mean volume of tumors reached about 100 mm3, treatment experiments similar to 4T1 tumor treatment were executed.
In vivo toxicity assay
PBS, MSN, FeCO-TPP, FeCO-TPP@MSN, and FeCO-TPP@MSN@HA groups were intravenously injected into healthy BALB/c mice (n = 5) at a high dosage (equal to 0.71 mg of FeCO-TPP per milliliter or 2 mg of MSN per milliliter, once a week for three times). The standard biochemistry test was executed to evaluate the liver/kidney functions related indicators including alanine aminotransferase, alkaline phosphatase, aspartate aminotransferase, nephric blood urea nitrogen, and creatinine, along with the blood panel parameters including white blood cells, red blood cells, red blood cell distribution width-SD, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, and lymphocyte.
Supplementary Material
Acknowledgments
We greatly appreciate the help of Z. Mao and W. Zhang (SZU) in cell energy metabolism, H. Yao (SIC, CAS) in TEM measurement, W. Gong (SZU) in organic synthesis, and the Instrumental Analysis Center of Shenzhen University (Xili campus) for assistance in material characterizations. Funding: This work was supported by the National Natural Science Foundation of China (51872188), Shenzhen Basic Research Program (JCYJ20170302151858466 and JCYJ20170818093808351), and Special Funds for the Development of Strategic Emerging Industries in Shenzhen (20180309154519685). Author contributions: Q.H. designed the project. Z.J. and J.M. performed the chemical characterization and biological experiments. P.Z. designed and built the cytotoxicity experiment. B.Z., Z.J., and M.F. designed and built the animal experiment for tumor therapy. Q.H. and J.M. analyzed and interpreted the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba1362/DC1
REFERENCES AND NOTES
- 1.Mura S., Nicolas J., Couvreur P., Stimuli-responsive nanocarriers for drug delivery. Nat. Mat. 12, 991–1003 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Zheng Y., Ji X., Yu B., Ji K., Gallo D., Csizmadia E., Zhu M., Choudhury M. R., De La Cruz L. K. C., Chittavong V., Pan Z., Yuan Z., Otterbein L. E., Wang B., Enrichment-triggered prodrug activation demonstrated through mitochondria-targeted delivery of doxorubicin and carbon monoxide. Nat. Chem. 10, 787–794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H., Zhang W., Zhu G., Xie J., Chen X., Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2, 17024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wegiel B., Gallo D., Csizmadia E., Harris C., Belcher J., Vercellotti G. M., Penacho N., Seth P., Sukhatme V., Ahmed A., Pandolfi P. P., Helczynski L., Bjartell A., Persson J. L., Otterbein L. E., Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. 73, 7009–7021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motterlini R., Otterbein L. E., The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9, 728–743 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Szabo C., Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 15, 185–203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumura D., Kashiwagi S., Jain R. K., The role of nitric oxide in tumour progression. Nat. Rev. Cancer 6, 521–534 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Otterbein L. E., Otterbein S. L., Ifedigbo E., Liu F., Morse D. E., Fearns C., Ulevitch R. J., Knickelbein R., Flavell R. A., Choi A. M., MKK3 mitogen-activated protein kinase pathway mediates carbon monoxide-induced protection against oxidant-induced lung injury. Am. J. Pathol. 163, 2555–2563 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He Q., Precision gas therapy using intelligent nanomedicine. Biomater. Sci. 5, 2226–2230 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zheng D.-W., Li B., Li C.-X., Xu L., Fan J.-X., Lei Q., Zhang X.-Z., Photocatalyzing CO2 to CO for enhanced cancer therapy. Adv. Mater. 29, 1703822–1703830 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ling K., Men F., Wang W.-C., Zhou Y.-Q., Zhang H.-W., Ye D.-W., Carbon monoxide and its controlled release: Therapeutic application, detection, and development of carbon monoxide releasing molecules (CORMs). J. Med. Chem. 61, 2611–2635 (2017). [DOI] [PubMed] [Google Scholar]
- 12.García-Gallego S., Bernardes G. J. L., Carbon-monoxide-releasing molecules for the delivery of therapeutic CO in vivo. Angew. Chem. Int. Ed. 53, 9712–9721 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Rimmer R. D., Pierri A. E., Ford P. C., Photochemically activated carbon monoxide release for biological targets. Toward developing air-stable photoCORMs labilized by visible light. Coordin. Chem. Rev. 256, 1509–1519 (2012). [Google Scholar]
- 14.Hou J., Pan Y., Zhu D., Fan Y., Feng G., Wei Y., Wang H., Qin K., Zhao T., Yang Q., Zhu Y., Che Y., Liu Y., Cheng J., Kong D., Wang P. G., Shen J., Zhao Q., Targeted delivery of nitric oxide via a ‘bump-and-hole’-based enzyme–prodrug pair. Nat. Chem. Biol. 15, 151–160 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Zheng D.-W., Chen Y., Li Z.-H., Xu L., Li C.-X., Li B., Fan J.-X., Cheng S.-X., Zhang X.-Z., Optically-controlled bacterial metabolite for cancer therapy. Nat. Commun. 9, 1680 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Q., Kiesewetter D. O., Qu Y., Fu X., Fan J., Huang P., Liu Y., Zhu G., Liu Y., Qian Z., Chen X., NIR-responsive on-demand release of CO from metal carbonyl-caged graphene oxide nanomedicine. Adv. Mater. 27, 6741–6746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin Z., Wen Y., Xiong L., Yang T., Zhao P., Tan L., Wang T., Qian Z., Su B.-L., He Q., Intratumoral H2O2-triggered release of CO from a metal carbonyl-based nanomedicine for efficient CO therapy. Chem. Commun. 53, 5557–5560 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Yu L., Hu P., Chen Y., Gas-generating nanoplatforms: Material chemistry, multifunctionality, and gas therapy. Adv. Mater. 30, e1801964 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Fujita K., Tanaka Y., Sho T., Ozeki S., Abe S., Hikage T., Kuchimaru T., Kizaka-Kondoh S., Ueno T., Intracellular CO release from composite of ferritin and ruthenium carbonyl complexes. J. Am. Chem. Soc. 136, 16902–16908 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Chaves-Ferreira M., Albuquerque I. S., Matak-Vinkovic D., Coelho A. C., Carvalho S. M., Saraiva L. M., Romão C. C., Bernardes G. J. L., Spontaneous CO release from RuII(CO)2-protein complexes in aqueous solution, cells, and mice. Angew. Chem. Int. Ed. Engl. 54, 1172–1175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Queiroga C. S. F., Almeida A. S., Vieira H. L. A., Carbon monoxide targeting mitochondria. Biochem. Res. Int. 2012, 749845 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schallner N., Otterbein L. E., Friend or foe? Carbon monoxide and the mitochondria. Front. Physiol. 6, 17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S., Lei Q., Qiu W.-X., Liu L.-H., Zheng D.-W., Fan J.-X., Rong L., Sun Y.-X., Zhang X.-Z., Mitochondria-targeting "nanoheater" for enhanced photothermal/chemo-therapy. Biomaterials 117, 92–104 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Chen W.-H., Luo G.-F., Qiu W.-X., Lei Q., Liu L.-H., Zheng D.-W., Hong S., Cheng S.-X., Zhang X.-Z., Tumor-triggered drug release with tumor-targeted accumulation and elevated drug retention to overcome multidrug resistance. Chem. Mater. 28, 6742–6752 (2016). [Google Scholar]
- 25.Yuan F., Li J.-L., Cheng H., Zeng X., Zhang X.-Z., A redox-responsive mesoporous silica based nanoplatform for in vitro tumor-specific fluorescence imaging and enhanced photodynamic therapy. Biomater. Sci. 6, 96–100 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Chen W.-H., Luo G.-F., Qiu W.-X., Lei Q., Liu L.-H., Wang S.-B., Zhang X.-Z., Mesoporous silica-based versatile theranostic nanoplatform constructed by layer-by-layer assembly for excellent photodynamic/chemo therapy. Biomaterials 117, 54–65 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Liu L.-H., Qiu W.-X., Li B., Zhang C., Sun L.-F., Wan S.-S., Rong L., Zhang X.-Z., A red Light activatable multifunctional prodrug for image-guided photodynamic therapy and cascaded chemotherapy. Adv. Funct. Mater. 26, 6257–6269 (2016). [Google Scholar]
- 28.Luo G.-F., Chen W.-H., Liu Y., Lei Q., Zhuo R.-X., Zhang X.-Z., Multifunctional enveloped mesoporous silica nanoparticles for subcellular co-delivery of drug and therapeutic peptide. Sci. Rep. 4, 6064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W.-H., Xu X.-D., Luo G.-F., Jia H.-Z., Lei Q., Cheng S.-X., Zhuo R.-X., Zhang X.-Z., Dual-targeting pro-apoptotic peptide for programmed cancer cell death via specific mitochondria damage. Sci. Rep. 3, 3468 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X., Long L., Wang H., Chen L., Liu X., Diiron hexacarbonyl complexes as potential CO-RMs: CO-releasing initiated by a substitution reaction with cysteamine and structural correlation to the bridging linkage. Dalton Trans. 43, 9968–9975 (2014). [DOI] [PubMed] [Google Scholar]
- 31.He Q., Gao Y., Zhang L., Zhang Z., Gao F., Ji X., Li Y., Shi J., A pH-responsive mesoporous silica nanoparticles-based multi-drug delivery system for overcoming multi-drug resistance. Biomaterials 32, 7711–7720 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Chen W.-H., Luo G.-F., Qiu W.-X., Lei Q., Hong S., Wang S.-B., Zheng D.-W., Zhu C.-H., Zeng X., Feng J., Cheng S.-X., Zhang X.-Z., Programmed nanococktail for intracellular cascade reaction regulating self-synergistic tumor targeting therapy. Small 12, 733–744 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Hui S., Ghergurovich J. M., Morscher R. J., Jang C., Teng X., Lu W., Esparza L. A., Reya T., Zhan L., Guo J. Y., White E., Rabinowitz J. D., Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng H., Xue M., Xia T., Ji Z., Tarn D. Y., Zink J. I., Nel A. E., Use of size and a copolymer design feature to improve the biodistribution and the enhanced permeability and retention effect of doxorubicin-loaded mesoporous silica nanoparticles in a murine xenograft tumor model. ACS Nano 5, 4131–4144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maeda H., Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliver. Rev. 91, 3–6 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Wang L., Huang J., Chen H., Wu H., Xu Y., Li Y., Yi H., Wang Y. A., Yang L., Mao H., Exerting enhanced permeability and retention effect driven delivery by ultrafine iron oxide nanoparticles with T1–T2 switchable magnetic resonance imaging contrast. ACS Nano 11, 4582–4592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/20/eaba1362/DC1