

SUMMARY
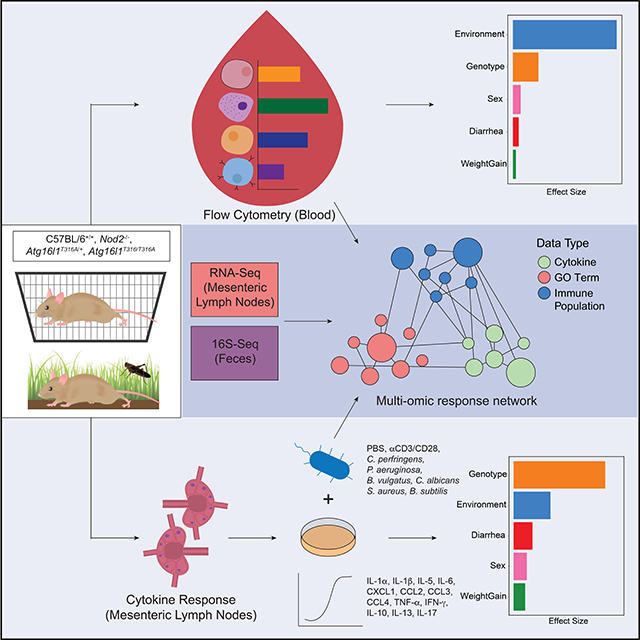
The relative contributions of genetic and environmental factors to variation in immune responses are poorly understood. Here, we performed a phenotypic analysis of immunological parameters in laboratory mice carrying susceptibility genes implicated in inflammatory bowel disease (Nod2 and Atg16l1) upon exposure to environmental microbes. Mice were released into an outdoor enclosure (rewilded) and then profiled for immune responses in the blood and lymph nodes. Variations of immune cell populations were largely driven by the environment, whereas cytokine production elicited by microbial antigens was more affected by the genetic mutations. We identified transcriptional signatures in the lymph nodes associated with differences in T cell populations. Subnetworks associated with responses against Clostridium perfringens, Candida albicans and Bacteroides vulgatus were also coupled with rewilding. Hence, exposing laboratory mice with genetic mutations to a natural environment uncovers different contributions to variations in microbial responses and immune cell composition.
eTOC blurb
The impact of genetics verses environment on immunity is incompletely understood. By releasing laboratory mice carrying IBD susceptibility genes into the outdoors, Lin, et al. find that exposure to environmental microbes promotes variation in immune cell populations, whereas cytokine responses to microbial stimulation are affected more by genetic IBD susceptibility.
Graphical Abstract
INTRODUCTION
Variation in the magnitude of the immune response is an important determinant of susceptibility to pathogen infections, as well as a predisposition to autoimmunity and cancer (Brodin and Davis, 2017; Schirmer et al., 2018). However, immunological studies with laboratory mice typically aim to control variation in order to focus on genetic factors that alter components of both the adaptive and innate immune system. Recent advances in technology and throughput have facilitated a systems immunology approach towards deciphering the relative genetic and environmental contributors to variation of the human immune system (Bakker et al., 2018; Brodin et al., 2015; Li et al., 2016; Schirmer et al., 2016; Ter Horst et al., 2016). By altering the environment in which laboratory mice are conventionally housed, combined with analyses of immune responses and gut microbial compositions acquired from the environment, we describe here a resource dataset that we used to probe the role of genetic mutations in two susceptibility genes (Nod2 and Atg16l1), implicated in the development of inflammatory bowel diseases, versus environmental drivers of heterogeneity in the immune system. Such controlled experiments on specific groups with the same genetic mutations exposed to a new environment would be difficult to conduct in human studies.
Recently, there has been a growing effort to characterize wild mice and pet shop mice, which appear to share greater similarity to the immunological state and microbial challenges of humans than laboratory mice (Abolins et al., 2017; Beura et al., 2016; Rosshart et al., 2019; Rosshart et al., 2017). We have developed an outdoor enclosure facility that enables us to reintroduce laboratory mice into a more natural environment, a process termed “rewilding” (Leung et al., 2018). The enclosures include soil and vegetation as well as barnlike structures but no other mammals (e.g., no wild conspecifics from whom lab-bred mice might acquire infections). Over the course of several weeks, mice dig burrows and otherwise adapt to their new environment (Supplemental Video). Previously, we have observed C57BL/6 mice in this environment exhibit a decreased type 2 and increased type 1 response to intestinal nematode (Trichuris muris) infection and greater susceptibility to heavier worm burdens (Leung et al., 2018). This facility provides us with a unique opportunity to rapidly alter the housing environment of laboratory mice that carry genetic mutations and be able to recover them for subsequent detailed immunological and gut microbial profiling.
Gene-bacteria associations have been identified in both human and wild mice indicating host genetic determinants of the gut microbial composition across mammals in natural environments (Suzuki et al., 2019). To interrogate the impact of host genetic variants on microbiota and inflammatory bowel disease (IBD) susceptibility in response to environmental changes, we investigated Atg16l1 and Nod2 variants which are among the highest risk factors for IBD associated with a dysregulated immune response to gut microbes (Wlodarska et al., 2015; Wong and Cadwell, 2018). Atg16l1 is a key component of the autophagy pathway, and Nod2 is involved in bacterial sensing. We and others previously demonstrated that mice harboring mutations in Atg16l1 or Nod2 develop signs of inflammation in a manner dependent on exposure to infectious entities (Biswas et al., 2010; Cadwell et al., 2010; Lavoie et al., 2019; Matsuzawa-Ishimoto et al., 2017; Pott et al., 2018; Ramanan et al., 2016; Ramanan et al., 2014). Mice with these particular mutations are sensitive to the presence of murine norovirus (MNV), Helicobacter species, and other commensal-like agents that are found in some animal facilities and excluded in others (Biswas et al., 2010; Cadwell et al., 2010; Caruso et al., 2019; Kernbauer et al., 2014; Pott et al., 2018; Ramanan et al., 2014). Since mice mutant in Atg16l1 or Nod2 can exhibit dysregulated immune responses to colonization by organisms that are otherwise commensals in wild type animals, we reasoned that they could be an interesting model system to investigate gene-environment interactions in the outdoor enclosure.
We hypothesized that by introducing laboratory mice carrying IBD susceptibility mutations into the outdoor enclosure, the drastic change in environment may trigger alterations in the immune response and the microbiota, which would have greater adverse effects on mutant mice than wildtype mice. Hence, to study the immunological consequences in response to environmental challenges after rewilding, we released Atg16l1T3I6A/T3I6A, Atg16l1T316A/+, Nod2−/− mice in addition to C57BL/6J wild type (WT) mice to determine whether mutations in these genes alter the immune response to microbial exposure in a natural environment. Here we present a detailed systems immunology profile of both rewilded and laboratory mice to investigate variation in microbial responses and the relationship with environment and genetic mutations.
RESULTS
Study design for immune profiling of rewilded and laboratory mice
From 116 mice released into the outdoor enclosure for 6–7 weeks we recovered 104 mice (25 WT, 28 Nod2−/−, 27 Atg16l1T316A/+, and 24 Atg16l1T316A/T316A) in time for analysis and compared them to 80 matched controls (19 WT, 19 Nod2−/−, 20 Atg16l1T316A/+, 22 Atg16l1T316A/T316A) maintained under specific pathogen free (SPF) conditions (herein referred to as lab mice) (Figure 1A). Blood samples were collected at the time of sacrifice and analyzed by flow cytometry with a lymphocyte panel (Table S1). Additionally, cytokine production in the plasma was assayed (Figure 1A). Fecal samples and cecal contents were collected for microbial profiling, and for reconstitution experiments (Companion study, Yeung et. al.). Mesenteric lymph node (MLN) cells were isolated for flow cytometry analysis with the same lymphocyte panel as the blood and an additional myeloid cell panel (STAR METHODS). Immune activity in the MLN was measured through transcriptional profiles by RNA-seq and cytokine production assays in response to microbial stimulations (STAR METHODS). All of the lab mice were sacrificed in one week, whereas the rewilded mice were sacrificed over a 2-week period based on trapping frequency. This collection of multiparameter datasets from nearly 200 mice provides an opportunity to examine inter-individual variation in innate and adaptive immune cell populations by flow cytometry analysis, evaluate cytokine responses to microbial stimulation, and integrate highly dimensional transcriptomics and microbiota data. In order to perform multi-omic analysis, 81 mice were completely profiled with measurements from all four data types; flow cytometry, MLN RNA-Seq, fecal microbiota and MLN restimulation and cytokine profiles. A systems level analysis also enabled us to quantify effects of environmental influences (i.e. microbiota acquired from environment) and IBD susceptibility gene mutations, Nod2 and Atg16l1, on the heterogeneity of immunological parameters among individual mice.
Figure 1. Environmental change drives inter-individual variation in immune cell populations.

(A) 25 C57BL/6+/+, 28 Nod2−/−, 27 Atg16l1T316A/+, 24 Atg16l1T3I6A/T3I6A mice (total=104) were housed in the wild enclosure (Rewilded) for 6–7 weeks and successfully trapped for flow cytometry analysis of lymphoid and myeloid cell populations in the blood and mesenteric lymph nodes (MLNs). Plasma was also collected for cytokine profiles. Age matched 19 C57BL/6+/+, 19 Nod2−/−, 20 Atg16l1T316A/+, 22 Atg16l1T316A/T316A mice (total=80) housed under SPF conditions (Lab) were analyzed as controls. Feces were collected for 16S rRNA sequencing. MLN cells were cultured with indicated stimulates for 48 hours and supernatants were collected for cytokine profiling. Samples that fail quality control are not included in downstream analyses. (B and C) UMAP projections of ~180,000 CD45+, ~110,000 CD19+, and ~36,000 CD4+ cells on flow cytometry data of the blood from lab and rewilded mice. Events are color-coded according to CD19, CD44 (B), CD4, and CD62L (C) fluorescence level. Box plots show the abundance of CD44hi CD19+ cells (B) and CD62Lhi CD4+ cells (C) in the blood of individual lab and rewilded mice. (D) Principal component analysis (PCA) of gated immune cell populations in the blood and the density of each population along the principal component (PC). Right panel indicate biplots of the gated immune cell populations that are projected onto PC1 and PC2. (E) Box plot of pairwise Euclidean distance measures based on blood immune cell population in the blood of lab versus lab, rewilded versus rewilded and lab versus rewilded mice. (F) Bar plot showing the pseudo R2 measure of effect size on the entire distance matrix used to calculate the PCA of immune cell populations in the blood (D). ***P < 0.001 by Kruskal-Wallis test between groups (B, C) and ***P < 0.001 by Mann-Whitney-Wilcoxon test between groups (E).
Variation of immune cell populations in the blood is largely driven by the environment
In line with previous studies exposing lab mice to feral and pet shop mice (Beura et al., 2016), we observed major differences in CD44hiCD62Ll° or CD44hiCD62Lhi T cells after rewilding (Companion study, Yeung et. al.). To visualize the overall landscape of flow cytometry data in an unbiased manner, phenotypic heterogeneity was analyzed on total CD45+ cells per mouse across the blood lymphocyte panel (Table S1) in lab and rewilded mice. We down sampled to 1,000 CD45+ cells from each mouse for nearly 180,000 single CD45+ cell events and single cell flow cytometry data was visualized by uniform manifold approximation and projection (UMAP) (Becht et al., 2019). UMAP analysis was superior to t-distributed stochastic neighborhood embedding (t-SNE) (Becht et al., 2019) (Figure S1A) for identifying major immune cell subsets (Figures 1B and 1C; Figures S1B–E). Unexpected clusters of CD44hiCD19+ cells were increased in the rewilded mice compared to lab mice (Figure 1B), while lab mice had more CD62LhiCD4+ cells (Figure 1C). This unsupervised visualization strategy was useful for identifying unexpected differences (e.g. in the CD19+ compartment and other populations not investigated by (Beura et al., 2016; Rosshart et al., 2019; Rosshart et al., 2017)) in cell clusters of the peripheral blood at a single cell level, which might have been overlooked by traditional flow cytometry gating.
In order to quantify known immune cell changes after rewilding, we employed a traditional gating strategy to systematically quantify the frequencies of immune cell subsets with a lymphocyte panel (Figures S2A–S2E; Table S2). The blood lymphocyte panel identified 13 lymphocyte populations, a total myeloid cell population (CD11b/CD11c/DX5), and a total CD45+ cell population. From the proportions of these immune cell populations, we examined the inter-individual variation in lab and rewilded mice by principal component analysis (PCA) (Figure 1D). As shown by the separation of lab and rewilded mice along principle component (PC) 1 and 2, there is a strong effect of the environment on lymphocyte populations, and major differences between these populations can be attributed to CD44hiCD62Lhi CD4 and CD44hiCD62Lhi CD8 T cells as inferred by examining loading factors along PC1 and PC2 (Figure 1D). The reduced proportion of CD44loCD62Lhi T cells is consistent with some previous reports on wild mice (Abolins et al., 2017; Beura et al., 2016), although we find that major mouse pathogens are absent in these rewilded mice. We also quantified the extent of variability between lab and rewilded mice by measuring the Euclidean distances between mice in our PCA. Notably, the within group distances in lab mice were significantly less than rewilded mice (Figure 1E) suggesting that the natural environment increases variability in immune cell populations. We then evaluated several population factors to identify covariates with the largest effect sizes on immune cell populations (STAR METHODS) (Figure 1F). As expected, the composition and activation state of immune cell populations as determined by cell surface marker expression in the peripheral blood are driven most strongly by environmental differences between the SPF facility and the outdoor enclosure. These differences far outweighed any effects of genetic deficiency in Nod2 or Atg16l1 in both environments (Figure 1F). Sex has a relatively small effect, although there is some indication that the environment may have different effects on male and female mice (Figures 1F, S3A, and S3B). These findings are consistent with the concept that non-heritable influences (i.e. environmental microbial challenges) explain the majority of the variation in the proportion of immune cell subsets as reported in twin studies (Brodin et al., 2015).
Variation of cytokine production in response to microbial stimulations is driven more by genetic mutations than by the environment
Cytokine production capacity in humans has been reported to be more strongly influenced by genetic than environmental factors (Li et al., 2016). To investigate the relative roles of Nod2/Atg16l1 deficiency and environment on induced responses against bacterial and fungal antigens, we performed ex vivo stimulation experiments on total MLN cells from rewilded and lab mice. To examine cytokine responses to microbial antigens, we stimulated cells under eight conditions; with six different microbes (C. perfringens, P. aeruginosa, B. vulgatus, B. subtilis, S. aureus and C. albicans), αCD3/CD28 beads to activate T cells (as a positive control), as well as PBS as a negative control (Figures 1A and 2A). Despite no significant differences for PBS control samples between lab and rewilded mice (data not shown), we chose to normalize cytokine production per mouse by expressing cytokine data as a fold change levels over PBS to further minimize batch effects (STAR METHODS). A broad array of 13 cytokines was measured: IFN-γ, IL-1α, IL-1β, IL-13 IL-5, IL-10, IL-17, IL-6, CCL2, CXCL1, CCL3, CCL4 and TNF-α. Average fold changes in cytokine production as compared to PBS controls for both lab and rewilded mice indicate increases in IFN-γ and IL-17 after αCD3/CD28 activation, consistent with the production of these cytokines by T cells in the MLNs (Figure 2A). In general, bacterial antigens induced CCL3, IFN-γ, TNF-α, CCL4, and IL-6 production, while the fungus C. albicans induced the regulatory cytokine IL-10 (Figure 2A). Bacterial antigens also induced CXCL1, which was not induced by C. albicans or αCD3/CD28 (Figure 2A). As expected αCD3/CD28 beads only was one of the most potent inducers of cytokine production, especially for IFN-γ, CCL4, TNF-α and IL-5, and rewilded mice produced higher levels of these cytokines than lab mice post stimulation (Figure 2B). IL-1α, IL-1β, IL-13 and CCL2 were not appreciably altered by any stimulation (Figure 2A).
Figure 2. The genetic contribution towards variation in cytokine production in response to microbial stimulation is masked when mice are rewilded.

(A to D) MLN cells from lab and rewilded mice were ex vivo cultured with B. subtilis, S. aureus, C. perfringens, B. vulgatus, P. aeruginosa, C. albicans, and αCD3/CD28 beads for 48 hours and the supernatants were assayed for 13 cytokines. (A) Heat map of average fold changes compared to cytokine levels of MLN cells cultured with PBS controls from all mice. (B) Box plot showing the top 10 log2 fold changes of cytokine responses to stimulation for MLN cells from lab and rewilded mice. (C) PCA plot of of fold change cytokine stimulation profiles with histograms of the total variance explained by each PC indicated on the axis. Right panel indicates the loading factor of each stimulated cytokine profile that contributes to each PC. (D) Bar plot showing the pseudo R2 measure of effect size on the entire distance matrix used to calculate the PCA of cytokine profiles (C). * P <0.05, ** P <0.01, *** P < 0.001 by Mann-Whitney-Wilcoxon test between groups (B).
We next used PCA to assess the sources of variation in stimulated cytokine production for both lab and rewilded mice (Figure 2C). Lab and rewilded mice do not clearly segregate along PC1 or PC2 suggesting that environment is not a major source of variation (Figure 2C). Effect size measures based on the Euclidean distance indicated genotype as a much stronger source of variation (Figure 2D). The differences in genotype are driven by IL-10 and IL-6 production as indicated by the PCA loading factors (Figure 2C). More specifically, the rewilded Nod2−/− mice appear to cluster distinctly from other genotypes, suggesting key differences in microbial stimulated cytokine production of rewilded Nod2−/− mice (Figure 2C). Genotype also had the greatest effect size on variation in plasma cytokine levels of individual mice (Figures S3C and S3D).
Rewilded Nod2−/− mice exhibit increased microbial stimulated cytokine responses compared to wild type rewilded mice
To better understand how Nod2 and Atg16l1 deficiency was affecting microbial stimulated cytokine production, we performed two-way comparisons and calculated a p-value for each comparison to determine which cytokine producing conditions were most significantly different between WT mice and mice with Nod2 and Atg16l1 deficiencies from laboratory and rewilded environments (Figures 3A and 3B). Compared to the lab mice, the rewilded mice had many more significant changes in stimulated cytokine production across Nod2 and Atg16l1 mutant mice (Figure 3A and 3B). These changes were largely driven by significant differences in rewilded Nod2−/− mice against rewilded WT mice (Figure 3A). Specifically, the Nod2−/− mice exhibited increases in IL-17, IL-5 and IL-10 in response to C. perfringens suggesting an increased responsiveness to this bacterial stimulant that was only observed in the rewilded condition (Figures 3A and 3C). Production of IL-10 in response to C. albicans was also elevated in Nod2 and Atg16l1 mutant mice, although Nod2−/− did not exhibit greater fungal colonization than WT mice (Figure 3C) (see companion study Yeung. et.al.). Despite these differences in cytokine responses, the rewilded Nod2−/− did not shown any signs of increased intestinal inflammation by histology or changes in goblet cell numbers (data not shown), which we have previously shown to be associated with susceptibility to B. vulgatus colonization (Ramanan et al., 2016; Ramanan et al., 2014). Together these results indicate that exposure to the outdoor environment (including wild microbial challenges) per se that induces elevated cytokine responses is not a sufficient hit by itself to trigger intestinal inflammation. Additional insults (i.e. a multi-hit model) may be needed to trigger full blown pathology (Ramanan et al., 2016).
Figure 3. Rewilded Nod2−/− mice display greater changes in cytokine production compared to rewilded WT mice.

(A to C) MLN cells from lab and rewilded mice were ex vivo cultured with B. subtilis, S. aureus, C. perfringens, B. vulgatus, P. aeruginosa, C. albicans, and αCD3/CD28 beads for 48 hours and the supernatants were assayed for 13 cytokines. (A and B) Heatmap of significant p-values from Mann-Whitney-Wilcoxon test between group results comparing fold changes of stimulated cytokine production in rewilded (A) and lab (B) WT mice against Nod2−/−, Atg16l1T316A/+ and Atg16l1T316A/T316A mice respectively. Within each cytokine condition the order of stimulation remains constant as follows; B. subtilis, B. vulgatus. C. albicans, aCD3/CD28, C. perfringens, P. aeruginosa, S. aureus. (C) Representative box plots of the most significant changes between WT and Nod2−/− mice. * P <0.05, ** P <0.01, *** P < 0.001 by Mann-Whitney-Wilcoxon test between groups (C).
An integrated classification model identifies features predictive of environment and genotype-specific effects
In addition to stimulated cytokine profiles, MLN cells were assessed for immune cell populations by flow cytometry analysis (Figures S2F–S2K and Table S2) and gene expression by RNA-seq analysis (Figures S4A and S4B; Table S2). Consistent with the peripheral blood, immune cell frequencies in MLNs showed a strong effect of environment (Figures S4C–S4E). Interestingly, a group of rewilded mice all sacrificed at week 7 of rewilding clustered separately in the PCA analysis of lymphocyte populations (Figure S4E). Between week 6 and 7 of rewilding it is possible specific events in the enclosure (e.g., a severe thunderstorm, high humidity and fungal blooms observed during that week), could have resulted in changes in T cell activation. Similar to blood lymphocyte populations, RNA-seq analysis (Figures S4A and S4B) and 16S rRNA profiling (Figures S4F–S4I; see companion study by Yeung et al) showed some environmental effects on variations. This large amount of data prompted us to use a multi-omic classification model that could prioritize features predictive of environment and genotype-specific effects, as well as identify relationships between these features.
We integrated multi-omic data from flow cytometry populations, gene expression, cytokine, and microbial profiles (Table S2) in a subset of mice (n=81) which had measures for all four data types. A concatenated matrix (Table S3) of these features was used to build a random forest classification model (STAR METHODS) to distinguish samples by environment or genotype (Figure 4). The contribution of each feature to the model was also assessed to identify features predictive of environment or genotype-specific effects (Figures 4A and 4C). When classifying the environment, a hub of transcription factors (including Zfp36, Atf4, Fos, several Jun and Klf family members) and Cd69 are strong predictive features of environment (Figure 4A). Pairwise correlation analysis between these top features also reveals associations to CD44hiCD62Lhi populations for CD4+ and CD8+ T cells in the blood and MLN, as well as IL-5 and TNF-α production in response to anti CD3/CD28 stimulation (Figure 4B). For the genotype model, our classification was not as accurate as the environment, as measured by area under the receiver operator curve (AUC) of 0.78 versus 1 on the test set (STAR METHODS). Pairwise correlation analysis revealed fewer associations between these features and overall less connected nodes (Figure 4D). Instead of cytokines i associated with T cell activation by αCD3/CD28 beads only, IL-17 responses to microbial antigen (B. vulgatus and P. aeruginosa) stimulation were correlated (Figures 4B and 4D). Interestingly, expressions of a number of ribosomal proteins (Rps and Rpl proteins) were also predictive of genotype (Figures 4C and 4D). In summary, we identified a network of transcription factor associated with T cells in the MLNs based on transcriptional profiling data that is strongly predictive of the effects of the different environments.
Figure 4. Multi-omic classification models identify predictive features of environment and genotype.

(A) Bar plot of the top 40 features predictive of the environment and (B) interaction network analysis of correlations (R2 > 0.6 or R2 < −0.6) between features (genes, immune cell populations, OTUs or cytokines) that are predictive of environmental changes. (C) Bar plot of the top 40 features predictive of the genotype and (D) network analysis of the associations between features that are associated with genotype differences. Data types are colored according to cytokine profiles (green), genes (red), immune populations (blue), and OTUs (purple). The size of the nodes in B and D are scaled proportionally to the number of connections with other nodes in each network.
Integrative network analysis identifies co-regulated modules in rewilded mice
To evaluate the interactions between data types, we next utilized an unsupervised sparse partial least squares (sPLS) regression model to integrate the different data types into a multi-component network (Figures 5A and S5). To condense the size of our network, we collapsed our gene expression profiles according to known Gene Ontologies (STAR METHODS) (2015; Ashburner et al., 2000) for immune system processes in Mus musculus, which also focuses our attention on the most relevant genes. In short, we compared our gene expression profiles to all child terms of the Gene Ontology term “immune system process” (GO:0002376) and collapsed our genes into modules based on these pre-defined ontologies. 954 genes were used to generate 91 specific gene ontologies from our gene expression data all related to the parent gene ontology “immune system process”. These modules were used as inputs alongside the cytokine profiles, microbial profiles and flow cytometry populations to generate an unsupervised multi-omic network. sPLS-regression models were built pairwise between each data type to generate a 188-node covariance network with 577 total connections between the four different types of features (Figures 5A and S5). In the resulting model, all microbial taxa fell below our 0.6 covariance threshold and were therefore removed (Table S4). This indicated that bacterial composition, determined by 16S sequencing alone, was not an important component of the network. Perhaps other strategies to assess microbial function (e.g. transcriptomics, metagenomics or metabolomics) would have yielded more connections to immune function.
Figure 5. An unsupervised multi-component model identifies a C. albicans and C. perfringens response network associated with environmental changes.

(A) Data matrices from flow cytometry data, transcriptional profiles, microbial profiles and stimulated cytokine profiles were used as inputs for a sPLS-regression model to generate a covariance network. (B) The most connected nodes, Blood CD4 T-cells CD44hi CD62Lhi, IL-5 in response to C. perfringens, and GO:0061844 (antimicrobial humoral immune response mediated by antimicrobial peptides) were evaluated for a difference between lab and rewilded mice populations. (C) The IL-5 C. perfringens subnetwork was derived from the network and recalculated to highlight the connections most related to IL-5 production. (D) GO: 0030593 (neutrophil chemotaxis) was identified as a significant connection in the subnetwork and the genes found in that GO term were plotted as a heatmap. Color bars indicate environment and genotype. The size of the nodes in (A) and (C) are scaled proportionally to the number of connections with other nodes in each network. Data types are colored according to cytokine profiles (green), GO term (red), immune populations (blue). * P <0.05, ** P <0.01, *** P < 0.001 by Mann-Whitney-Wilcoxon test between groups (B).
While total CD4+ and CD8+ T cell populations in the MLN were among the most connected nodes of the network specific gene ontologies, cytokine responses and other immune populations were also strongly interconnected (Figure 5A). As expected from earlier analysis (Figure 1), the blood CD44hiCD62Lhi CD4 T cell population was strongly interconnected and enriched in rewilded mice (Figures 5A and 5B). Additionally, by collapsing our gene expression profiles into annotated gene ontologies, we identified a module (GO:0061844: antimicrobial humoral immune response mediated by antimicrobial peptides) as the most connected gene module that was enriched in rewilded mice (Figures 5A and 5B). Interestingly, IL-5 production in response to C. perfrigens stimulation was the most connected cytokine response and was found to be higher in rewilded mice (Figures 5A and 5B; Figure S5). This node is part of a 15-node subnetwork connected to total CD4 and CD8 T cell populations in the MLN, several other cytokine responses and the GO:0030593: neutrophil chemotaxis module (Figure 5C). This module is of particular interest because of the increased neutrophilia observed in the rewilded mice (Yeung et.al. companion paper), which is driven by increased fungal colonization. Indeed, this module is tightly linked with cytokine responses to C. albicans antigen stimulation (Figure 5C). While expressions of genes in this module are increased by the rewilding environment, there does not appear to be genotype specific differences regulating expression of the genes in this module (Figure 5D). This multi-omics network analysis therefore substantiated the omics-by-omics inferences (Figures 1 and 2) but also provided insight by identifying an important role for C. albicans responses during rewilding, which is more fully addressed in the companion study (Yeung et.al. companion paper).
DISCUSSION
In summary, we found that changing the environment profoundly contributes to the inter-individual variations on immune cell frequencies, whereas cytokine responses to pathogen stimulation were more affected by genetic deficiencies in the IBD susceptibility genes Nod2 and Atg16l1. While we have only examined the effects of two genes, these observations are reminiscent of human twin studies whereby the majority of cell population influences (measured by flow and mass cytometry) are determined by non-heritable factors (Brodin et al., 2015), whereas cytokine production capacity in response to stimulation may be more strongly influenced by genetic factors (Li et al., 2016). Another large human study indicated that features of innate immunity were more strongly controlled by genetic variation than lymphocytes, which were driven by environmental effects (Patin et al., 2018). Thus, although studies of lab mice, by design, often emphasize genetic effects on immune phenotype (including cell population distributions), here we show that even brief exposure to a natural environment renders predictors of immune phenotype in mice a better match for predictors of human immune phenotypes. Additionally, our dataset described here will be a useful resource for other investigators to delve into the immunological consequences and wild microbial acquirements of rewilding.
An attractive hypothesis arising from these findings is that perhaps environment is a primary driver of the composition of the immune system, but genetics is a stronger driver of per-cell responsiveness. However, in this study we only investigated how two genes (Nod2 and Atg16l1) influenced the immunological phenotypes that we measured. Additionally, we only measured immune phenotypes in the peripheral blood and the mesenteric lymph nodes. This was because it was not possible logistically to assess more tissues from the large number of mice that were analyzed at the same time. In future studies, examination of other peripheral tissues such as the intestinal tract, lungs, skin and brain, as well as immune organs such as the spleen and bone marrow may provide a more complete assessment of the total immunological effect of rewilding.
To test the above hypothesis further would require us to release mice from diverse genetic backgrounds into the outdoor enclosure in a similar experiment. This experiment only examined the effects of Nod2 and Atg16l1 deficiency in the C57BL/6 background. For example, analysis of macrophage activation from five different strains of mice that provided genetic variation on the order of the human population yielded considerable insights into how genetic variation affects transcriptional regulation mechanisms (Link et al., 2018). Alternatively, mice from the collaborative cross may enable high-resolution genome mapping for such complex traits as cytokine production in response to stimulation (Noll et al., 2019). Studies of wild animal populations may also determine if per cell cytokine responses show a higher genetic variance than do immune cell population distributions. Hence, the combination of new environmental challenge strategies such as rewilding of mouse models, combined with modern multi-omic approaches in fully wild systems, should enable us to better define determinants of immune variation at a molecular level.
This study was initially designed to test the hypothesis that Nod2 and Atg16l1 mutant mice may respond to wild microbial exposure from the re-wilding environment in an adverse way. Our previous studies had found that B. vulgatus colonization of Nod2 and norovirus colonization of Atg16l1 mutant mice predisposed them to intestinal inflammation. Hence, we wanted to examine if these mice with mutations in genes associated with the development of IBD would be associated with environmental or wild microbial triggering of intestinal inflammation. However, we did not observe significant differences in intestinal inflammation based on histology for either the Nod2 or Atg16l1 mutant mice, although we find an elevated production of cytokines in the Nod2 mutant mice in response to the rewilding environment (including wild microbial challenges). While there were not significant differences in intestinal inflammation, it is possible that additional insults (i.e. a multi-hit model) are needed to trigger intestinal inflammation that is sufficiently severe to be discernable by histologic examination. In our previous studies, an additional piroxicam (for the Nod2 mutant mice (Ramanan et al., 2016) or dextran sodium sulfate (DSS) (for the Atg16l1 mutant mice (Matsuzawa-Ishimoto et al., 2017)) insult was required to drive pathogenesis of the B. vulgatus /norovirus colonized mice.
A systems level analysis identified interconnected networks of transcriptional signatures, immune cell populations and cytokine profiles to microbial stimulation, highlighting a potentially important role for fungal stimulation from rewilding (Companion study, Yeung et. al.). This unanticipated result may have been missed through conventional comparisons. Increased colonization by commensal fungi is perhaps the most striking environmental effect of the rewilding experiment that we performed. While Nod2 deficiency did not affect fungal colonization, Atg16l1 mutant mice are more susceptible (Companion study, Yeung et. al.). Future studies with mice from more genetically diverse backgrounds will better define the genetic basis of host susceptibility to commensal fungi colonization through rewilding and determine how this may affect the innate and adaptive immune responses.
Hence, rewilding laboratory mice of different genetic backgrounds and careful monitoring of other non-heritable influences (e.g. differential behavior via which divergent immunological experience of individuals may accrue, by analogy with causes of divergence in neurological phenotype (Freund et al., 2013) observed during rewilding (Cope et al., 2019) could be a powerful approach towards dissecting drivers of immune response heterogeneity under homeostatic as well as during disease settings or infectious challenges.
STAR METODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Dr. P’ng Loke (Png.Loke@nih.gov). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and wild enclosure
All animal work was approved by NYU Langone IACUC (#IA16-0087 and #IA16– 00864). All 6–10 week old mice on both genders were used for experiments. All mouse lines were bred onsite in an MNV/Helicobacter-free specific pathogen free (SPF) facility at NYU School of Medicine to generate littermates from multiple breeding pairs that were randomly assigned to either remain in the institutional vivarium (lab mice) or released into the outdoor enclosures (rewilded mice) to control for the microbiota at the onset of the experiment. Nod2−/− and Atg16l1T316A/T316A mice on the C57BL/6J background were previously described (Matsuzawa-Ishimoto et al., 2017; Ramanan et al., 2014). Atg16l1T316A/T316A mice, Atg16l1T316A/+, and wild-type (WT) control mice were generated from Atg16l1T316A/+ breeder pairs, and Nod2−/− mice were generated from Nod2−/− breeder pairs. Additional C57BL/6J mice were purchased from Jackson Laboratory and bred onsite to supplement WT controls for experiments. 16S microbial diversity at the conclusion of the experiment did not show appreciable differences in microbial composition within the lab populations (Supplementary Figure S5). Outdoor enclosures were previously described (Budischak et al., 2018; Leung et al., 2018) and the protocols for releasing the laboratory mice into the outdoor enclosure facility were approved by Princeton IACUC (#1982–17).
The enclosures consist of replicate outdoor pens, each measuring about 180 m2 and fenced by 1.5-m high, zinced iron walls that are buried >80 cm deep and topped with electrical fencing to keep out terrestrial predators. Aluminum pie plates are strung up to deter aerial predators. A (180 × 140 × 70 cm) straw-filled shed is provided in each enclosure, along with two watering stations and a feeding station, so that the same mouse chow used in the laboratory (PicoLab Rodent Diet 20) was provided ad libitum to all mice. Mice outdoors, however, also had access to food sources found within the enclosures, including berries, seeds, and insects. 26–30 mice of mixed genotypes but the same sex were housed in each enclosure for 6–7 weeks. Longworth traps baited with chow were used to catch mice approximately 2 weeks and 4 weeks after release and again 6–7 weeks after release; for each trapping session, two baited traps were set per mouse per enclosure in the early evening, and all traps were checked within 12 hours. For subsequent microbiome assessment, a fresh stool sample was collected directly from the caught mice, flash frozen on dry ice, and stored at −80°C until further analysis. Mice were weighed with a spring balance.
30 WT, 29 Nod2−/−, 31 Atg16l1T316A/+, and 26 Atg16l1T316A/T316A laboratory mice (Total=116) were released into the outdoor enclosure. 19 WT, 19 Nod2−/−, 20 Atg16l1T316A/+, 22 Atg16l1T316A/T316A matched littermates (Total=80) were maintained in the institutional vivarium for comparison. For rewilded mice, traps were set regularly until the remaining mice were caught and were sampled for fecal microbiota. 25 WT, 28 Nod2−/−, 27 Atg16l1T316A/+, and 24 Atg16l1T316A/T316A rewilded mice (Total=104) were caught in the final trapping for terminal analyses. All lab control mice were recovered. Euthanasia was performed by CO2 asphyxiation, and blood, MLNs, and intestinal tissue were harvested. Two Atg16lr3 rewilded mice failed quality control and were not included in downstream analyses. One Atg16lrT316A/+ lab mouse was not appropriately processed and excluded in the final meta data table (Table S2; N=79 in lab mice and N=102 in rewilded mice).
METHOD DETAILS
Flow cytometry analysis
At harvesting, MLNs were removed and the single-cell suspensions were prepared in FACS buffer (HBSS containing 1% BSA, 1mM EDTA, 20mM HEPES, and 1mM sodium pyruvate). The whole blood were also collected in a heparin containing tube and after centrifuging at 2000 rpm for 5 minutes, the designated plasma from supernatant was removed and stored at −80°C until all samples were collected and analyzed together. After two rounds of red blood cell lysis with 1x RBC lysis buffer for 5 minutes and wash with FACS buffer, the single-cell suspensions of whole blood cells were ready for the following staining procedure. MLN and whole blood cells were stained for live/dead with blue reactive dye and cell surface markers were labeled with the following antibody panels: Lymphoid panel: CD49b Pacific Blue, CD11b Pacific Blue, CD11c Pacific Blue, CXCR3 Brilliant Violet 421, CD27 Brilliant Violet 510, KLRG1 Brilliant Violet 605, CD3 Brilliant Violet 786, CD127 Brilliant Violet 711, PD1 PerCP/Cy5.5, CD4 APC/Cy7, CD19 PE/Dazzle594, CD8 Brilliant Violet 650, CD43 Alexa Fluor 488, CD62L APC, CD44 PE, CD69 Alexa Fluor 700, CD45 Buv395, CD25 PE/Cy7. Myeloid panel: B220 Pacific Blue, CD86 Brilliant Violet 510, CD3 Brilliant Violet 605,CD69 Brilliant Violet 786, CD40 Alexa Fluor 488, Ly6G PerCP/Cy5.5, PDL2 APC, IA/IE APC/Cy7, PDL1 PE, CD64 PE/Dazzle594, F4/80 Alexa Fluor 700, CD11c Brilliant Violet 650, Siglec-F Brilliant Violet 421, CD103 Brilliant Violet 711, Ly6C PE/Cy7, CD11b Buv395. FACS analyses were performed in a ZE5 cell analyzer (BIO-RAD) and recorded FACS data were analyzed by Flowjo v10.4.2.
MLN cell stimulation and cytokine profiling
Single cell suspension of MLN cells were reconstituted in RPMI at 2 × 106 cells/mL, and 0.1 mL was cultured in 96-well microtiter plates that contained 107 cfu/mL UV-killed microbes, 105 αCD3/CD28 beads, or PBS control. Overnight microbial cultures were reconstituted at 108 cfu/mL prior to irradiation. The stimulated microbes are as following: Staphylococcus aureus (Maurer et al., 2015), Pseudomonas aeruginosa (PAO1) (kindly provided by Dr. Andrew Darwin, NYU) (Srivastava et al., 2018), Bacillus subtilis (ATCC 6633), Clostridium perfringens (NCTC 10240), Bacteroides vulgatus (ATCC 8482), and Candida albicans (UC820, kindly provided by Dr. Stefan Feske, NYU). Supernatants were collected after 2 days and stored at −80°C. Concentrations of IL-1α, IL-1β, IL-4, IL-5, IL-6, IL-10, IL-13, IL-17A, CCL2, CCL3, CCL4, CXCL1, IFN-γ, and TNF-α in supernatants were measured using a custom mouse LEGENDplex assay (Biolegend) according to the manufacturer’s instructions. Plasma concentrations of IL-1α, IL-1β, IL-6, IL-10, RANTES, CCL2, CCL3, CCL4, CCL20, CXCL1, CXCL10, TNFα, GM-CSF were measured using a second custom mouse LEGENDplex assay (Biolegend).
16S library preparation and sequencing
DNA was isolated from stool samples using the NucleoSpin Soil Kit (Macherey-Nagel). Bacterial 16S rRNA gene was amplified at the V4 region using primer pairs and paired-end amplicon sequencing was performed on the Illumina MiSeq system as previously described (Neil et al., 2019). Sequencing reads were processed using the DADA2 pipeline in the QIIME2 software package. Taxonomic assignment was performed against the Silva v132 database. Differential abundance taxa was identified using discrete false-discovery rate (DS-FDR) methodology in different biological groups at a threshold DS-FDR score of 30 (Jiang et al., 2017).
MLN cell RNA preparation and sequencing
Frozen samples of single cell suspensions from MLN of lab or rewilded mice were thaw to isolate RNA from approximately 106 cells by RNeasy Plus Mini Kit according to manufacturer’s instructions. CEL-seq2 were performed to do RNA sequencing on samples with good RNA qualities (RNA integrity number ≧ 5)
QUANTIFICATION AND STATISTICAL ANALYSIS
FACS data visualizations by t-distributed stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP)
Flowjo v10.4.2 software and plugin were installed per manufacturer’s instructions. The DownSample function in plugin was applied to filter out 1,000 cells on total gated CD45+ cells in blood lymphoid panel staining and the raw channel values from each staining marker were exported to make a FACS value matrix (16 X 1,000) per mouse. FACS data from 2 mice were not passing the data filtrations and 180 FACS value matrixes were concatenated into a giant matrix to perform the downstream t-SNE (fitsne v1.0.1 package (https://arxiv.org/abs/1712.09005), Python v3.6.5) or UMAP (umap-learn v0.3.7 package (https://arxiv.org/abs/1802.03426), Python v3.6.5analysis for the immune cell cluster visualizations. The thresholds were set up for each marker base on expressing distributions across total cells.
Principal component analysis and effect size measures
In all cases principal component analysis was performed with the ape (https://www.springer.com/gp/book/9781461417422) v5.2 package in R v3.4.1. Euclidean distances were calculated using base R functions and the subsequent distance matrix was used to determine principle components (PC). Biplots were constructed by projecting the weighted averages of each input feature (immune cell population, cytokine level etc.,) along PC1 and PC2 derived from the biplot.pcoa function from the ape package. Effect size measures were determined using the MDMR v0.5.1 (https://link.springer.com/artide/10.1007/s11336-016-9527-8) package in R.
Per mouse normalization of cytokine production levels
For each mouse profiled for cytokine production in response to microbial stimuli a PBS control was also sampled. In order to normalize per mouse cytokine production, we calculated the fold change of each cytokine measure to the PBS control. There was no overall difference in baseline cytokine production for any cytokine in response to PBS between lab and rewilded mice.
Machine learning models for environment and genotype classification
For multi-omic classification modeling gene expression values, cytokine production levels, immune cell populations and operational taxonomic unit (OTU) counts were normalized by log2-transformation and concatenated together. From ~15,000 genes only the 200 most variable genes, as measured by total variance across all samples, were utilized for modeling. 576 features (200 genes, 104 cytokine measures, 36 immune cell populations and 236 OTUs) from 40 lab and 41 rewilded mice with around 20 mice from each of the four genotypes were supplied to the models. Two random forest models were trained with caret v6.0–80 (R v3.4.1) (5) on a 75% split (61 of 81 mice) with 5-fold cross validation repeated 10 times. The first model was built to classify environment and the second to classify genotype. Both models were evaluated by area under the receiver operator curve (AUC) on the remaining 25% split (20 of 81 mice) left out from the training process. Due to data limitations we did not build an additional model to classify both environment and genotype simultaneously. Feature importance was assessed by the built-in variable importance function varImp within caret.
Unsupervised multi-omic network model built by sPLS
To further assess multi-omic relationships between data features (cytokines, immune cell populations, genes and OTUs) an unsupervised network model was constructed. The same 81 mice used to build the classification models were also used to generate this network. The same four data types used to build the classification models were also used with the exception of the 200 most variable genes. Instead we queried the Mus musculus Gene Ontology for biological processes related to “immune system process”. From all subsequent child terms, we generated a list of all genes relevant to these processes. Using our gene expression data, we were able to match 954 genes and generate 91 immune system specific gene ontologies. The average expression of all the genes in each ontology were averaged together and used as input into our multi-component sPLS network. In order to generate a multi-omic network of interactions we performed pairwise sparse partial least squares regression as demonstrated by Li et al. (Li et al., 2017) between each of the four data types. After filtering for a covariance threshold of 0.6 in either direction our network consisted of 188 nodes and 577 edges across three data types. None of the OTUs passed this threshold to be included in the network.
DATA AND CODE AVAILABILITY
Raw sequence data from 16S, ITS, and RNA sequencing experiments are deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA559026 and gene expression omnibus (GEO) accession number GSE135472. All processing was performed in R and analysis scripts can be found on Github at https://github.com/ruggleslab/RewildedMice
Supplementary Material
Table S4. Data metrics on all data types of gene ontology, MLN stimulated cytokine levels, blood lymphocyte populations, MLN lymphocyte and myeloid populations, and bacterial operational taxomic units (OTUs). Data related to Figure 5.
Table S2. Data metrics on metadata, blood lymphocyte populations, MLN lymphocyte and myeloid populations, MLN stimulated cytokine levels, and plasma cytokine levels from all lab and rewilded mice. Data related to Figure 2.
Table S3. Data metrics on metadata, MLN gene expression, and bacterial operational taxomic units (OTUs) from lab and rewilded mice. Data related to Figure 4.
Supplemental Video. Overall mouse activities. Related to STAR METHODS
Highlights.
Immune and microbial phenotyping of lab and rewilded Nod2 and Atg16l1 mutant mice.
Environmental differences drive variation in population frequencies of immune cells.
Cytokine responses to microbial stimulation are affected more by genetic mutations.
Multi-omic models identify responses to specific microbes driven by rewilding.
Acknowledgements
We wish to thank William Craigens, Daniel Navarrete Prado, Allison Lee, and Veena Chittamuri for assistance with trapping and husbandry in the field, the PU Lab Animal Resources staff for logistical support. We wish to thank the NYU School of Medicine Flow Cytometry and Cell Sorting, Microscopy, Genome Technology, and Histology Cores for use of their instruments and technical assistance (supported in part by National Institute of Health (NIH) grant P31CA016087, S10OD01058, and and S10OD018338).
Funding: This research was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) and also NIH grants DK103788 (K.C. and P.L.), AI121244 (K.C.), HL123340 (K.C.), DK093668 (K.C.), AI130945 (P.L. and K.C.), R01 HL125816 (K.C.), R01 AI140754 (K.C.), HL084312, AI133977 (P.L.), research station and research rebate awards from PU EEB (A.L.G.), pilot award from the NYU CTSA grant UL1TR001445 from the National Center for Advancing Translational Sciences (NCATS) (K.C., P.L.), pilot award from the NYU Cancer Center grant P30CA016087 (K.C.), AI100853 (Y.C.), and DK122698 (F.Y.). This work was also supported by the Department of Defense grant W81XWH-16–1-0256 (P.L.), Faculty Scholar grant from the Howard Hughes Medical Institute (K.C.), Crohn’s & Colitis Foundation (K.C.), Merieux Institute (K.C.), Kenneth Rainin Foundation (K.C.), Stony-Wold Herbert Fund (K.C.), and Bernard Levine Postdoctoral Research Fellowship in Immunology (Y.C.). K.C. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases.
Declaration of interests: K.C. receives research funding from Pfizer and Abbvie and P.L. receives research funding from Pfizer. K.C. has consulted for or received an honorarium from Puretech Health, Genentech, and Abbvie. P.L. consults for and has equity in Toilabs. K.C. has a provisional patent, U.S. Patent Appln. No. 15/625,934. P.L. is a federal employee.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43(Database issue), D1049–1056. Published online 2014/11/28 DOI: gku1179 [pii] 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abolins S, King EC, Lazarou L, Weldon L, Hughes L, Drescher P, Raynes JG, Hafalla JCR, Viney ME, and Riley EM (2017). The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun. 8, 14811 Published online 2017/05/04 DOI: ncomms14811 [pii] 10.1038/ncomms14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 25(1), 25–29. Published online 2000/05/10 DOI: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker OB, Aguirre-Gamboa R, Sanna S, Oosting M, Smeekens SP, Jaeger M, Zorro M, Vosa U, Withoff S, Netea-Maier RT, et al. (2018). Integration of multi-omics data and deep phenotyping enables prediction of cytokine responses. Nat Immunol. 19(7), 776–786. Published online 2018/05/23 DOI: 10.1038/s41590-018-0121-3 10.1038/s41590-018-0121-3 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2019). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. Published online 2018/12/12 DOI: nbt.4314 [pii] 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, et al. (2016). Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 532(7600), 512–516. DOI: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL, and Kobayashi KS (2010). Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A. 107(33), 1473914744 Published online 2010/08/04 DOI: 1003363107 [pii] 10.1073/pnas.1003363107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, and Davis MM (2017). Human immune system variation. Nat Rev Immunol. 17(1), 21–29. Published online 2016/12/06 DOI: nri.2016.125 [pii] 10.1038/nri.2016.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, et al. (2015). Variation in the human immune system is largely driven by non-heritable influences. Cell. 160(1–2), 37–47. DOI: 10.1016/j.cell.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budischak SA, Hansen CB, Caudron Q, Garnier R, Kartzinel TR, Pelczer I, Cressler CE, van Leeuwen A, and Graham AL (2018). Feeding Immunity: Physiological and Behavioral Responses to Infection and Resource Limitation. Front Immunol. 8, 1914 Published online 2018/01/24 DOI: 10.3389/fimmu.2017.01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, and Virgin HW (2010). Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 141(7), 1135–1145. Published online 2010/07/07 DOI: S0092-8674(10)00545-3 [pii] 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso R, Mathes T, Martens EC, Kamada N, Nusrat A, Inohara N, and Nunez G (2019). A specific gene-microbe interaction drives the development of Crohn’s disease-like colitis in mice. Sci Immunol. 4(34). Published online 2019/04/21 DOI: 4/34/eaaw4341 [pii] 10.1126/sciimmunol.aaw4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope EC, Opendak M, LaMarca EA, Murthy S, Park CY, Olson LB, Martinez S, Leung JM, Graham AL, and Gould E (2019). The effects of living in an outdoor enclosure on hippocampal plasticity and anxiety-like behavior in response to nematode infection. Hippocampus. 29(4), 366–377. Published online 2018/09/27 DOI: 10.1002/hipo.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund J, Brandmaier AM, Lewejohann L, Kirste I, Kritzler M, Kruger A, Sachser N, Lindenberger U, and Kempermann G (2013). Emergence of individuality in genetically identical mice. Science. 340(6133), 756–759. Published online 2013/05/11 DOI: 340/6133/756 [pii] 10.1126/science.1235294. [DOI] [PubMed] [Google Scholar]
- Jiang L, Amir A, Morton JT, Heller R, Arias-Castro E, and Knight R (2017). Discrete False-Discovery Rate Improves Identification of Differentially Abundant Microbes. mSystems. 2(6). Published online 2017/11/29 DOI: 10.1128/mSystems.00092-17mSystems00092-17 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernbauer E, Ding Y, and Cadwell K (2014). An enteric virus can replace the beneficial function of commensal bacteria. Nature. 516(7529), 94–98. DOI: 10.1038/nature13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie S, Conway KL, Lassen KG, Jijon HB, Pan H, Chun E, Michaud M, Lang JK, Gallini Comeau CA, Dreyfuss JM, et al. (2019). The Crohn’s disease polymorphism, ATG16L1 T300A, alters the gut microbiota and enhances the local Th1/Th17 response. Elife. 8 Published online 2019/01/23 DOI: 10.7554/eLife.3998239982 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JM, Budischak SA, Chung The H, Hansen C, Bowcutt R, Neill R, Shellman M, Loke P, and Graham AL (2018). Rapid environmental effects on gut nematode susceptibility in rewilded mice. PLoS Biol. 16(3), e2004108 Published online 2018/03/09 DOI: 10.1371/journal.pbio.2004108pbio.2004108 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Sullivan NL, Rouphael N, Yu T, Banton S, Maddur MS, McCausland M, Chiu C, Canniff J, Dubey S, et al. (2017). Metabolic Phenotypes of Response to Vaccination in Humans. Cell. 169(5), 862–877 e817. Published online 2017/05/16 DOI: S0092-8674(17)30477-4 [pii] 10.1016/j.cell.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Oosting M, Smeekens SP, Jaeger M, Aguirre-Gamboa R, Le KTT, Deelen P, Ricano-Ponce I, Schoffelen T, Jansen AFM, et al. (2016). A Functional Genomics Approach to Understand Variation in Cytokine Production in Humans. Cell. 167(4), 1099–1110 e1014. Published online 2016/11/05 DOI: S0092-8674(16)31400-3 [pii] 10.1016/j.cell.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell. 173(7), 1796–1809 e1717. Published online 2018/05/22 DOI: S0092-8674(18)30511-7 [pii] 10.1016/j.cell.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR, Lazrak A, Marinis JM, et al. (2017). Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 214(12), 3687–3705. Published online 2017/11/02 DOI: jem.20170558 [pii] 10.1084/jem.20170558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer K, Reyes-Robles T, Alonzo F 3rd, Durbin J, Torres VJ, and Cadwell K (2015). Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe. 17(4), 429–440. Published online 2015/03/31 DOI: S1931-3128(15)00116-X [pii] 10.1016/j.chom.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil JA, Matsuzawa-Ishimoto Y, Kernbauer-Holzl E, Schuster SL, Sota S, Venzon M, Dallari S, Galvao Neto A, Hine A, Hudesman D, et al. (2019). IFN-I and IL-22 mediate protective effects of intestinal viral infection. Nat Microbiol. Published online 2019/06/12 DOI: 10.1038/s41564-019-0470-1 10.1038/s41564-019-0470-1 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll KE, Ferris MT, and Heise MT (2019). The Collaborative Cross: A Systems Genetics Resource for Studying Host-Pathogen Interactions. Cell Host Microbe. 25(4), 484–498. Published online 2019/04/12 DOI: S1931-3128(19)30161-1 [pii] 10.1016/j.chom.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, Alanio C, Scepanovic P, Hammer C, Jonsson F, et al. (2018). Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat Immunol. 19(3), 302–314. Published online 2018/02/25 DOI: 10.1038/s41590-018-0049-7 10.1038/s41590-018-0049-7 [pii]. [DOI] [PubMed] [Google Scholar]
- Pott J, Kabat AM, and Maloy KJ (2018). Intestinal Epithelial Cell Autophagy Is Required to Protect against TNF-Induced Apoptosis during Chronic Colitis in Mice. Cell Host Microbe. 23(2), 191–202 e194. Published online 2018/01/24 DOI: S1931- 3128(17)30553-X [pii] 10.1016/j.chom.2017.12.017. [DOI] [PubMed] [Google Scholar]
- Ramanan D, Bowcutt R, Lee SC, Tang MS, Kurtz ZD, Ding Y, Honda K, Gause WC, Blaser MJ, Bonneau RA, et al. (2016). Helminth infection promotes colonization resistance via type 2 immunity. Science. 352(6285), 608–612. Published online 2016/04/16 DOI: science.aaf3229 [pii] 10.1126/science.aaf3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan D, Tang MS, Bowcutt R, Loke P, and Cadwell K (2014). Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity. 41(2), 311–324. DOI: 10.1016/j.immuni.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, McCulloch JA, Anastasakis DG, Sarshad AA, Leonardi I, et al. (2019). Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. 365(6452). Published online 2019/08/03 DOI: 365/6452/eaaw4361 [pii] 10.1126/science.aaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, Hickman HD, McCulloch JA, Badger JH, Ajami NJ, et al. (2017). Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell. 171(5), 1015–1028 e1013. Published online 2017/10/24 DOI: S0092-8674(17)31065-6 [pii] 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, Kumar V, Netea MG, and Xavier RJ (2018). The causes and consequences of variation in human cytokine production in health. Curr Opin Immunol. 54, 50–58. Published online 2018/06/19 DOI: S0952-7915(17)30173-5 [pii] 10.1016/j.coi.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Horst RT, Jansen T, Jacobs L, Bonder MJ, et al. (2016). Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell. 167(7), 1897 Published online 2016/12/17 DOI: S0092-8674(16)31658-0 [pii] 10.1016/j.cell.2016.11.046. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Seo J, Rimal B, Kim SJ, Zhen S, and Darwin AJ (2018). A Proteolytic Complex Targets Multiple Cell Wall Hydrolases in Pseudomonas aeruginosa. MBio. 9(4). Published online 2018/07/19 DOI: mBio.00972-18 [pii] 10.1128/mBio.00972-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki TA, Phifer-Rixey M, Mack KL, Sheehan MJ, Lin D, Bi K, and Nachman MW (2019). Host genetic determinants of the gut microbiota of wild mice. Mol Ecol. 28(13), 3197–3207. Published online 2019/05/30 DOI: 10.1111/mec.15139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter Horst R, Jaeger M, Smeekens SP, Oosting M, Swertz MA, Li Y, Kumar V, Diavatopoulos DA, Jansen AFM, Lemmers H, et al. (2016). Host and Environmental Factors Influencing Individual Human Cytokine Responses. Cell. 167(4), 1111–1124 e1113. Published online 2016/11/05 DOI: S0092-8674(16)31401-5 [pii] 10.1016/j.cell.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Kostic AD, and Xavier RJ (2015). An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe. 17(5), 577–591. DOI: 10.1016/j.chom.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, and Cadwell K (2018). There was collusion: Microbes in inflammatory bowel disease. PLoS Pathog. 14(9), e1007215 DOI: 10.1371/journal.ppat.1007215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. Data metrics on all data types of gene ontology, MLN stimulated cytokine levels, blood lymphocyte populations, MLN lymphocyte and myeloid populations, and bacterial operational taxomic units (OTUs). Data related to Figure 5.
Table S2. Data metrics on metadata, blood lymphocyte populations, MLN lymphocyte and myeloid populations, MLN stimulated cytokine levels, and plasma cytokine levels from all lab and rewilded mice. Data related to Figure 2.
Table S3. Data metrics on metadata, MLN gene expression, and bacterial operational taxomic units (OTUs) from lab and rewilded mice. Data related to Figure 4.
Supplemental Video. Overall mouse activities. Related to STAR METHODS
Data Availability Statement
Raw sequence data from 16S, ITS, and RNA sequencing experiments are deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA559026 and gene expression omnibus (GEO) accession number GSE135472. All processing was performed in R and analysis scripts can be found on Github at https://github.com/ruggleslab/RewildedMice