

Abstract
Over the past decade, few reports suggested that the drug thalidomide (HbF inducer) may be of value in a subset of transfusion-dependent and non-transfusion dependent thalassemia patients. A cohort of 37 patients with symptomatic β-thalassemia syndrome [14 transfusions dependent thalassemia (TDT), and 23 Non-transfusion dependent Thalassemia (NTDT)], who were unable to pursue conventional therapy with transfusion and chelation, were recruited over 3 years in a center in Iraqi Kurdistan. After taking informed consent, patients were put on low dose Thalidomide (2–10 mg/kg), with regular follow up after that for a minimum of 8 months for a response. Patients with TDT were considered responders if their yearly transfusion requirement dropped by 25% or more, while NTDT responders were those who had a hemoglobin raise of 1 g m/dL or more. The median age of enrolled patients was 10 years (range 3–43) and included 21 males and 16 females. After a mean of 1.7 months (SD 0.76), responses were documented in 28 patients (75.7%). Among NTDT patients, a significant increase in hemoglobin from a mean of 7.83 (SD 1.07) to 9.96 g/dL (SD 1.11 g m/dL) was documented. While among TDT patients, there was a significant drop in yearly transfusions from 27 (SD 17.7) to 7.79 (SD 7.5) blood unit per year. The response in both categories was sustained after a median follow up of 15 months (8–36 m). Only minimal side effects were documented throughout in the form of constipation and only one patient developed extramedullary hemopoietic abdominal masses. A significant response to thalidomide was documented in the majority of TDT and NTDT patients, a response which was obtained after a mean of 1.7 months, and the response was sustained with limited side effects. The results support a possible role for this medication in a subset of thalassemia patients.
Keywords: Thalassemia, Thalidomide, Hemoglobin F inducer
Introduction
Beta thalassemia is an autosomal recessive inherited disorder of hemoglobin, due to a defect involving the beta globin gene. The resultant phenotype ranges from transfusion dependent thalassemia major (Transfusion dependent thalassemia [TDT]), to the asymptomatic thalassemia minor. In between these two extremes lies the phenotype of thalassemia intermedia, which is usually transfusion independent (NTDT) [1]. Both syndromes are at risk of complications of chronic hemolytic anemia, extramedullary hemopoiesis, iron overload and the risk of acquiring transfusion-transmitted infections [2].
Thalidomide is a synthetic glutamic acid derivative, which has been introduced in the late fifties of the last century, and became accessible for the management of morning sickness in pregnant women, but it was later abandoned because of its teratogenic effect, which led to one of the worst medically induced tragedies in history, when more than 10,000 babies were born with major crippling malformations [3]. Several decades later, the drug found new uses, particularly in the management of multiple myeloma, and leprosy [3]. The first report on its use in thalassemia appeared in 2008 [4], this was followed by few other case reports and small case series over the past decade, though no controlled, double-blind trial results were reported yet [5–10].
Iraq has a large population of patients with symptomatic beta-thalassemia [11], and the country passed over the past few years in periods of instability, and restriction of the availability of health services in some areas. Many of the thalassemic patients became internally displaced, and lost their access to proper health care and suffered extreme financial hardship. This eventually led to conventional thalassemia care being either unavailable or to families declining pursuit of these services, and the need to provide non-conventional approach is these unusual situations presented itself as a challenge to physicians managing these patients in this part of the world. Therefore we investigated the role of thalidomide in this particular category of patients with transfusion-dependent or symptomatic non-transfusion dependent thalassemia patients, after proper ethical approval and informed consent of patients and their guardians.
Patients and Methods
In the period from October 2015 to October 2018, 37 patients with confirmed symptomatic β-thalassemia, including 14 transfusion-dependent (TDT) (37.8%) and 23 Non-transfusion dependent thalassemia patients (62.2%) were recruited. Patients were recruited either because they declined transfusion therapy and chelation therapy, or these services were not readily available in the cities of their residence.
Patients with liver, renal, cardiac, pulmonary, neurological deficits or history of thrombotic episodes were excluded. For all females in the childbearing age, pregnancy tests were performed to exclude pregnancy, and they were advised not to contemplate pregnancy while on thalidomide and for 6 months after that.
Following full clinical evaluation and record review, Thalidomide was given so that the dose will be in the range of 2–10 mg/kg at any time throughout therapy. Initially however, a dose of 3 mg/kg adjusted to nearest 50 mg was used. If no response in 2 months then the dose was escalated accordingly. After that, patients were followed up for response and any adverse effects. Patients who could continue on iron chelation by Deferasirox or Deferoxamine were encouraged to do so, as per guidelines of such therapy per the thalassemia center.
Criteria for Response
Patients were followed for the response, blood counts, RFT, LFT and any adverse effects, initially on monthly bases for the first 3 months, then every other month after that; if no response within 12 months, thalidomide was discontinued and the patient advised to resume conventional management.
Patient with NTDT was considered responsive if their Hb rises by 1 g m or more. The response was categorized as useful if Hb increased by 1–1.9 g m/dL and very good if Hb rise was 2 g or more from baseline Hb level. Patients with TDT were considered responsive if their transfusion requirement dropped by 25% or more from pre-therapy status (6 months before vs. 6 months after). The response was deemed to be significant if the patient became transfusion independent.
Ethical approval was granted from the ethical committee in the college of medicine-Hawler medical university.
Statistical Analysis
Data were analyzed using the Statistical Package for Social Sciences (SPSS, version 22). Chi square test of association was used to compare proportions. Fisher’s exact test was used when the expected count of more than 20% of the cells of the table was less than 5. Student’s t test of two independent samples was used to compare two means. Paired sample t-test was used to compare means before and after treatment. A p value of ≤ 0.05 was considered statistically significant.
Results
The median age of the enrolled patients was 10 years (range 3–43 years) and included 21 males and 16 female. They included 14 patients with TDT and 23 with NTDT. Seven patients were splenectomized (18.9%).
After a mean time of 1.7 months (SD 0.76) of initiating thalidomide, a total of 28/37 (75.7%) of patients showed a response to therapy as defined in materials and methods, including 17/23 in NTDT (73.9%) and 11/14 in TDT (78.5%) (Tables 1, 2). The difference in response rate between TDT and NTDT was not significant (P = 0.94). The mean hemoglobin increased in NTDT from 7.83 g/dL (SD 1.07) prior therapy to a mean 9.96 g/dL (SD 1.11) at last follow up (P < 0.001). Transfusion requirement in TDT, on the other hand, dropped from 27 annually (SD 17.7) prior therapy, to 7.79 annually (SD 7.5) at last follow up (P = 0.001) (Table 1).
Table 1.
NTDT and TDT Response to Thalidomide as measured by Hemoglobin increment in NTDT (upper part) and drop in transfusion requirement in TDT (lower part)
| No. (%) | ||
|---|---|---|
| Hb difference (Hb2–Hb1) g/dL in NTDT | ||
| < 1 | 6 (26.1) | |
| 1–1.9 | 5 (21.7) | |
| ≥ 2 | 12 (52.2) | |
| Total | 23 (100.0) | |
| Reduction of transfusion requirements (% of baseline) in response to thalidomide in TDT. | ||
| < 25% | 3 (21.4) | |
| 25–50% | 2 (14.3) | |
| > 50% (but not transfusion independent) | 4 (28.6) | |
| Transfusion independent | 5 (35.7) | |
| Total | 14 (100.0) | |
Table 2.
Mean percentage of ferritin change after treatment by response to treatment
| Response | N | Mean percentage of ferritin change after treatmenta | (± SD) | p |
|---|---|---|---|---|
| Yes | 27 | 29.869 | (± 36.968) | 0.012 |
| No | 8 | − 7.770 | (± 27.993) |
aEquals: (Ferritin before treatment–ferritin after treatment)/ferritin before treatment
Figures 1 and 2 show the changes in hemoglobin and transfusion requirements in patients with NTDT and TDT, respectively, with treatment. The median follows up of all the patients was 15 months (range 8–36 months), with 17 of the 28 responders having a follow up more than 12 months. As demonstrated in both figures, the responses attained initially were sustained throughout subsequent follow up.
Fig. 1.
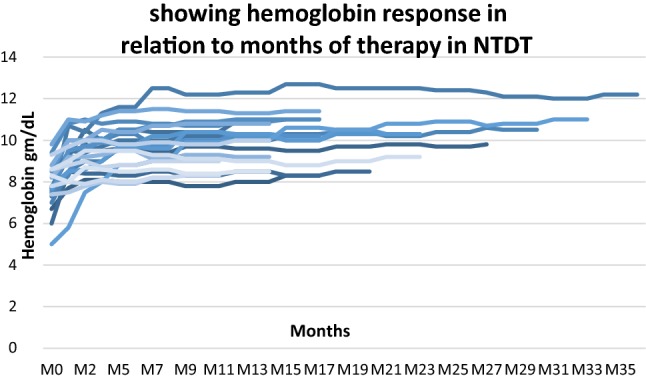
Showing hemoglobin response in relation to months of therapy in NTDT
Fig. 2.
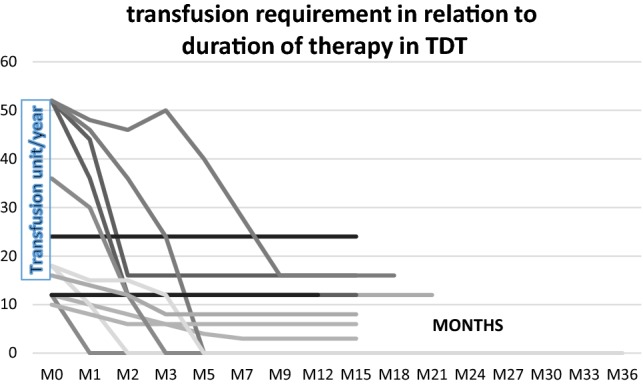
Transfusion requirement in relation to duration of therapy in TDT
Our results showed no significant association between the response rate to thalidomide with the following variables: group (NTDT/TDT) (P > 0.999), age (P = 0.214), gender (P > 0.999), splenectomy (P = 0.656).
It is evident in Table 2 that the mean percentage of ferritin change after treatment was 29.86% among patients who responded to treatment compared with − 7.77% among non-responders (P = 0.012).
Among our patients seven were using Deferasirox, five of them were among responders and tow were non responders.
Regarding the adverse effects during therapy, ten patients developed constipation of whom three required laxatives while seven responded to dietary measures. One patient developed extramedullary hemopoiesis in the form of multiple abdominal masses, and thalidomide was stopped and Hydroxyurea therapy was initiated after which masses disappeared and with a continued response and no deterioration in blood indices, LFT or RFT was recorded.
Discussion
Symptomatic Beta thalassemia is a crucial health problem in the Eastern Mediterranean region (EMR) and its management in most of these countries is based on a combination of transfusion and chelation therapy. Bone marrow transplantation remains the only curative approach to these syndromes [12], though its adoption is limited in the developing countries because of its vast expense and need of expertise in the setting of dedicated centers. Transfusions and chelation therapy thus remain the mainstay of management in EMR currently and are usually provided in the context of specialized thalassemia center, which is an expensive practice to run, and is to a great extent dependent on the governmental sponsorship and is quite influenced by literacy and dedication of the patients’ families.
Several non-conventional approaches to the management of NTDT and a lesser extent TDT have been proposed. They include the Hb F inducer Hydroxyurea, which was found to be quite useful in sickle cell disease, was used in Thalassemia with variable results [13]. Other non-selective agents, including cytotoxic and hypomethylating drugs, short-chain fatty acids and erythropoietin has also been investigated [14].
Thalidomide is another non-conventional management option, the mechanism of action of which is not precisely known, though some studies have found that this may occur through Hb F induction [6, 10]. Furthermore, in vitro experiments have suggested that this drug induces the expression of Gamma Globin chains through increased reactive oxygen species-mediated p38 MAPK signaling and histone H 4 acetylation [15]. The induced increased gamma chain production would eventually reduce α: β chain imbalance, and thus the toxic effect of deposited excess α-chains on the red cell precursors, which is the most critical mechanism explaining the pathogenesis of beta-thalassemia [16].
Though few reports suggest a promising role for thalidomide in symptomatic beta-thalassemia, its use has been debated, and there is yet no substantial justification for its use in the absence of published controlled trials, except if conventional therapy cannot be provided [17]. The latter situation was the case in our series, wherein the face of extreme hardship, such treatment was offered. The current study which included one of the largest cohorts studied yet, with a relatively long follow up, and it revealed that around three-quarters of Iraqi patients treated with this medication responded significantly, whether TDT or NTDT. Earlier case reports or small series also documented a significant response in thalassemia intermedia/NTDT [5, 8] and TDT [7, 18], although at least one case report on an Indian TDT failed to elicit such a response [19]. The rate of response appears much better than rates reported by the more popular HbF inducer “Hydroxyurea” in NTDT and TDT [13], this observation was also suggested by an earlier small series of Chinese Thalassemia intermedia reported recently. The time to response in the current study was 1.7 months, which is consistent with previous reports suggesting a time to response ranging from 2 weeks to 3 months [4, 5, 7, 8]. Furthermore, the response was sustained even after long follow up reaching up to 3 years in the current study (median 15 months), and again this is shared by previous observers [5, 8].
Limited complications were reported in the current series, and thrombosis, which is one of the most feared complications, was not documented in any patients. Despite that reports of deep venous thrombosis and stroke have been documented in thalidomide treated thalassemia significant patients in previous studies [20] and physicians contemplating its use should be vigilant to its occurrence and should take preventive measure.
Conclusion
The current series has demonstrated a significant response to thalidomide in more than three-quarters of symptomatic beta-thalassemia patients, whether TDT and NTDT. These observations support a role for this medication in a subset of these patients when conventional therapy is declined or is unavailable, until such a time that controlled trials results confirm these observations.
Compliance with Ethical Standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
Ethical approval have been done by the ethical committee of medical college hawler medical university
Informed Consent
Informed signed written consent was taken from the patient involved.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Shawky RM, Kamal TM. Thalassemia intermedia: an overview. Egypt J Med Hum Genet. 2012;13:245–255. [Google Scholar]
- 2.Origa R. β-thalassemia. Genet Med. 2017;19(6):609–619. doi: 10.1038/gim.2016.173. [DOI] [PubMed] [Google Scholar]
- 3.Vargesson N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today. 2015;105(2):140–156. doi: 10.1002/bdrc.21096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguilar-Lopez LB, Delgado-Lamas JL, Rubio-Jurado B, Perea FJ, Ibarra B. Thalaidomide therapy in patients with thalassemia major. Blood Cell Mol Dis. 2008;4(1):136–137. doi: 10.1016/j.bcmd.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Fozza C, Pardini S, Giannico DB, Targhetta C, Di Tucci AA, Dessalvi P, Angelucci E, Dore F. Dramatic erythroid response to low-dose thalidomide in two patients with transfusion independent thalassemia and severe post-transfusional alloimmune hemolysis. Am J Hematol. 2015;90(7):141. doi: 10.1002/ajh.24030. [DOI] [PubMed] [Google Scholar]
- 6.Ricchi P, Costantini S, Spasiano A, De Dominicis G, Di Matola T, Cinque P, Ammirabile M, Marsella M, Filosa A. The long-term and extensive efficacy of low dose thalidomide in a case of an untransfusable patient with non-transfusion-dependent thalassemia. Blood Cells Mol Dis. 2016;57:97–99. doi: 10.1016/j.bcmd.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Masera N, Tavecchia L, Capra M, Cazzaniga G, Vimercati C, Pozzi L, Biondi A, Masera G. Optimal response to thalidomide in a patient with thalassaemia major resistant to conventional therapy. Blood Transfus. 2010;8(1):63–65. doi: 10.2450/2009.0102-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li YS, Ren Q, Zhou Y, Li P, Lin W, Yin X. Thalidomide has a significant effect in patients with thalassemia intermedia. Hematology. 2018;23(1):50–54. doi: 10.1080/10245332.2017.1354427. [DOI] [PubMed] [Google Scholar]
- 9.Ramanan V, Kelkar K. Role of thalidomide in treatment of beta thalassemia. J Blood Disord Med. 2017 [Google Scholar]
- 10.Ren Q, Zhou Y, Wang L, Chen YS, Ma YN, Li P, et al. Clinical trial on the effect of thalidomide on hemoglobin synthesis in patients with moderate thalassemia intermedia. Ann Hematol. 2018;97:1933–1936. doi: 10.1007/s00277-018-3395-5. [DOI] [PubMed] [Google Scholar]
- 11.Kadhim KA, Baldawi KH, Lami FH. Prevalence, incidence, trend, and complications of thalassemia in Iraq. Hemoglobin. 2017;41(3):164–168. doi: 10.1080/03630269.2017.1354877. [DOI] [PubMed] [Google Scholar]
- 12.Manglani MV, Kini PS. Management of β-thalassemia-consensus and controversies. Pediatric Hematol Oncol J. 2017;2:94–97. [Google Scholar]
- 13.Kosaryan M, Zafari M, Alipur A, Hedayatizadeh-Omran A. The effect and side effect of hydroxyurea on patients with beta thalassemia: a systemic review to December 2012. Hemoglobin. 2014;38(4):262–271. doi: 10.3109/03630269.2014.927770. [DOI] [PubMed] [Google Scholar]
- 14.De Dreuzy E, Bhukhai K, Leboulch P, Payen E. Current and future alternative therapies for beta-thalassemia major. Biomed J. 2016;39:24–38. doi: 10.1016/j.bj.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers G. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;110(8):2864–2871. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fard AD, Hosseini SA, Shahjahani M, Salari F, Jaseb K. Evaluation of novel fetal hemoglobin inducer drugs in treatment of β-hemoglobinopathy disorders. Int J Hematol Oncol Stem Cell Res. 2013;7(3):47–54. [PMC free article] [PubMed] [Google Scholar]
- 17.https://thalassaemia.org.cy/news/tif-on-thalidomide
- 18.Jiskani SA, Memon S. Effect of thalidomide in patients with β–thalassemia major. Hematol Transfus Int J. 2018;6(6):234–236. [Google Scholar]
- 19.Manas K, Virender K, Amita T, Amita M. Thalidomide in transfusion dependent thalassemia: hope or hype. J Pediatr Hematol Oncol. 2017;39(6):485. doi: 10.1097/MPH.0000000000000900. [DOI] [PubMed] [Google Scholar]
- 20.Gunaseelan S, Prakash A. thalidomide-induced stroke in a child with thalassemia major. Pediatr Hematol Oncol. 2017;39(8):e519–e520. doi: 10.1097/MPH.0000000000000860. [DOI] [PubMed] [Google Scholar]