

Abstract
In cucurbits, CmWIP1 is a master gene controlling sex determination. To bring new insight in the function of CmWIP1, we investigated two Arabidopsis WIP transcription factors, AtWIP1/TT1 and AtWIP2/NTT. Using an inducible system we showed that WIPs are powerful inhibitor of growth and inducer of cell death. Using ChIP-seq and RNA-seq we revealed that most of the up-regulated genes bound by WIPs display a W-box motif, associated with stress signaling. In contrast, the down-regulated genes contain a GAGA motif, a known target of polycomb repressive complex. To validate the role of WIP proteins in inhibition of growth, we expressed AtWIP1/TT1 in carpel primordia and obtained male flowers, mimicking CmWIP1 function in melon. Using other promoters, we further demonstrated that WIPs can trigger growth arrest of both vegetative and reproductive organs. Our data supports an evolutionary conserved role of WIPs in recruiting gene networks controlling growth and adaptation to stress.
Subject terms: Plant sciences, Flowering
Maria V Gomez Roldan, Farhaj Izhaq et al. study the role of WIP transcription factors in Arabidopsis thaliana. Using an inducible system, they generate ChIP-seq and RNA-seq data that reveal how WIP proteins repress development and growth. Through ectopic expression of WIP in Arabidopsis carpels, they obtain male flower, mimicking sex determination in melon.
Introduction
Flowers are the reproductive organs of angiosperms, which lead to the production of fruits and seeds. Most angiosperms bear hermaphrodite flowers, developing both male and female organs in the same flower. For the rest of the flowering plants, the selective abortion of the female or male organ primordia of initially hermaphroditic flower meristems results in unisexual flowers1.
The production of unisexual flowers and plants is instrumental in plant breeding, as they ease the production of F1 hybrid seeds. In Cucurbitaceae, sex determination is wide spread2. Out of 800 inspected species, 460 are monoecious and 340 are dioecious3, which makes this plant family a practical model to investigate the molecular mechanisms controlling sex determination. In melon, three sex genes, androecious (A), andromonoecious (M), and gynoecious (G), have been identified to play important roles in the development of unisexual flowers and plants. The A and M genes encode for the limiting enzymes in the ethylene biosynthesis pathway, CmACS11 and CmACS7, respectively4,5. The G gene encodes a zinc finger transcription factor, CmWIP16. Expression of CmACS7 in carpel primordia inhibits, through an unknown non-cell-autonomous mechanism, the development of stamen primordia and leads to unisexual female flowers. CmWIP1 is a central integrator of the transcriptional networks leading both to inhibition of carpel development and the control of the expression of the stamina inhibitor, CmACS7. Still the identities of the transcription networks recruited by CmWIP1 to cause organ-specific abortion are mostly unknown.
CmWIP1 belongs to subclass A1d of zinc finger transcription factors, characterized by a highly conserved C-terminal domain and two conserved motifs in the N-terminal7,8. In the C-terminal, there are three conserved amino acids, tryptophan (W), isoleucine (I), and proline (P) on which the protein family is named WIP, followed by four C2H2 zinc finger (ZF) motifs. WIP proteins are land plants specific transcription factor, present in one copy in the liverwort Marchantia9, two copies in the moss Physcomitrella, three copies in the genome of the lycophyte Selaginella moellendorffii and six copies in most diploids (such as Arabidopsis, tomato, and melon) and higher copies in polyploids angiosperm. So far, little is known about the molecular function for WIP proteins and no transcriptomics analysis have been done that allows identification of the main gene targets of this family of transcription factors.
As melon is not a practical model system to investigate the molecular mechanisms underlining the function of a given transcription factor, we used Arabidopsis thaliana to bring new insight to transcription networks recruited by orthologues of CmWIP1 in Arabidopsis. In Arabidopsis, there are six WIP transcription factor (C2H2 family, subclass A1d) family members10. TT1/AtWIP1 (TRANSPARENT TESTA 1) is expressed in the seed coat endothelium, where most pro-anthocyanins (PAs) are accumulated. Loss-of-function tt1 mutant shows yellow seeds due to a reduced PAs accumulation8. NTT/AtWIP2 (NO TRANSMITTING TRACT) is expressed in the transmitting-tract during silique development, and ntt loss-of-function mutants display a half-filled fertilized silique phenotype, due to the malformation of transmitting tract in the carpel11. A recent study has demonstrated that NTT, AtWIP4, and AtWIP5 are redundantly expressed in the hypophysis and are required for the distal stem cell fate within the root meristem12. Interestingly, the function of AtWIPs in Arabidopsis is partially conserved/overlapping as the expression of the coding sequence of any AtWIP gene under the TT1 promoter is able to restore the tt1 yellow seed phenotype10. These results suggested that all WIPs in Arabidopsis have highly conserved domains and may only differ in their spatial and temporal expression patterns. However, it is not clear whether WIP genes are functionally conserved across different plant species.
Ectopic expression of TT1 and NTT under the control of the 35S CaMV promoter showed severe phenotypic aberrations with rosette leaves deeply serrated8,13. Here, we show that 35S:CmWIP1 transgenic Arabidopsis plants have identical phenotypes, indicating a conserved growth repression role for WIP proteins. To bring new insight into how WIP proteins accomplish growth inhibition, we expressed TT1 and NTT genes using a dexamethasone inducible (Dex) system and performed Chromatin ImmunoPrecipitation with sequencing (ChIP-seq) and RNA sequencing (RNA-seq) analysis. This combined analysis allowed us to identify genes that are collectively regulated by two representative WIP proteins in Arabidopsis. Overexpression of WIP genes activated several genes involved in programed cell death and in hormone signaling related to stress responses, such as ethylene and jasmonic acid (JA), and repressed many genes involved in organ and plant development.
To test whether the WIP-mediated inhibition of growth observed in transient expression in Arabidopsis could mimics CmWIP1 inhibition of carpels in melon, we expressed TT1 under the control of a carpel specific promoter. As did CmWIP1 in melon, expression of TT1 in carpel produced male flowers. To test whether the inhibition of growth and development is a general mechanism of WIP proteins we ectopically expressed TT1 in two more flower organs, stamina and petals primordia, and in two vegetative tissues, trichrome and lateral roots. Our results showed that WIPs are able to inhibit both vegetative and reproductive organ growth. Our data also shows that several molecular pathways controlled by WIP proteins are conserved between plant species and their role is probably determined by their spatio-temporal expression during development. Our data also point toward the use of WIP proteins as a biotechnology tool to engineer male or female plants in other species.
Results
Functional annotation of WIP genes in Arabidopsis and Melon
To better understand the molecular conservation of WIP proteins, we first examined the protein sequences and the expression profiles of WIPs in Arabidopsis (Supplementary Figs. 1a and 2). A phylogenetic tree analysis revealed that TT1/AtWIP1 clustered together with AtWIP3, AtWIP6, whereas NTT/AtWIP2 clustered together with AtWIP4 and AtWIP5 (Fig. 1a). To investigate the conservation of WIP genes between Arabidopsis and melon, we searched, by BLAST protein, the recently sequenced Cucumis melo L. genome14, for sequences orthologues to AtWIPs. This resulted in the identification of six CmWIP genes which were further analyzed according to their gene structures. As in Arabidopsis, melon WIP genes are composed of two exons, a N-terminal region with two conserved motifs of unknown function (n1 and n2), and a highly conserved C-terminal region on which four zinc fingers (ZFs) and nuclear localization signals (NLSs) are located (Supplementary Fig. 1b). Alignment analysis showed that two WIP proteins from melon (MELO3C004740 and MELO3C003009) were clustered with TT1, whereas CmWIP1 and three others related proteins (MELO3C009657, MELO3C023544, and MELO3C009030) were part of the NTT clade (Fig. 1a).
Fig. 1. WIP transcription factors in Arabidopsis and Melon.

a Phylogenetic tree with the six WIP genes from Arabidopsis and melon. The closest homologue of CmWIP1 in Arabidopsis is NTT, involved in transmitting tract development in Arabidopsis. NTT together with WIP4 and WIP5 are required for initiation of the root meristem. b Rosettes and leaves of WT Col-0 and 35S:CmWIP1 transgenic lines, 35S:CmWIP1 shown serrated leaves, as previously described in 35S:TT1 and 35S:NTT8,13. Scale bar = 0.5 cm. c Vanillin staining of Col-0, tt1 mutant, and transgenic immature seeds using TT1 and CmWIP1 under the control of TT1 promoter (pTT1) to complement yellow seeds phenotype of tt1 mutants. Scale bar = 20 µm. d Quantification of pro-anthocyanidins (PA) in Col-0, tt1 mutant, and transgenic mature seeds. Bars indicate standard errors of two biological repeats and three technical repeats. Asterisks show significant differences compared to tt1 mutant (Student t-test, p < 0.05, n = 6 biologically independent samples, each corresponding to a pool of 10 mg mature seeds).
To test whether WIP proteins are functionally conserved between melon and Arabidopsis, we first overexpressed CmWIP1, under the control of cauliflower mosaic virus 35S promoter (35S), in Arabidopsis. The obtained transgenic plants (35S:CmWIP1) strongly expressed CmWIP1 and phenocopied plants overexpressing TT1 (35S:TT1) and NTT (35S:NTT) (Supplementary Fig. 3a–c)8,13. 35S:CmWIP1 transgenic plants also displayed smaller rosettes, serrated leaves, dwarf plants, defective floral organs, and reduced plant fertility (Fig. 1b). Root elongation, internode, and silique sizes were also all reduced compared to WT Col-0 plants (Supplementary Fig. 3d–g). This similarity in the observed phenotypes suggests that common molecular processes are initiated by CmWIP1 in Arabidopsis compared to its orthologues TT1 and NTT.
It has been previously demonstrated that all AtWIP genes are able to restore pro-anthocyanins (PA) accumulation in the seed coat of tt1 mutant by using a promoter swap approach10. To test whether CmWIP1 is able to fulfill the function of AtWIPs in seed coat development, we expressed the coding sequence of CmWIP1 under the TT1 promoter (2 Kb) in the tt1 mutant (Supplementary Fig. 4a). As a control, we expressed coding sequence of TT1 under its own promoter (2 kb) in tt1 mutant. Two independent transgenic lines that showed a high CmWIP1 expression, were further characterized (Supplementary Fig. 4b). We analyzed the accumulation of PA in seed coat by vanillin staining in immature seeds (Fig. 1c) and cyanidin quantification from the mature seeds (Fig. 1d) for each transgenic line. Similar to the WT control, PA accumulation was fully complemented by pTT1:TT1 construct in tt1 mutant. Seeds from tt1 lines complemented by the pTT1:CmWIP1 construct showed a significant increase of PA accumulation when compared to tt1 mutant; however WT levels were not attended. Similar complementation levels of the tt1 lines were obtained by Appelhagen et al.10 when using pTT1:NTT. Taken together, our results demonstrate that WIP proteins share both conserved protein structure and function, between melon and Arabidopsis. Thus, characterization of WIP proteins in Arabidopsis will likely bring new insight in the function of WIP proteins in melon.
Overexpression of AtWIPs trigger senescence and cell death
The cell-type-specific and transient expression pattern of WIP genes in Arabidopsis (TT1 in endosperm cells of the seed and NTT in transmitting tract and quiescent center cells of root tips) may be a technical limitation to explore their molecular function. Furthermore, expression of WIP genes under the control of the 35S promoter led to recovery of lines with strong growth inhibitions that correlate with expression of the transgene. Inducible gene expression systems permit synchronizing target gene expression at particular developmental stages and in particular tissues. Hence, we generated transgenic lines that express TT1 and NTT under the control of dexamethasone-inducible (Dex) system (Supplementary Fig. 5a, b)15. To test the transient expression of TT1 and NTT on germination, seeds of Dex:NTT and Dex:TT1 lines were sown on MS medium containing 1 µM of Dex, and compared to the control Dex:GR lines (only 35S:LhGR vector). At this concentration germinated seedlings, of both Dex:NTT and Dex:TT1 lines, stop growing once radicle and cotyledons emerged whereas the control Dex:GR line germinate and grow normally (Fig. 2a). However, in contrast to Dex:NTT lines, Dex:TT1 lines displayed a more severe growth inhibition. This difference in the severity of growth inhibition, observed in all generated independent transgenic lines following Dex-treatment, could be explained by 3-fold more accumulation of TT1 mRNA compared to NTT (Fig. 2b).
Fig. 2. Overexpression of TT1 and NTT induces growth arrest and leaf senescence.

a Phenotype of control (Dex:GR), Dex:TT1, and Dex:NTT transgenic lines grown on MS medium containing 1 µM of dexamethasone (Dex), scale = 0.5 mm. b Expression of TT1 and NTT in Dex:TT1 and Dex:NTT transgenic lines, respectively, relative to expression in Dex:GR after Dex induction (1 µM) during 8 h. Phenotype of 5-week-old Dex:TT1 (c) and Dex:NTT (d) plants 4 days after spray with different concentrations of dexamethasone (1 nM to 1 µM) and compared to Dex:GR control plants (scale = 3.5 cm). Ion leakage measured on Dex:TT1 (e) and Dex:NTT (f) at different time points on leaf disks of plants sprayed with 1 µM of Dex (compared to corresponding Dex:GR). Asterisks show significant differences between overexpressing lines and controls (Student t-test, *p < 0.05, **p < 0.005, n = 4, independent treated plants, from each plant three leave disks were pooled for each time point).
To investigate the effect of TT1 and NTT expression on growth at later developmental stages, 5-week-old plants were sprayed with different concentrations of Dex, from 1 nM to 1 µM. Plant phenotypes were evaluated 4 days after treatment. Cell death and leaves senescence were observed in Dex:TT1 lines even at very low concentration of Dex (50 nM) (Fig. 2c). In contrast, Dex:NTT plants showed leaves senescence only at 1 µM Dex treatment (Fig. 2d). No phenotypic changes were observed on the controls plants (Dex:GR) sprayed with 1 µM of Dex, neither on Dex:TT1 and Dex:NTT plants treated with low concentration of Dex (1 nM).
Cell death and senescence is characterized by cell membrane damage and electrolyte leakage16. To further test the effect of WIPs on cell death and senescence, leaves disks of Dex:TT1, Dex:NTT, and Dex:GR were collected after spray with 1 µM of Dex, and the changes of ion leakage were monitored at different intervals. The electrolyte leakage (K+ efflux from plant cells) was measured in the suspension solution (water) at 1, 2, 4 and 24 h after Dex treatment. The obtained results indicate that overexpression of both TT1 and NTT significantly increased ion leakage. However, the ion leakage in the Dex:TT1 plants is much higher compared to Dex:NTT (Fig. 2e, f) and confirm the more severe leaf senescence phenotype observed in Dex:TT1 plants. Taken together, these results suggest that TT1 and NTT are powerful transcription factors controlling plant growth and cell death.
Genome-wide identification of WIP binding sites
To identify binding motifs and potential target genes of WIP transcription factors, we generated Dex:TT1-GFP or Dex:NTT-GFP transgenic plants and performed ChIP-seq analysis. Following Dex treatment, Dex:TT1-GFP and Dex:NTT-GFP transgenic lines accumulate TT1 and NTT mRNA transcript as well as a strong GFP fluorescence signal (Supplementary Fig. 5c–e). Moreover, induced Dex:TT1-GFP and Dex:NTT-GFP lines show similar growth arrest phenotypes than lines without the GFP-tag (Fig. 2a) confirming that the GFP tag do not impair the TT1 or NTT function. Immunoprecipitation was done using an anti-GFP antibody on 7 days old seedlings treated with 1 µM Dex during 8 h. High-depth sequencing of the Illumina ChIP-seq libraries were generated and mapped (Supplementary Figs. 6a and 7). Peaks were distributed across the whole genome but strongly enriched in genic regions, especially in promoters, for both TT1 and NTT, comparing to input (Fig. 3a, b and Supplementary Fig. 8a). Most of the peaks were also overlapping and located around the transcription start sites (TSS) (Fig. 3c). Analysis of the ChIP-seq data led to the identification of 7349 and 8847 peaks associated with TT1 and NTT binding, respectively (Fig. 3d). To validate the ChIP-seq data we analyzed a common tagged gene of TT1 and NTT using ChIP-PCR, WSIP2 (WUS-INTERACTING PROTEIN 2). Consistent with the ChIP-seq data, specific enrichment was obtained with primers at the ChIP-seq peak (Supplementary Fig. 8b, c). Comparative analysis of TT1 and NTT peaks led to the identification of 5914 genes with overlapping binding sites (Fig. 3d and Supplementary Data 1). The strong overlap between TT1 and NTT target genes (80 and 67%, respectively) confirms the robustness of the ChIP-seq data and point out toward TT1 and NTT conserved functions. This is consistent with NTT and other WIPs complementing tt1 mutant (Fig. 1c)10. This is also in line with the WIP overexpression lines sharing similar phenotypes (Fig. 2a) and the reported WIP functional redundancy (NTT, WIP4, and WIP5)12.
Fig. 3. Genome-wide identification of WIPs binding sites.

a Comparison between TT1-GFP and NTT-GFP peak density in the region ±2 kb around the Transcription Start Site (TSS). b Representation of the distribution of TT1-GFP and NTT-GFP binding peaks identified by ChIP-seq in four different genomic regions (intergenic, promoter, exon, intron). Peaks between −1000 bp and the TSS were considered as located on the promoter region. Peaks located on 5′UTR and 3′UTR regions were considered as part of the exon of the corresponding gene. The percentages of binding sites on each region are indicated. c Density plot showing overlap of Dex:TT1 and Dex:NTT using hexagonal binning routine. Each point represents the distance of the nearest peak to the TSS of the corresponding gene. d The Venn diagram shows the overlap of TT1-GFP and NTT-GFP ChIP-seq target genes.
A motif search analysis on the common binding sites of TT1 and NTT using Homer software17 was then performed. Top three most significant binding motifs were identified as GAGA-repeat, bZIP (ABI5), and W-box (WRKY) motifs (Supplementary Fig. 8d). Interestingly a binding motif, targeted by a third WIP protein, AtWIP5, identified by DNA affinity purification sequencing (DAP-seq), was also listed among them on fifth position (Supplementary Fig. 8e)18.
Identification of the early target genes of WIP proteins
To determine the gene regulatory networks recruited by WIP proteins to control growth, 7 days old seedlings of Dex:TT1, Dex:NTT, and the control line, Dex:GR, were treated with 1 µM Dex and used for gene expression analysis. High-depth sequencing of the Illumina RNA-seq libraries were generated and mapped. Normalized reads counts were analyzed using DEseq2 package (Supplementary Figs. 6b and 7). As expected, the expression of TT1 and NTT was strongly induced after Dex treatment in the Dex:TT1 and Dex:NTT lines, respectively, confirming the efficiency of the Dex treatment (Supplementary Data 2). Furthermore biological replicates formed distinct clusters from the control samples in the PCA-plot (Supplementary Fig. 9a).
Statistical RNA-Seq analysis identified 3538 and 3188 differentially expressed genes (DEGs) in Dex:TT1 and Dex:NTT, respectively (FDR < 0.05, FC log2 ± 2.0). RT-qPCR confirmed the expression level of selected DEGs, showing that our RNA-seq data were reliable (Supplementary Fig. 9b). To identify co-regulated genes we compared the DEGs from both RNA-seq data. We found 1363 DEGs sharing similar differential expression profiles in Dex:TT1 and Dex:NTT lines compared to Dex:GR control lines. Out of this, 761 genes were up-regulated and 602 genes were down-regulated on both lines. Interestingly, some genes (40 + 23) showed antagonistic expression in TT1 and NTT (Supplementary Fig. 9c and Supplementary Data 2).
In order to identify the set of primary WIP targets, we screened the DEGs for association with TT1 and NTT ChIP-Seq peaks. We observed significant overlap between those two different gene sets (Fig. 4a, b and Supplementary Data 3). Among the putative TT1-direct targeted genes, a slight higher number of genes were up-regulated (872, 24% of total DEG) comparing to those down-regulated (687, 19% of total DEGs); whereas on NTT, targeted genes were slightly more down-regulated (870, 27% of total DEGs) comparing to up-regulated genes (701, 21% of total DEGs). All together this data suggest that WIP proteins can act as a transcriptional activator or repressor.
Fig. 4. Integration of WIPs ChIP-seq and RNA-Seq data.

a The Venn diagram shows the overlap between targets and differential expressed genes identified after induction of TT1 with Dex. b The Venn diagram shows the overlap between targets and differential expressed genes identified after induction of NTT with Dex. c GO term enriched clustering analysis representing classification of genes directly targeted by TT1 and NTT depending on their expression profiles (up- or down-regulated). Color intensities represent significant values of each GO term. d Most highly enriched DNA-element identified in TT1 and NTT up-regulated target genes. e Most highly enriched DNA-element identified in TT1 and NTT down-regulated target genes. Representative binding profiles based on the IGB (Integrated Genome Browser) for Dex:TT1-GFP (green) and Dex:NTT-GFP (red) ChIP-seq data for selected genes whose expression was induced (f) or repressed (g) in response to Dex. A cartoon of JAZ5, JAZ8, ABO3, KAN, ULT1, and BLH4 genes is shown below each panel, with boxes corresponding to exons and bars to intron. Y-axis corresponds to the height of the peak scaled to different values for each gene. The bars at the bottom of the peaks represent the motif locations determined by MACS2. WRKY and GAGA motifs are present on the promoter and exon regions of represented genes (blue and red boxes, respectively). Coordinates of each locus on the TAIR10 Arabidopsis Genome is indicated.
To further investigate the main biological process in which WIP proteins are involved, we first performed Gene Ontology (GO) analysis, on shared DEGs that were also bound by TT1 and NTT. Consistent with WIP proteins sharing conserved function, most of the enriched GO terms, identified using clusterProfiler19, were collectively common, in the TT1 and NTT-up and in the TT1 and NTT-downregulated genes lists, respectively (Supplementary Data 4). GO terms related to meristem growth and maintenance, cell differentiation, ion homeostasis, and response to brassinosteroids were collectively overrepresented in the TT1 and NTT downregulated genes list (Fig. 4c). In contrast, GO terms related to pathogen resistance, including cell death, immunity, leaf senescence, and stress responses to hormones such as ethylene, JA, and salicylic acid (SA) were collectively overrepresented in the TT1 and NTT up-regulated genes list (Fig. 4c). We also performed GO analysis on TT1 and NTT-specific gene list. Consistent with the analysis of the shared gene list, all the downregulated genes were enriched in GO terms related to development and the upregulated genes were enriched in genes related to defense (Supplementary Fig. 10). For instance, GO terms related to seed dormancy process, plant ovule development, and adaxial/abaxial axis specification were significantly enriched in the TT1 down-regulated genes list (e.g., AGAMOUS-LIKE 14, AGL14; PETAL LOSS, PTL). On the other site, NTT down-regulated genes list was enriched in terms that relate to trichoblast differentiation, pigment accumulation and response to auxin (e.g., LEUCINE-RICH REPEAT/EXTENSIN 1, LRX1; AUXIN-REGULATED GENE INVOLVED IN ORGAN SIZE, ARGOS; PIN-FORMED 3 and PIN7).
Collectively, the expression profiles and GO analysis of TT1 and NTT targets point out on the role of WIP TFs in repressing development and growth by inducing plant defense response via hormonal variations.
WRKY and GAGA motifs are predominant in WIP target genes
Next we performed a motif search analysis on the binding sites of genes regulated by TT1 and NTT (combined ChIP-seq and RNA-seq analysis). Strikingly, we found that TT1 and NTT occupy overlapping binding sites, at probability far greater than what could have occurred by chance. The majority of the up-regulated targeted genes bound by TT1 (37%) and NTT (22%) were found to contain a TTGACY W-box (WRKY) motif with the statistical p-value of 1e−39 and 1e−25, respectively (Fig. 4d). W-box motifs are known targets of WRKY TFs, associated with stress and hormone signaling. As WRKY proteins, TT1 and NTT are also able to induce JA responsive genes such as JASMONATE-ZIM-DOMAIN (JAZ) PROTEIN 5 and JAZ8 by recognizing W-box motif in their promoters (Fig. 4f)20. WIP proteins also bind to the promoters of several WRKY genes such as ABA OVERLAY SENSITIVE MUTANT 3 (ABO3), which contain also W-box motifs that allow to autoregulate its own expressions, via a positive feedback loop21.
In the other hand, the majority of the down-regulated targeted genes bound by TT1 (27%) and NTT (33%) contains GAGA-repeat motifs with the statistical p-value of 1e−31 and 1e−60, respectively (Fig. 4e). GAGA-motif binding factors (GAFs) could be divers, Trithorax-like and Pipsqueak protein families in animals, BASIC PENTACYSTEINE (BPC) protein family in plants, but all have a central function in growth and development22–24. It is also intriguing, that both animal and plant GAFs recruit members of the Polycomb Repressive Complex (PRC) to the Polycomb repressive DNA-elements (PREs) to silence the bound genes23,25. Similarly, genes involved on developmental process, such as KANADI (KAN), ULTRAPETALA 1 (ULT1), and BEL1-LIKE HOMEODOMAIN 4 (BLH4) were bound by TT1 and NTT, suggesting that WIPs act as transcriptional repressors through recognition of GAGA-motifs on their promoters (Fig. 4g).
In sum, integration of RNA-seq and ChIP-seq analysis reveals the interaction of WIP TFs with GAGA motif to repress the expression of growth and development promoting genes, and interaction with W-box motif to activate expression of genes associated with stress signaling pathways.
Ectopic expression of TT1 in Arabidopsis carpel leads to male flowers
To test the hypothesis that WIP proteins are general inhibitor of growth we ectopically expressed WIP proteins in two reproductive tissues, the carpel and the stamina primordia, and in two vegetative tissues, the trichome and lateral root primordia. Transgenic plants were generated and analyzed for organ growth inhibition.
We ectopically expressed TT1 in Arabidopsis carpel primordia using the tissue-specific CRABS CLAW (pCRC) promoter. CRC is a member of the YABBI family required for carpel development in Arabidopsis and its expression starts at stage 6 when the gynoecium primordium becomes distinct, disappearing at stage 10–1126. We obtained several independent pCRC:TT1 transgenic lines developing normal sepals, petals and stamens but with rudimentary arrested carpels (Fig. 5a, b and Supplementary Fig. 11a). Toluidine blue staining and scanning microscopy analysis showed normal development of carpel primordia at early developmental stages (stage 5), but from stage 8 carpel morphology was affected in pCRC:TT1 lines, given place to a rudimentary structure at later stages (stage 11), which finally leads to female-sterile flowers that mimic unisexual male flowers in melon (Fig. 5d, e and Supplementary Fig. 12a–h). Quantification of TT1 expression in young flowers of pCRC:TT1 transgenic lines correlated with the inhibition of carpel growth (Supplementary Fig. 11a).
Fig. 5. Ectopic expression of TT1 in reproductive and vegetative organs of Arabidopsis.

Phenotype of Col-0 (a), pCRC:TT1 (b), and pAP3:TT1 (c) transgenic flowers (scale = 0.5 mm). Toluidine blue staining of Col-0 (d), pCRC:TT1 (e), and pAP3:TT1 (f) flowers at late developmental stage (stage 10). Rudimentary carpel on pCRC:TT1 is designed with dotted line (s = sepals, p = petal, c = carpel, st = stamen; scale = 200 µm). Phenotype of Col-0 (g) and pGL1:TT1 (i) transgenic plants, which shows serrated leaves. Electronic microscopic analysis of leaves surfaces of Col-0 (h) and pGL1:TT1 (j) which shows reduced number of trichomes (scale = 2 mm). k Phenotype of Col-0 and pSLR:TT1 transgenic plants (scale = 1 mm). l Quantification of primary root length and lateral root density in Col-0 and pSLR:TT1 transgenic lines (n = 8, biologically independent seedlings).
To test whether ectopic expression of WIP gene can inhibit the development of other flower organs, we expressed TT1 under the control of APETALA3 promoter (pAP3). AP3 is a B class floral homeotic gene specifically expressed in the petal and stamens27. Again, in pAP3:TT1 lines a severe growth inhibition of stamen and petals was observed (Fig. 5a, c). Toluidine blue staining and scanning microscopy analysis showed that both petal and stamen development were completely inhibited in the pAP3:TT1 lines (Fig. 5d, f and Supplementary Fig. 11i–l). As expected, inhibition of growth correlated with the expression of TT1 in flowers of pAP3:TT1 transgenic lines (Supplementary Fig. 11b).
To test whether ectopic expression of WIP gene can also inhibit the development of vegetative tissues, we expressed TT1 under the control of the GLABRA-1 (pGL1) and the SOLITARY ROOTS (pSLR) promoters (Supplementary Fig. 11c, d). GL1 is a R2R3 MYB-type transcription factor expressed in young leaf primordia and developing trichomes28. Loss of function of GL1 leads to leaves without trichomes. Independent pGL1:TT1 transgenic lines were generated and all displayed serrated leaves with few or no trichomes (Fig. 5g–j). Inhibition of trichomes development also correlated with the expression of TT1 in leaves of pGL1:TT1 transgenic lines (Supplementary Fig. 11c). SOLITARY ROOTS gene encodes the IAA14 protein, a member of the Aux/IAA gene family that promotes lateral roots formation29. Loss of function of SLR leads to mutants that completely lack lateral roots and reduced root gravitropism. Three independent pSLR:TT1 transgenic lines were generated and none of them showed lateral root development after 4 weeks. As expected primary root elongation in the pSLR:TT1 lines was not affected (Fig. 5k, l). As for leaf phenotype and trichomes inhibition, lateral roots inhibition correlated with the expression of TT1 in lateral root primordia of pSLR:TT1 transgenic lines (Supplementary Fig. 11d).
In conclusion, our results demonstrate that TT1/AtWIP1 transcription factor could act as strong suppressor of organ development, as did CmWIP1 in carpel primordia of male melon flowers6. These data also suggest that WIP proteins could be recruited to inhibit growth of both vegetative and reproductive organs.
Discussion
WIP genes, C2H2 ZF transcription factors have different roles in land plants. In Marchantia polymorpha, MpWIP controls the development of the air pore complex9. In Brassica napus, BnTT1 is involved in the flavonoids and fatty acid biosynthesis30. In Gerbera hybrida, GhWIP2 controls petal and petiole’s growth via regulation of cell expansion31. In C. melo, CmWIPl is a central integrator of expression networks leading to sex determination. Expression of CmWIP in carpel primordia inhibits carpel development, leading to unisexual male flowers6. In Arabidopsis, there are six WIP genes which show a strong amino acid conservation and were found to be functionally redundant10. WIP genes in Arabidopsis are involved in the accumulation of PA in the seed coat, development of replum and transmitting tract; and root meristem development10–12.
Based on their sequence homology, we hypothesized that WIP genes might show conserved function across species and using a heterologous system might help to understand the molecular background of carpel arrest regulated by CmWIP1 in the non-model species, melon. We observed a significant restoration of the accumulation of PA in tt1 mutants by expressing CmWIP1, suggesting that basic molecular functions are shared between these proteins (Fig. 1). Partial restoration was also obtained by Appelhagen et al.10 in some cases (promTT1:NTT and promTT1:AtWIP3), however PA accumulation was less than 50% compared to WT seeds.
The transient and divers spatio-temporal expression patterns of WIP proteins during development have been a handicap for the characterization of the gene networks involving WIP proteins. Our results using an inducible promoter system, allowed us to synchronize the expression of two WIP proteins, NTT and TT1, in a particular tissue. Phenotyping of the Dex lines at seed germination and seedling stages demonstrated that WIP proteins are strong inhibitors of growth and triggers of cell death.
To identify gene regulatory networks recruited by WIP proteins we used RNA-seq and ChIP-seq analysis. By search for enriched GO terms in TT1 and NTT data, we found several genes, associated with hormones stress signaling, up-regulated. This includes ethylene, ABA and JA signaling pathways, showing that hormone crosstalk mechanisms may be timely orchestrated by WIP proteins to limit defense-associated fitness costs. In contrast several genes involved in auxin transport (PIN3, PIN4, and PIN7) and gibberellins were downregulated by WIP. In a recent work, it has been shown that constitutive overexpression of GhWIP2 (homologue of NTT in G. hybrida) caused major developmental defects associated with cell expansion including dwarfism, short petals, and petioles31. These phenotypes were also correlated with downregulation of gibberellins and auxin signaling genes and upregulation of abscisic acid signaling genes in GhWIP2 overexpressing plants. All findings on different plant species converge into a conserved function of WIP proteins in integrating contrasting hormonal signals to regulate organ growth.
ChIP-seq analysis allowed us to identify two regulatory elements, W-box and GAGA motif, as binding sites of WIP transcription factors. Further integration of ChIP-seq, RNA-seq, and GO analysis revealed how WIP proteins could play a role as activators or repressors of transcription. We showed that WIP interact with GAGA motif to repress the expression of the growth and development promoting genes. It will be interesting to see whether WIP proteins also interact with of the Polycomb Repressive Complex to silence the bound genes, as described for GAFs in plants and animals23,25. W-box motif, known target of WRKY proteins that controls the expression of genes involved in defense-related phytohormones, such as SA or JA, imply a similar role for WRKY and WIP genes in plant immunity (Fig. 6). From this we can also hypothesize that CmWIP1 in male flowers inhibits carpel development, through interaction with GAGA and W-box motifs. As melon is recalcitrant to genetic transformation it was difficult to tag CmWIP1 protein with an epitope that could be used for immunoprecipitation, required for ChIP-seq analysis. In silico screening of the promoter of genes differentially expressed between male and female flower, for the presence of GAGA and W-box motifs will be one way to identify CmWIP1 direct targets. Such targets could be further analyzed for differential expression in gynoecious plant compared to androecious plants and validated using genetic approaches.
Fig. 6. Schematic model of how WIP TFs control plant growth.
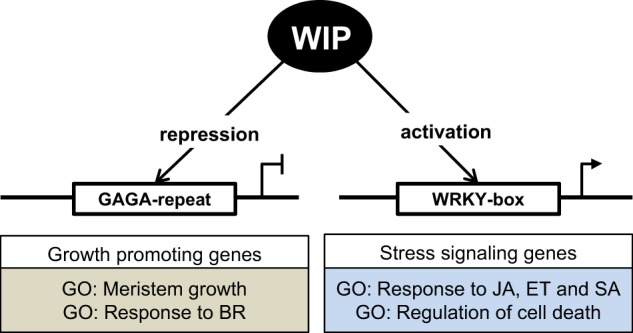
WIP TFs bound GAGA motif to repress the expression of the growth and development promoting genes, and W-box motif to activate expression of genes associated with stress signaling pathways.
In melon and related Cucurbitaceae species, CmWIP1 and two ethylene biosynthesis limiting enzymes, CmACS11 and CmACS7 play a central role in sex determination. Expression of CmACS7 requires carpel development and inhibits stamina development, leading to female flowers5. Expression of CmWIP1 is under the negative control of ethylene produced by CmASC11 in flowers developing in young vines. The non-expression of CmACS11 permits the expression of CmWIP1 which inhibits the carpel development, leading to male flowers. To test whether expression of WIP proteins can also lead to male flowers in Arabidopsis, we disconnected the control of the expression of the WIP protein of the ethylene signaling, using CRC carpel specific promoter. In melon, sex determination occurs at stage 4–6 of flower development4. The choice of CRC to drive the expression of WIP protein in Arabidopsis is justified by its early expression during carpel development, stage 6, and because it is very specific for nectary and carpel development. When TT1 was ectopically expressed under CRC promoter in carpel primordia in Arabidopsis, it led to male flower formation, mimicking CmWIP1 function in melon.
Interestingly, ectopic expression of TT1 in stamina primordia, as well as in trichomes and lateral roots, demonstrated that TT1 inhibition of growth is not only confined to the carpel, but was also to other reproductive and vegetative organs. In contrast, it was previously shown that ectopic expression of NTT under the control of the ASYMETRIC LEAVES 1 (AS1) promoter promote formation of roots from cotyledons primordia instead of causing growth arrest as we observed with TT112. Whether, TT1 and NTT have different functions or the level of expression obtained by the chosen promoters (extremely low or extremely high) is different between those experiments, we may consider WIP genes as regulators of cell fate and differentiation. Indeed, in our RNA-seq data, we have identified a set of genes that are significantly different with only one of the AtWIP, suggesting that those genes have contributed to acquire specific roles of TT1 and NTT during evolution. For instance, several genes involved in cytokinin pathway showed opposite behavior after TT1 and NTT induction.
Our results have highlighted the evolutionary biology of WIP genes. Throughout the species, WIPs genes tend to shape development through several pathways. As we have observed, following expression of WIP proteins, up-regulation of stress inducible genes and down-regulation of growth and development associated genes, this study has initiated another debate over the complex function of WIPs. This study has also highlighted the role of WIP proteins in crosstalk between hormones associated with stress and the one associated with growth. The difference of genetic regulation of the WIP transcription factors emphasizes on the difference of their particular role in the plant development. A substantial work is still required to complete the puzzle of functional characterization of the WIP transcription factors in the plant kingdom. Still because we were able to engineer unisexual flowers in Arabidopsis, using discoveries in melon, our work highlight how the WIP proteins could be used as biotechnology tool to control crossing and production of F1 hybrids seeds in general.
Methods
Plant material
Arabidopsis (A. thaliana) genotypes (accession Columbia-0, Col-0) were grown under a 16-h-light/8-h-dark condition at 22 °C in a growth room. For seed germination, sterilized seeds were incubated at 4 °C for 2 days and sown on Murashige and Skoog (MS) medium plates containing 1% sucrose and 0.8% agar. tt1-3 mutants (SALK_026171) (Col-0 background) were previously characterized10.
Plasmid construction and Arabidopsis transformation
To construct the binary vector 35S:CmWIP1, CmWIP1 cDNA from C. melo was amplified with iProof® DNA polymerase (Bio-Rad) and cloned into pDONR-207 and then inserted in the binary vector pGWB2 using the Gateway cloning system with LR clonase II and transformed in Arabidopsis using Agrobacterium tumefaciens strain C58C1. Primers used are listed in Supplementary Data 5.
For both, the promoter swap and the ectopic expression experiments, the protocol used was the following: 2 kb of TT1 promoter, 727 bp of AP3 promoter, 3.8 kb of CRC promoter, 2 kb of SLR promoter, and 1.5 kb of GL1 promoters were amplified using genomic DNA as template. For all of them, PCR was done with a reverse primer having SpeI restriction site. After digestion with SpeI (New England, Biolabs) the promoters were cloned into pIJPB binary vector32 and confirmed by sequencing. The AtTT1 CDS was amplified with iProof® DNA polymerase (Bio-Rad) from cDNA and cloned into pDONR-207. AtTT1 or CmWIP1 CDS were then introduced into the pIJPB plasmid by Gateway® system (Invitrogen™). Transformation of Arabidopsis was done using A. tumefaciens strain C58C1 by floral dip method. Primers used for cloning and validation of these lines are listed in Supplementary Data 5.
Dex:TT1 and Dex:NTT constructs were produced as follows: TT1 and NTT CDS were cloned into the donor vector (pDONR221) (Life Technology). TT1-GFP and NTT-GFP sequences were generated by chimeric PCR and then cloned into the donor vector. TT1, NTT, TT1-GFP, and NTT-GFP were then introduced into the Gateway destination vector pEN-L4-pOp6M2-R1 which contain the pOp6 promoter that carry six copies of a lac operator sequence33. pOp6:TT1, pOp6:TT1-GFP, pOp6:NTT, and pOp6:TT1-GFP constructs were transformed in Arabidopsis seedlings carrying the 35S::LhG4 vector and T1 generation seeds were selected for Basta and Kanamycin resistance. Primers used for cloning and validation of these lines are listed in Data S5.
Phylogenetic analysis
Phylogenetic analysis of WIP proteins from A. thaliana and C. melo was performed using the software MEGA (Molecular Evolutionary Genetic Analysis; version 7.0.20)34. Sequences were aligned with ClustalW method, and phylogenetic tree was obtained by neighbor-joining (NJ).
Conductivity assays
Senescence and associated cell death were estimated as previously described35, with minor modifications. Briefly, ion leakage was measured from 5-week-old plants of Dex:TT1, Dex:NTT, and control (Dex:GR) sprayed with different concentrations of Dex (0.001, 0.05, 0.1, and 1 µM). Three leave disks were collected after 24 h treatment and placed in 2 mL sterile water at room temperature and conductivity (Ci) was measured at 0, 1, 2, 4, and 24 h using an ion conductivity meter (B-173, Horiba, Kyoto, Japan). To calculate final electrolyte conductivity (Ct), leave disks were then placed at −20 °C for 1 h, and at 70 °C for 3 h to induce 100% ion leakage. Relative ion leakage was calculated for each time point and expressed as a percentage of the total ion leakage (% = Ci/Ct × 100).
Vanillin staining and proanthocyanidins quantification
Immature seeds (heart stage of embryogenesis) of Arabidopsis were stained with 1% vanillin (4-Hydroxy-3-methoxybenzaldehyde) (Sigma-Aldrich) in 6 N Hydrochloric acid for 30 min and observed with Leica macroscope. PA quantification was carried out according to Appelhagen et al.10. Approximately 10 mg of mature seeds were grinded with 200 μL of acidic methanol. 1200 µL acidic BuOH was added followed by 40 µL “ferric reagent” mix. Tubes were boiled at 95 °C for 20 min and then cooled for 5 min on ice. Spinning was done at 13,000 rpm for 4 min. Supernatant was used to measure the UV absorption (550 nm) and compared to Cyanidin Chloride standard (Sigma-Aldrich) curve for quantification.
ChIP-seq and ChIP-PCR analysis
ChIP assays were performed as described by Jégu et al.36. Briefly, 7 days old seedlings of Dex:TT1-GFP and Dex:NTT-GFP lines were transferred to MS liquid medium supplied with 1 µM Dex. After 8 h treatment, seedlings were crosslinked with 1% formaldehyde and the reaction was stopped with glycine (2 mM). Chromatin was sonicated to an average length of 300 bp and immunoprecipitated (overnight at 4 °C) with an anti-GFP antibody (Clontech 632592). The chromatin was then reverse-crosslinked (RNase and proteinase K digestion) and DNA was extracted with phenol-chloroform. Precipitated DNA (10 ng of IP or input DNA) was used for ChIP-seq library construction using a NEBNext® Ultra DNA Library Prep Kit for Illumina®. Single-end sequencing of IP and input samples were performed using GAIIx Illumina® (read length of 50 bp). In total six libraries were sequenced, one for Dex:TT1-GFP and one for its input and two for Dex:NTT-GFP samples and two for its corresponding inputs. Reads were mapped with BWA (Burrows–Wheeler Aligner) onto Arabidopsis genome release 43 from Ensembl database37, then only reads with mapQ > 30 and uniquely mapped were kept using Samtools (http://www.htslib.org/). Reads identified as PCR duplicates were discarded using Picard Toolkit (https://broadinstitute.github.io/picard/).
The MACS2 (version 2.1.2) software was used to identify significantly enriched regions (q-value < 10e−10)38. Visualization and analysis of genome-wide enrichment profiles were done with IGB (Integrated Genome Browser, https://bioviz.org/). Peak annotations were assigned using HOMER, keeping only peaks in an interval of [−1500; +150] around the TSS (Transcription Start Site). Heatmaps were drawn using deeptools software39; hexbin plot with R40 and ggplot2 library41. Putative binding peaks obtained from ChIP-seq analysis were confirmed by ChIP-qPCR assays using anti-GFP (Clontech 632592) and anti-IgG antibody (Millipore) antibodies. The primers used for ChIP-qPCR assays are listed in Supplementary Data 5.
RNA-Seq analysis
Seven days old seedlings of Dex:TT1, Dex:NTT, and Dex:GR lines were treated with Dex (1 µM during 8 h), then grinded and total RNA was extracted with RNAeasy plant kit and treated with DNaseI (Qiagen, Germany). Eight samples were used for library preparation (two biological replicates per genotype), comprising Dex:TT1 and its corresponding control (Dex:GR) lines, and Dex:NTT and its corresponding control (Dex:GR) lines. Multiplex Illumina sequencing using DNA barcoding libraries were prepared using the TruSeq RNA Library Preparation Kit Illumina® according to the manufacturer’s recommendations. Libraries were sequenced on the HiSeq2000 platform Illumina®, and between 30 and 50 million read pairs per sample were obtained. Quality was assessed using FastQC (version 0.11). Tophat2 (version 2.1.0) has been used to generate the mapping files and RSeQC aligned reads above 94% of read pairs of each sample correctly to TAIR (version 10). The mapped reads were assigned to genes with featureCount (v1.5.0-p3). DESeq2 (version 1.10.1) analysis was employed for differentially expressed gene calling (p-value adjusted > 0.05 and log2 FC < −2 and >2).
GO analysis, motif search, and gene network construction
Motifs search was performed by HOMER with TAIR10 motif database17. GO enrichment analysis was carried out with Rstudio (http://www.rstudio.com/) and ClusterProfiler package19. Euler diagram were computed with R and eulerr package42. Heatmap was drawn with R package Pheatmap (https://rdrr.io/cran/pheatmap/).
Microscopy
Plant tissues were fixed in the fixation buffer (2% paraformaldehyde, 1% glutaraldehyde, 1% caffeine, 0.1 M phosphate buffer, pH 7). Samples were dehydrated with 70, 95, and 100% ethanol and embedded in resin Technovit® 7100 (Kulzer). 10 μm sections were cut with a microtome Leica RM2165 equipped with cutting glass, mounted on the glass slide and stained with 0.05% (w/v) toluidine blue. Fresh leaf samples and inflorescence were observed with Tabletop SEM SH-1500MB electron microscope. Arabidopsis roots were stained with 0.1 mM propidium iodide for 15 min as previously described43 and analyzed in a Leica TCS-SP2-AOBS spectral confocal laser scanning microscope (Leica Microsystems).
Expression analysis
For RT-qPCR, total RNA was extracted with RNeasy plant kit and treated with DNase I according to the manufacturer’s instructions (Qiagen, Germany). Reverse transcription was done with SuperScript II RT (Invitrogen, Germany). Quantitative PCR was done with Bio-Rad CFX96 Real-time PCR system using SYBR® Green no ROX (Eurogentec). Relative expression for all genes was normalized to AtACT2 and as internal reference. Relative expression was calculated with the relative −ΔΔCt method. Primers used are listed in Supplementary Data 5.
Statistics and reproducibility
For phenotypic analysis, at least two independent transgenic lines for each construct were analyzed. For PA quantification, six measurements for each line were realized using 10 mg of mature seeds. For ion leakage measurements two independent experiments were performed. For each experiment, four plants per genotype and per treatment were recorded. Per time point a pool of 3 leave disks were collected per plant. T-test was calculated using the two-tailed test function in excel.
For RNA-seq and ChIP-seq, pools of 7 days old seedlings have been used as biological replicate. Pools contained around 100 seedlings grown on different Petri dishes randomly distributed in the growth chamber and treated independently with dexamethasone. Two biological replicates were performed for RNA-seq. DEseq2 algorithm was used to define the significant transcripts for each genotype (Dex:TT1 and Dex:NTT) against its corresponding controls. Three ChIP-seq experiments were performed, two biological replicates for Dex:NTT and one for Dex:TT1. Only peaks that were discovered above the background (corresponding to input) were considered for further analysis.
For RT-qPCR analysis, the same RNAs used for RNA-seq were analyzed. The expression values correspond to the average of two biological replicates, with three technical replicates. For ChIP-PCR, immunoprecipitated chromatin was prepared from new samples, and the average corresponds to three technical replicates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This work was supported by the European Research Council (ERC-SEXYPARTH, 341076), the ANR (EPISEX, ANR-17-CE20-0019), and the LabEx Saclay Plant Sciences-SPS (ANR-10-LABX-40-SPS). M.V.G.R. was supported by the Intra-European Fellowships for Career Development (IEF) (Grant PIEF-GA-2012-330908). The authors thank Ikram Blilou for fruitful discussions, the research facilities provided by the Institute of Plant-Science Paris-Saclay (IPS2, France) and the Pôle Scientifique de Modélisation Numérique of the ENS (Lyon, France) for computing resources.
Author contributions
M.V.G.R., A. Boualem, and A. Bendahmane designed the experiments and supervised the study. M.V.G.R., F.I., P.C., A.H., V.S., J.E., D.L., T.J., M.V., and J.S. performed and/or analyzed the data. M. Benhamed and M. Bendahmane helped with the interpretation of the data. M.V.G.R., A. Boualem, and A. Bendahmane wrote the manuscript. All authors revised the draft manuscript and read and approved the final manuscript.
Data availability
The raw RNA-seq and ChIP-seq data described in this study have been deposited to the NCBI Short Read Archive (SRA) database under the BioProject ID PRJNA608903. Sequence data from CmWIP1 can be found in the GenBank data library under accession number GQ870274.1. Sequence data from TT1/AtWIP1 (AT1G34790), NTT/AtWIP2 (AT3G57670), AtSTOP1 (AT1G34370), CRC (AT1G69180), AP3 (AT3G54340), GL1 (AT3G27920), and SLR (AT4G14550) can be found at The Arabidopsis Information Resource (TAIR) database.
Code availability
The code and software sources from previously published papers are listed in the “Methods”. All other details of the code used in this study are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Maria Victoria Gomez Roldan, Farhaj Izhaq.
Supplementary information
Supplementary information is available for this paper at 10.1038/s42003-020-0969-2.
References
- 1.Irish EE, Nelson T. Sex determination in monoecious and dioecious plants. Plant Cell. 1989;1:737–744. doi: 10.2307/3868981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renner, S. & Schaefer, H. Phylogeny and evolution of the Cucurbitaceae. In Plant Genetics and Genomics: Crops and Models (eds Grumet, R., Katzir, N. & Garcia-Mas, J.) vol 20 (Springer, Cham, 2016).
- 3.Kocyan A, Zhang L-B, Schaefer H, Renner SS. A multi-locus chloroplast phylogeny for the Cucurbitaceae and its implications for character evolution and classification. Mol. Phylogenet. Evol. 2007;44:553–577. doi: 10.1016/j.ympev.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 4.Boualem A, et al. A conserved mutation in an ethylene biosynthesis enzyme leads to andromonoecy in melons. Science. 2008;321:836–838. doi: 10.1126/science.1159023. [DOI] [PubMed] [Google Scholar]
- 5.Boualem A, et al. A cucurbit androecy gene reveals how unisexual flowers develop and dioecy emerges. Science. 2015;350:688–691. doi: 10.1126/science.aac8370. [DOI] [PubMed] [Google Scholar]
- 6.Martin A, et al. A transposon-induced epigenetic change leads to sex determination in melon. Nature. 2009;461:1135–1138. doi: 10.1038/nature08498. [DOI] [PubMed] [Google Scholar]
- 7.Englbrecht CC, Schoof H, Böhm S. Conservation, diversification and expansion of C2H2 zinc finger proteins in the Arabidopsis thaliana genome. BMC Genomics. 2004;5:39. doi: 10.1186/1471-2164-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sagasser M, Lu GH, Hahlbrock K, Weisshaar B. A. thaliana TRANSPARENT TESTA 1 is involved in seed coat development and defines the WIP subfamily of plant zinc finger proteins. Genes Dev. 2002;16:138–149. doi: 10.1101/gad.212702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones VAS, Dolan L. MpWIP regulates air pore complex development in the liverwort Marchantia polymorpha. Development. 2017;144:1472–1476. doi: 10.1242/dev.144287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appelhagen I, et al. Weird fingers: functional analysis of WIP domain proteins. FEBS Lett. 2010;584:3116–3122. doi: 10.1016/j.febslet.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Crawford BCW, Ditta G, Yanofsky MF. The NTT gene is required for transmitting-tract development in carpels of Arabidopsis thaliana. Curr. Biol. 2007;17:1101–1108. doi: 10.1016/j.cub.2007.05.079. [DOI] [PubMed] [Google Scholar]
- 12.Crawford B, et al. Genetic control of distal stem cell fate within root and embryonic meristems. Science. 2015;347:655–659. doi: 10.1126/science.aaa0196. [DOI] [PubMed] [Google Scholar]
- 13.Chung KS, Lee JH, Lee JS, Ahn JH. Fruit indehiscence caused by enhanced expression of NO TRANSMITTING TRACT in Arabidopsis thaliana. Mol. Cells. 2013;35:519–525. doi: 10.1007/s10059-013-0030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruggieri V, et al. An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci. Rep. 2018;8:8088. doi: 10.1038/s41598-018-26416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craft J, et al. New pOp/LhG4 vectors for stringent glucocorticoid-dependent transgene expression in Arabidopsis. Plant J. 2005;41:899–918. doi: 10.1111/j.1365-313X.2005.02342.x. [DOI] [PubMed] [Google Scholar]
- 16.Demidchik V, et al. Stress-induced electrolyte leakage: the role of K+-permeable channels and involvement in programmed cell death and metabolic adjustment. J. Exp. Bot. 2014;65:1259–1270. doi: 10.1093/jxb/eru004. [DOI] [PubMed] [Google Scholar]
- 17.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Malley RC, et al. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell. 2016;165:1280–1292. doi: 10.1016/j.cell.2016.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phukan UJ, Jeena GS, Shukla RK. WRKY transcription factors: molecular regulation and stress responses in plants. Front. Plant Sci. 2016;7:760. doi: 10.3389/fpls.2016.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banerjee A, Roychoudhury A. WRKY proteins: signaling and regulation of expression during abiotic stress responses. Sci. World J. 2015;2015:807560. doi: 10.1155/2015/807560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sangwan I, O’Brian MR. Identification of a soybean protein that interacts with GAGA element dinucleotide repeat DNA. Plant Physiol. 2002;129:1788–1794. doi: 10.1104/pp.002618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santi L, et al. The GA octodinucleotide repeat binding factor BBR participates in the transcriptional regulation of the homeobox gene Bkn3. Plant J. 2003;34:813–826. doi: 10.1046/j.1365-313X.2003.01767.x. [DOI] [PubMed] [Google Scholar]
- 24.Meister RJ, et al. Definition and interactions of a positive regulatory element of the Arabidopsis INNER NO OUTER promoter. Plant J. 2004;37:426–438. doi: 10.1046/j.1365-313X.2003.01971.x. [DOI] [PubMed] [Google Scholar]
- 25.Hecker A, et al. The Arabidopsis GAGA-binding factor BASIC PENTACYSTEINE6 recruits the POLYCOMB-REPRESSIVE COMPLEX1 component LIKE HETEROCHROMATIN PROTEIN1 to GAGA DNA motifs. Plant Physiol. 2015;168:1013–1024. doi: 10.1104/pp.15.00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowman JL, Smyth DR. CRABS CLAW, a gene that regulates carpel and nectary development in Arabidopsis, encodes a novel protein with zinc finger and helix-loop-helix domains. Development. 1999;126:2387–2396. doi: 10.1242/dev.126.11.2387. [DOI] [PubMed] [Google Scholar]
- 27.Jack T, Brockman LL, Meyerowitz EM. The homeotic gene apetala3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens. Cell. 1992;68:683–697. doi: 10.1016/0092-8674(92)90144-2. [DOI] [PubMed] [Google Scholar]
- 28.Larkin J, Oppenheimer D, Pollock S, Marks M. Arabidopsis GLABROUS1 gene requires downstream sequences for function. Plant Cell. 1993;5:1739–1748. doi: 10.2307/3869690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukaki H, Tameda S, Masuda H, Tasaka M. Lateral root formation is blocked by a gain-of-function mutation in the SOLITARY-ROOT/IAA14 gene of Arabidopsis. Plant J. 2002;29:153–168. doi: 10.1046/j.0960-7412.2001.01201.x. [DOI] [PubMed] [Google Scholar]
- 30.Lian J, et al. Silencing of BnTT1 family genes affects seed flavonoid biosynthesis and alters seed fatty acid composition in Brassica napus. Plant Sci. 2017;254:32–47. doi: 10.1016/j.plantsci.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 31.Ren G, et al. GhWIP2, a WIP zinc finger protein, suppresses cell expansion in Gerbera hybrida by mediating crosstalk between gibberellin, abscisic acid, and auxin. New Phytol. 2018;219:728–742. doi: 10.1111/nph.15175. [DOI] [PubMed] [Google Scholar]
- 32.Himmelbach A, et al. A set of modular binary vectors for transformation of cereals. Plant Physiol. 2007;145:1192–1200. doi: 10.1104/pp.107.111575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karimi M, Bleys A, Vanderhaeghen R, Hilson P. Building blocks for plant gene assembly. Plant Physiol. 2007;145:1183–1191. doi: 10.1104/pp.107.110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song Y, et al. Age-triggered and dark-induced leaf senescence require the bHLH transcription factors PIF3, 4 and 5. Mol. Plant. 2014;7:1776–1787. doi: 10.1093/mp/ssu109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jégu T, et al. The Arabidopsis SWI/SNF protein BAF60 mediates seedling growth control by modulating DNA accessibility. Genome Biol. 2017;18:1–16. doi: 10.1186/s13059-017-1246-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramírez F, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.R Core Team. R: A Language and Environment for Statistical Computing (2014).
- 41.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016. [Google Scholar]
- 42.Larsson, J. eulerr: Area-Proportional Euler and Venn Diagrams with Ellipses. R Package Version 6.0.0 (2019).
- 43.Wuyts N, et al. High-contrast three-dimensional imaging of the Arabidopsis leaf enables the analysis of cell dimensions in the epidermis and mesophyll. Plant Methods. 2010;6:1–14. doi: 10.1186/1746-4811-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The raw RNA-seq and ChIP-seq data described in this study have been deposited to the NCBI Short Read Archive (SRA) database under the BioProject ID PRJNA608903. Sequence data from CmWIP1 can be found in the GenBank data library under accession number GQ870274.1. Sequence data from TT1/AtWIP1 (AT1G34790), NTT/AtWIP2 (AT3G57670), AtSTOP1 (AT1G34370), CRC (AT1G69180), AP3 (AT3G54340), GL1 (AT3G27920), and SLR (AT4G14550) can be found at The Arabidopsis Information Resource (TAIR) database.
The code and software sources from previously published papers are listed in the “Methods”. All other details of the code used in this study are available from the corresponding author upon request.