

Abstract
Introduction
Hereditary (variant) transthyretin amyloidosis (ATTRv) with polyneuropathy (ATTR-PN) is a rare genetic disorder that causes progressive autonomic and sensorimotor neuropathy, severe disability, and death within 10 years of onset. Previous studies have primarily focused on how baseline cardiac characteristics affect mortality, but the impact of non-cardiac baseline characteristics is less defined.
Methods
We systematically searched PubMed/Medline (1990–2019) to identify studies that assessed the impact of baseline ATTR-PN characteristics on survival. Outcomes were first summarized descriptively. Extracted survival data were then disaggregated, and parametric mixture models were used to assess survival differences among patient groups defined by factors known to affect survival.
Results
The search yielded 1193 records, of which 35 were retained for analysis. Median survival ranged from 0.5 to > 25 years. The largest survival differences were between cohorts who underwent liver transplantation (LTx) versus those who did not. Among LTx cohorts, pre-LTx ATTR-PN disease duration ≥ 7 years, poor nutritional status, and late disease onset reduced median survival by 13, 12, and 10 years, respectively. Other prognostic survival factors included non-Val30Met genotype and baseline presence of urinary incontinence, erectile dysfunction, or muscle weakness.
Conclusion
Survival in patients with ATTR-PN is highly variable and affected by non-cardiac baseline characteristics, such as autonomic dysfunction, large fiber involvement, late-onset disease, and non-Val30Met mutation. Careful interpretation of these findings is warranted given that this synthesis did not control for differences between studies. Survival in patients with ATTR-PN remains poor among those who are untreated or with delayed diagnosis.
Electronic supplementary material
The online version of this article (10.1007/s40120-020-00183-7) contains supplementary material, which is available to authorized users.
Keywords: Amyloidosis, Mortality, Polyneuropathy, Prognosis, Survival, Transthyretin
Key Summary Points
| Hereditary transthyretin amyloidosis (ATTRv) with polyneuropathy (ATTR-PN) is a rare genetic disorder. |
| Survival in patients with ATTR-PN is largely characterized by and dependent on associated cardiac damage, but the impact of non-cardiac characteristics on survival are less defined. |
| This study sought to assess the impact of non-cardiac baseline characteristics on survival in ATTR-PN. |
| Survival in patients with ATTR-PN was highly variable and affected by non-Val30Met mutation and non-cardiac baseline characteristics, such as autonomic dysfunction, large fiber involvement, and late-onset disease. |
Introduction
Hereditary (variant) transthyretin amyloidosis (ATTRv) is a rare autosomal dominant disease whereby mutations in the transthyretin (TTR) gene destabilize the protein’s native homeotetrameric structure and cause it to disassociate into amyloidogenic TTR monomers. As a result, insoluble amyloid fibrils accumulate in the peripheral and autonomic nerves, heart, gastrointestinal tract, kidneys, eyes, and connective tissues [1, 2]. Three phenotypes predominate in ATTRv amyloidosis: one is almost exclusively characterized by restrictive cardiomyopathy, another predominantly by polyneuropathy (ATTR-PN), and the third is a mixed phenotype [3–5].
ATTR-PN prevalence has been estimated at approximately 10,000 to 40,000 persons globally, with endemic foci in Portugal, Sweden, and Japan [6, 7]. Notably, disease phenotypes and penetrance across these endemic areas and elsewhere are often different, even for the same genotype [8]. For example, inaugural symptoms of early-onset disease (< 50 years old) include spontaneous burning or sharp pain, non-painful sensations (paresthesia and restless leg syndrome), and/or autonomic symptoms (sexual dysfunction, impaired bladder function, constipation alternating with diarrhea, early satiety, orthostatic dizziness and syncope) associated with damaged distal small myelinated and unmyelinated nerve fibers [1]. Conversely, in late-onset disease, the combination of well-preserved unmyelinated nerve fibers and axonal sprouting is associated with impaired superficial and deep sensation, severe neuropathic pain, early distal motor involvement, but relatively mild autonomic symptoms at initial presentation [1, 9, 10]. Absent early intervention with liver transplantation (LTx) or disease-modifying pharmacological treatments, ATTR-PN symptoms generally follow an ascending, proximally-oriented pattern as patients experience progressive debilitating polyneuropathy and inexorable loss of motor and organ functions.
Among persons with the classic disease phenotype, death has been reported to occur within 10–15 years of symptom onset, often from cachexia and malnutrition due to gastrointestinal complications [11, 12]. Prognostic survival factors include cardiac dysautonomia [3] and the presence of a mutation other than the more common Val30Met (also referred to as p.Val50Me) tgenotype [11, 13]; both of which are associated with poorer survival.
Owing partly to its phenotypic heterogeneity but also to its rarity, our understanding of survival in ATTR-PN is limited and largely based on observations of persons with the Val30Met genotype. Furthermore, the progression of associated cardiac symptoms and their effects on survival have been the predominant focus in larger studies [14]. However, as our understanding has evolved to reflect a heterogeneous disease, so too should our understanding of how this heterogeneity affects survival. This systematic review and analysis were conducted to describe the impact of non-cardiac clinicopathologic factors on survival and to assess survival trends among ATTR-PN subgroups.
Methods
The relevant evidence was assessed in a two-step process consisting of a systematic literature review to identify relevant data and a descriptive synthesis of those data. The target disease population was limited to persons with ATTR-PN.
Record identification and selection
Records were identified via a systematic literature review per a pre-specified protocol and conducted according to modified American Academy of Neurology guidelines (AAN) [6, 15]. Structured electronic searches of PubMed/Medline and Scopus were conducted on 15 February 2019 to identify relevant primary studies without regard to language of origin or year of publication (See Electronic Supplementary Material Appendix A). Non-human studies and case reports (studies with < 10 ATTR-PN patients) were excluded. Narrative reviews, systematic reviews, and meta-analyses were retained separately so that they could be searched for relevant primary reports not yielded by the electronic search.
The electronic search was supplemented with a hand search of conference materials from the International Symposium on Amyloidosis for the three most recent years held (2014, 2016, and 2018). The rationale for evaluating this supplemental material was twofold. First, the consideration of unpublished scientific materials is indicated by the AAN guidelines as a prerequisite for a comprehensive review [15]. Second, materials presented at conferences may not have had time to be published while otherwise being relevant to the content of the present review. To avoid including duplicated information between published and conference sources, a conference record was included only when the information presented in the record was not also reported in an associated peer-reviewed manuscript authored by one or more of the same authors. If the same study was reported as a conference material and as a peer-reviewed manuscript, only the latter was considered for inclusion in this review.
Records yielded by the search strategy were de-duplicated and assessed for inclusion via three rounds of review (title, abstract, and full-text) based on a pre-specified rubric applied by two reviewers. Inconsistencies were adjudicated by consensus. Records were retained if they satisfied the following inclusion criteria:
Human studies of adults (aged ≥ 18 years).
Primary report.
Reported survival among persons with ATTR-PN.
Follow-up time for survival assessment was ≥ 5 years.
Sample size is ≥ 10.
Survival was assessed from a defined baseline event (symptom onset, diagnosis, or treatment initiation).
Reported whether patients received disease-modifying pharmacologic treatment or LTx.
Patients were symptomatic at the baseline event (assumed unless noted otherwise).
Records were also included in the descriptive analysis of clinicopathologic prognostic factors if they also included a multivariate assessment of the impact of clinicopathologic factors on survival.
Where sequential studies reported the same endpoints updated for the same cohort, the study with the longest follow-up time was retained, while those with shorter follow-up times were excluded—except where those with shorter follow-up times included relevant survival endpoints not reported in the more recent reports. Additionally, studies or outcomes for a given cohort were excluded if they included only patients with ATTRv variants associated with the phenotype predominantly characterized by cardiomyopathy (i.e., Val122Ile, Thr60Ala, Ser77Tyr, Ile84Ser, Val30Ala, and His90Asp). There was no a priori determination of which specific diagnostic methods or degree of documentation constituted a valid diagnosis of ATTR-PN. It was only required that the cohort in question was described as one comprised entirely of individuals with ATTR-PN.
Data Extraction and Synthesis
There were two survival outcome categories of interest for this study.
The first category included time-to-event survival outcomes, such as survival probabilities, survival percentages, failure probabilities, and failure percentages as measured from a baseline event (symptom onset, diagnosis, or treatment initiation). Where survival outcomes were reported in figures only, survival data were extracted using digitization software (Digitizeit, version 2.3; Braunschweig, Germany). Survival outcomes were pooled according to clinicopathologic characteristics reported in each study.
The impacts of clinicopathologic characteristics on survival were analyzed further. To mitigate the potentially biasing effects of uneven censoring and varying follow-up times among the extracted survival outcomes, patient-level survival data were reconstructed from reported aggregate-level outcomes using methods previously described [16– 18]. This dis-aggregation produced a register of patient-level survival observations, and those with “disease onset” as the baseline event (i.e., starting point from which survival was measured) were considered suitable for further analysis.
Modeling was used to derive survival curves from the aforementioned patient-level observations. Expecting latent heterogeneity within the dataset, we applied parametric mixture modeling that decomposed the overall survival curve into constituent curves. In other words, mixture models assume that the overall survival of a cohort is a function of two or more subgroups: each with its own survival curve that can be estimated with a standard parametric distribution. Specifically, the overall survival for a given cohort was specified as a weighted average () of the constituent subgroup survival curves (here and ; e.g., LTx and no-LTx) according to Eq. 1:
| 1 |
Parameter denotes the proportion of patients belonging to the latent subgroup 1. All model parameters (, , and ) are estimated using the flexsurv-library package of the R programing language for the calculation of the hazards and survival functions and the “nlm-command” for maximizing the likelihood function. Standard errors around the estimated parameters were calculated by taking the diagonal elements of the inverse Hessian at the maximum likelihood.
We stratified the survival data into three classes: (1) LTx versus no LTx; (2) early onset versus late onset; and (3) V30M (i.e, Val30Met) versus non-V30M genotype. Survival was estimated for each class via the method described above, and the mean survival in the class was calculated as the area under the curve. The mean [95% confidence interval (CI)] difference between classes was equal to the difference between survival means.
The second survival outcome category pertained to clinicopathologic prognostic survival factors. When assessed by univariate or multivariate analysis to determine their individual impacts on survival, these factors were recorded along with the directionality and magnitude of their effects and subsequently analyzed descriptively.
Retained studies were classified using the modified AAN guideline [15] as has been reported previously [6]. Evidence classifications ranged from class I (strongest evidence, low risk of bias) to class IV (weakest evidence, very high risk of bias). Risk of bias was evaluated primarily with respect to two considerations: the method by which the initial ATTR-PN diagnosis was made and the frequency of follow-up.
Compliance with Ethics Statement
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Results
The electronic search of the peer-reviewed literature yielded 979 unique records, and the hand search of conference materials yielded 1014 records. In total, 1993 records were assessed in full, of which 35 were retained (Fig. 1; Table 1). These reported survival outcomes assessed over 265,346 person-years [N = 11,116; median follow-up 10 (range 5–35) years]. Most of the retained studies were of a longitudinal, observational design conducted between 1990 and 2007. Risk of bias was low, and evidentiary quality of the retained studies was moderate overall (Table 1).
Fig. 1.
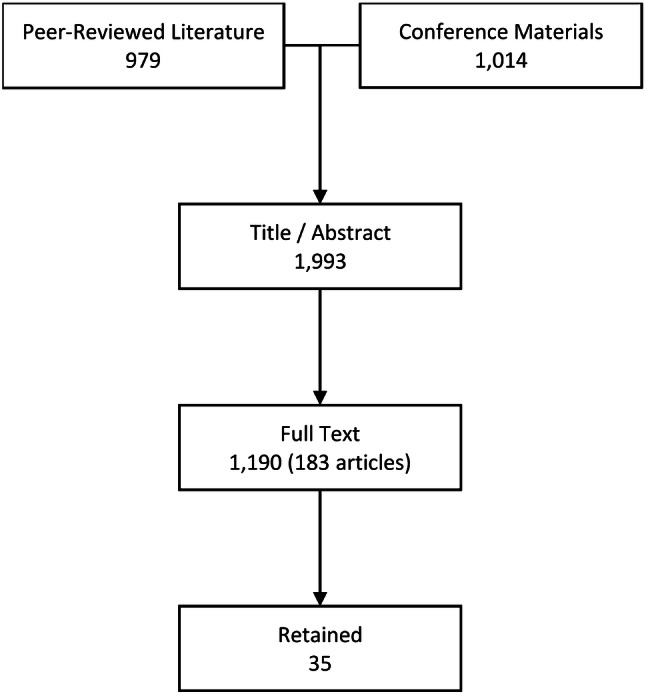
Flowchart summarizing the process of study adjudication
Table 1.
Summary of the 35 studies retained for analysis
| First author of study, year of publication [reference number] | N (size of cohort) | Country | Diagnostic method(s) | Baseline periodicitya | Follow-up (years)b | Evidence class, risk of biasc |
|---|---|---|---|---|---|---|
| Adams, 2000 [19] | 45 | Multiplei | Tissue biopsy + genetic testing | 1993–1999 | 6 | 1, Low |
| Adams, 2018 [20] | 225 | Multiple | NR | 2013–2016 | 7 | 1, Low |
| Algalarrondo, 2015/2016 [3, 21]d | 215 | France | Tissue biopsy + genetic testing | 1993–2011 | 10 | 1, Low |
| Arruda-Olson, 2013 [22] | 37 | USA | Tissue biopsy | 1980–2010 | 6 | 2, Low |
| Barrosso, 2017 [23] | 75 | Mixed | Tissue biopsy + genetic testing | NR–2014 | 10 | 1, Low |
| Bittencourt, 2002 [24] | 24 | Brazil | Tissue biopsy + genetic testingo | 1993–1999 | 7 | 2, Moderate |
| Carvalho, 2002 [25] | 35 | Portugal | Genetic testing | 1992–2000 | 5 | 2, Moderate |
| Coelho, 2018 [26] | 3160 | Portugal | Mixed | 1992–2016 | 24 | 2, Moderate |
| Coutinho, 2013 [27] | 106e | Portugal | Genetic testing | 1998–2010 | 13 | 1, Moderate |
| Coutinho, 2017 [27] | 232 | Portugal | NR | 1998–2015 | 15 | 1, Moderate |
| Ericzon, 2015 [28] | 1623 | Multiplej | NR | 1990–2010 | 20 | 2, High |
| Franz, 2013 [29] | 21 | Germany | Genetic testing | 1996–2011 | 10 | 2, Moderate |
| Gonzalez-Lopez, 2018 [30] | 118 | Spain | NR | NR | 25 | 3, High |
| Ines, 2018 [31] | 3160 | Portugalk | NR | NR–2016 | 35 | 3, High |
| Kawaji, 2014 [32] | 27 | Japan | Tissue biopsy + genetic testing | 1987–2011 | 7 | 1, Low |
| Lagarto, 2016 [33] | 116 | Portugal | Genetic testing | 2006–2014 | 7 | 2, Moderate |
| Loavenbruck, 2016 [34] | 41f | USA | Tissue biopsy + genetic testing | 1993–2013 | 20 | 1, Low |
| Lobato, 2004 [35] | 62 | Portugal | Tissue biopsy + genetic testingp | 1989–2000 | 9 | 1, Moderate |
| Mariani, 2015 [36] | 194 | Francel | Tissue biopsy + genetic testing | 1988–2010 | 25 | 2, Low |
| Munar-Ques, 2005 [37] | 102 | Spain | Tissue biopsy ± genetic testing | 1976–2003 | 29r | 3, Moderate |
| Ohya, 2011 [38] | 34 | Japan | Tissue biopsy ± genetic testing | 1994–2006 | 10 | 3, Moderate |
| Okamoto, 2011 [39] | 104 | Sweden | Tissue biopsy ± genetic testing | 1990–2008 | 25 | 1, Low |
| Okamoto, 2009 [40] | 141 | Sweden | Tissue biopsy ± genetic testing | 1990–2008 | 20 | 1, Low |
| Okumura, 2016 [41] | 65 | Japan | Genetic testing | 1990–2010 | 20 | 2, Moderate |
| Plante-Bordeneuve, 1998 [42] | 65 | France | Genetic testing ± tissue biopsy | 1988–1997 | 10 | 2, High |
| Suhr, 2016 [11] | 264 | Multiplej | NR | 1991–2012 | 10 | 2, High |
| Suhr, 2002 [43] | 51g | Sweden | Tissue biopsy + genetic testing | 1990–2000 | 20 | 2, Moderate |
| Swiecicki, 2015 [44] | 110 h | USA | Family history ± genetic testing | 1970–2013 | 8 | 4, High |
| Takei, 2005 [45] | 43 | Japan | Genetic testing | 1993–2004 | 10 | 2, Moderate |
| Tashima, 1998 [46] | 120 | Multiplem | Tissue biopsy + genetic testingq | 1973–1998 | 24 | 3, Moderate |
| Wixner, 2015 [47] | 115 | Sweden | Tissue biopsy + genetic testing | 1990–2011 | 9 | 1, Low |
| Yamamoto, 1998 [48] | 116 | Japan | Tissue biopsy + family history | 1974–1995 | 20 | 4, High |
| Yamamoto, 2007 [49] | 86 | Multiplem | Tissue biopsy + genetic testing | 1990–2005 | 16 | 1, Low |
| Yamashita, 2012 [50] | 80 | Multiplen | Tissue biopsy + genetic testing | 1990–2010 | 20 | 2, Low |
| Yamashita, 2018 [51] | 104 | Japan | Tissue biopsy ± genetic testing | 2000–2017 | 10 | 2, Low |
HTx Heart transplantation, KTx kidney transplantation, LTx liver transplantation, mBMI modified body mass index, NR not reported
aRefers to the time-period over which the study was conducted
bRefers to maximum number of years from the baseline event at which survival outcomes were reported
cEvidence grades range from 1 (best) to 4 (worst)
dNote that two studies are reported here
eRefers to the matched cohort of LTx (n = 53) ves. non-LTx (n = 53)
fRefers to the transthyretin amyloidosis cohort (n = 41) and excludes the primary amyloidosis cohort (n = 60)
gRefers to the transthyretin amyloidosis cohort (n = 51) and excludes the control cohort (n = 19)
hRefers to Val30Met (n = 42), Ser77Tyr (n = 15), and other mutations (n = 53) and excludes Thr60Ala (n = 68) and Val122Ile (n = 28)
iFrance, Germany, and Portugal
jPortugal, France, Sweden, USA, Brazil, Spain, Japan, UK, Germany, Italy, Australia, Switzerland, Netherlands, Belgium, Argentina, Canada, Denmark, China, and Singapore
kThe geographical origin of the patient cohort was not explicitly stated in the report. Portugal was assumed based on authors’ affiliations
lAll patients were treated in France, but n = 110 (57%) were Portuguese
mSweden and Japan
nJapan, Sweden, Australia, and USA
oRefers to definitive testing performed after LTx
pGenetic testing performed on first-degree relatives for n = 3 (5%) patients
qGenetic testing was performed only on patients living past 1997
rNot reported and so imputed as the year of publication (2005) minus the earliest baseline year (1976)
Survival was first assessed descriptively and by combining the aggregate-level data into groups according to distinguishing characteristics known to the authors to impact survival (i.e., genotype, whether one has received a liver transplant, and whether one has early- or late-onset disease). This preliminary assessment (figures not shown but available upon request) confirmed the need for mixture modeling, which was conducted using disaggregated data from the 12 studies (45 observations, N = 3348) with disease onset as the baseline timepoint.
Disaggregated survival data were analyzed with parametric mixture modeling to assess survival differences among patient groups defined by factors known to affect survival. The most pronounced survival difference (reported here and subsequently as mean years from symptom onset to death) was observed between LTx and non-LTx patients: 26.0 (95% CI 25.0–27.9) versus 12.0 (95% CI 11.2–13.0); difference 15.0 (95% CI 13.7–16.3) (Fig. 2). Also significant was the survival difference between early-onset versus late-onset patients: 24.7 (95% CI 23.4–26.0) versus 12.9 (95% CI 11.0–13.8); difference 11.8 (95% CI 10.1–13.4) (Fig. 3). Combining these factors demonstrated that LTx patients with early-onset disease had significantly longer survival than LTx patients with late-onset disease: 26.6 (95% CI 25.6–27.4) versus 13.1 (95% CI 11.6–14.7); difference 13.5 (95% CI 11.7–15.3) (Fig. 4). However, the same comparison among non-LTx patients yielded a non-significant difference (12.7 vs. 12.5).
Fig. 2.

Comparison of modeled survival curves for patients who underwent liver transplantation (LTx) with those who did not (No LTx) demonstrates the survival benefit associated with LTx
Fig. 3.

Cohorts with disease that was not early onset (Not Early onset) experienced markedly shorter survival than did cohorts with early-onset disease (Early onset)
Fig. 4.

Impact of timing of disease onset. Cohorts with LTx and no early-onset disease (LTx + NO early onset) are shown to have shorter modeled survival versus cohorts with LTx and early-onset disease (LTx + Early onset)
A comparison between V30M genotype versus non-V30M genotype indicated longer survival for V30M (22.2 vs. 10.4), but the trend was not significant owing to limited observations for non-V30M patients.
Of the 35 retained studies, eight reported the outcomes of multivariate survival analysis (Table 2). Other than LTx status, the clinicopathologic survival factors identified as having a statistically significant impact on survival included sex/gender, pre-LTx disease duration, and symptoms of disease progression [e.g., modified body mass index (mBMI), serum albumin levels at the time of LTx, and the presence/absence of urinary incontinence at the time of LTx]. These observations indicated that female sex/gender, ≤ 7-year disease duration by the time of LTx, mBMI > 600, and a lack of urinary incontinence at LTx were all associated with improved survival versus matched comparisons.
Table 2.
Non-cardiac baseline prognostic survival factors and associated effects assessed by multivariate analysis
| Domaina | Description | Survival difference (years) | Mitigating factor(s) |
|---|---|---|---|
| Genotype | |||
| Mariani, 2015 [36] | Val30Met vs. Non-Val30Met | 1 (8 vs. 7) | Among No LTx |
| Mariani, 2015 [36] | Val30Met vs. Non-Val30Met | 4 (17 vs. 13) | Among LTx |
| Sex/gender | |||
| Munar-Ques, 2005 [37] | Female vs. male | − 1 (9 vs. 10) | None reported |
| Okamoto, 2009 [40] | Female vs. male | ≥ 4 (≥ 25 vs. 21) | Among LTx |
| Okamoto, 2009 [40] | Female vs. male | ≥ 4 (≥ 25 vs. 21) | Among LTx with early onset |
| Okamoto, 2009 [40] | Female vs. male | ≥ 9 (≥ 20 vs. 11) | Among LTx with late onset |
| Yamamoto, 1998 [48] | Female vs. male | 1 (11 vs. 10) | Among no LTx |
| Pre-LTx disease duration | |||
| Bittencourt, 2002 [24] | < 7 years vs. ≥ 7 years | ≥ 6 (≥ 7 vs. 1) | None reported |
| Okamoto, 2009 [40] | < 7 years vs. ≥ 7 years | ≥ 4 (≥ 25 vs. 21) | None reported |
| Suhr, 2002 [43] | < 7 years vs. ≥ 7 years | 3 (14 vs. 11) | Lower baseline mBMI in < 7 years |
| Yamamoto, 2007 [49] | < 7 years vs. ≥ 7 years | ≥ 17 (≥ 20 vs. 3) | None reported |
| Biometrics/symptoms | |||
| Suhr, 2002 [43] | mBMI > 600 vs. < 600 (at LTx) | 3 (14 vs. 11) | Pre-LTx disease duration was < 7 vs. ≥ 7 years |
| Yamamoto, 2007 [49] | mBMI > 600 vs. < 600 (at LTx) | ≥ 17 (≥ 20 vs. 3) | Pre-LTx disease duration was < 7 vs. ≥ 7 years |
| Bittencourt, 2002 [24] | Serum albumin (at LTx) | ≥ 6 (≥ 7 vs. 1) | Among LTx |
| Adams, 2000 [19] | Urinary incontinence (at LTx) | ≥ 4 (≥ 5 vs. 1) | Among LTx |
mBMI Modified body mass index
aPresented as first author of study, year of publication [reference number]
Discussion
This synthesis of published evidence illustrates the heterogenous nature of survival among persons with ATTR-PN and highlights a number of patient subgroups for whom survival differs from the commonly-cited 10–15 years—in some cases markedly less [52]. For non-LTx patients, heterogeneity was most evident among those with non-Val30Met genotypes for whom median survival was poorer compared to persons with the ATTRv Val30Met (6.3 vs. 12.1 years after onset). Furthermore, those who underwent LTx had better survival than those who were untreated (21.8 vs. 9.6 years). Evidence of the impact of disease-modifying pharmacological treatments was limited but demonstrated survival benefits of tafamidis versus no treatment and potentially versus LTx.
Aside from genotype and treatment experience, other baseline non-cardiac clinicopathologic characteristics associated with shorter survival included longer disease duration before LTx, late-onset disease, and lower mBMI. Given the previously known associations of many of the non-cardiac clinicopathologic survival factors identified in this study with baseline disease progression and the negative relationship between baseline disease progression and survival time [53], some of our findings are unsurprising. There were, however, notable studies demonstrating the impact of competing prognostic factors on survival. For example, at 20 years of follow-up after ATTRv Val30Met onset, Okamoto et al. [40] reported a non-significant effect of sex on survival at 15 years of follow-up after disease onset for those with early-onset disease (76 vs. 78%, p = 0.33); in contrast, when the same comparison was evaluated among LTx patients with late-onset disease, the difference was significant [69% (females) vs. 37% (males); p = 0.02]. The significant effect of sex in late-onset patients—who typically present with more advanced cardiac symptoms—despite no such finding in early-onset patients highlights intra-genotype survival differences that may be mitigated by sex and disease onset. Another example was reported by Adams et al. [19] who demonstrated that the survival benefits of LTx can be entirely negated by advanced disease progression at baseline, as indicated by a Norris score ≤ 55.
We also assessed the risk of bias and evidentiary quality of the records retained in this review and, notably, both were within acceptable limits overall. These metrics were largely based on the definitiveness of the ATTRv amyloidosis diagnosis (i.e., genetic and histological testing) and the transparency by which diagnoses were reported. Given that many of the retained records reported LTx cohorts and that histological and genetic testing are often required for LTx [25, 53], it follows that the records retained for this review would be scored highly.
This study has limitations which should be noted. First, in 1996 there began a more concerted, evidence-driven effort to optimize patient selection for LTx globally [40, 53]. LTx-associated survival benefits reported here may have been downwardly biased due to our having grouped data from patients who underwent LTx in pre- and post-1996 time periods, whereas survival after LTx in a contemporary treatment setting may be longer. This implies a related limitation: that due to lack of evidence it was not possible to control for patients having been at different stages of disease progression at the time of LTx or starting other treatments. Third, this study is limited to patients who received no treatment, underwent LTx, or received tafamidis due to these being the only ATTR-PN patients with long-term survival outcomes (i.e., ≥ 5 years). There are other treatments available or currently being evaluated to treat ATTR-PN, but there are as yet insufficient survival follow-up data on these treatments. Lastly, survival is only one outcome—albeit a critical one—for evaluating ATTR-PN disease progression, whereas progression of neuropathy would also have been an informative endpoint had sufficient evidence been available. Similarly, our survival analysis did not consider the implications of cardiac sequelae, which have been reported extensively in the literature and are important because our objective was to look at the impact of non-cardiac factors.
Conclusion
Notwithstanding the aforementioned limitations, this study evaluates survival in individual ATTR-PN subgroups for whom survival has often been imprecisely defined. Our estimates indicate that 10- to 15-year survival is an overly-optimistic assumption for many patients. Median survival for untreated patients with non-Val30Met genotypes is considerably worse than that for Val30Met disease, with survival approximately 6 years from symptom onset in the former and approximately 12 years in the latter. Conversely, the 10- to 15-year survival estimate that is commonly used may underestimate the benefits of definitive treatment with LTx or disease-modifying treatment with medications. Median survival may range from 14 to 25 years from symptom onset in treated patients, depending on the genotype of the disease and the extent of disease progression by the time treatment is started. Nevertheless, the evidence presented here demonstrates that ATTR-PN is quite heterogenous and, consequently, that our understanding of the prognosis of these patients should be similarly nuanced.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Funding
This study, including the rapid service publication fee, was funded by Pfizer, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Leslie Amass and Michelle Stewart are employees of Pfizer Inc. and hold stock and/or stock options. Marc F Botteman and John A Carter are employees of Pharmerit International, which was funded by Pfizer Inc. for study design, execution, analysis, and manuscript development. Alejandra González‐Duarte previously received support from Alnylam Pharmaceuticals and Pfizer Inc. for her role as a principal investigator of studies sponsored by these entities. Isabel Conceição previously received support as principal investigator from Alnylam Pharmaceuticals, IONIS Pharmaceuticals, and Pfizer Inc. Alejandra González‐Duarte and Isabel Conceição serve on the THAOS advisory board, which is sponsored by Pfizer Inc.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Footnotes
Enhanced digital features
To view enhanced digital features for this article go to 10.6084/m9.figshare.11891175.
References
- 1.Conceicao I, Gonzalez-Duarte A, Obici L, et al. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9. doi: 10.1111/jns.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quintas A, Vaz DC, Cardoso I, Saraiva MJ, Brito RM. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem. 2001;276(29):27207–27213. doi: 10.1074/jbc.M101024200. [DOI] [PubMed] [Google Scholar]
- 3.Algalarrondo V, Antonini T, Théaudin M, et al. Cardiac dysautonomia predicts long-term survival in hereditary transthyretin amyloidosis after liver transplantation. JACC Cardiovasc Imaging. 2016;9(12):1432–1441. doi: 10.1016/j.jcmg.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Hawkins PN, Ando Y, Dispenzeri A, Gonzalez-Duarte A, Adams D, Suhr OB. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–638. doi: 10.3109/07853890.2015.1068949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coelho T, Maurer MS, Suhr OB. THAOS—the transthyretin amyloidosis outcomes survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi: 10.1185/03007995.2012.754348. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2017 doi: 10.1002/mus.26034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Duarte A, Soto KC, Martinez-Banos D, et al. Familial amyloidosis with polyneuropathy associated with TTR Ser50Arg mutation. Amyloid. 2012;19(4):171–176. doi: 10.3109/13506129.2012.712925. [DOI] [PubMed] [Google Scholar]
- 8.Waddington-Cruz M, Schmidt H, Botteman MF, et al. Epidemiological and clinical characteristics of symptomatic hereditary transthyretin amyloid polyneuropathy: a global case series. Orphanet J Rare Dis. 2019;14(1):34. doi: 10.1186/s13023-019-1000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conceicao I, Costa J, Castro J, de Carvalho M. Neurophysiological techniques to detect early small-fiber dysfunction in transthyretin amyloid polyneuropathy. Muscle Nerve. 2014;49(2):181–186. doi: 10.1002/mus.23901. [DOI] [PubMed] [Google Scholar]
- 10.Koike H, Hashimoto R, Tomita M, et al. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid. 2011;18(2):53–62. doi: 10.3109/13506129.2011.565524. [DOI] [PubMed] [Google Scholar]
- 11.Suhr OB, Larsson M, Ericzon B-G, Wilczek HE. Survival after transplantation in patients with mutations other than Val30Met: extracts from the FAP world transplant registry. Transplantation. 2016;100(2):373–381. doi: 10.1097/TP.0000000000001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coelho T, Maia LF, da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–2814. doi: 10.1007/s00415-013-7051-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herlenius G, Wilczek HE, Larsson M, Ericzon B-G. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the familial amyloidotic polyneuropathy world transplant registry. Transplantation. 2004;77(1):64–71. doi: 10.1097/01.TP.0000092307.98347.CB. [DOI] [PubMed] [Google Scholar]
- 14.Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 15.American Academy of Neurology . Clinical practice guideline process manual. St. Paul: American Academy of Neurology; 2011. [Google Scholar]
- 16.Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9. doi: 10.1186/1471-2288-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korn EL, McShane LM, Freidlin B. Statistical challenges in the evaluation of treatments for small patient populations. Sci Transl Med. 2013;5(178):178. doi: 10.1126/scitranslmed.3004018. [DOI] [PubMed] [Google Scholar]
- 18.Tingley K, Coyle D, Graham ID, et al. Using a meta-narrative literature review and focus groups with key stakeholders to identify perceived challenges and solutions for generating robust evidence on the effectiveness of treatments for rare diseases. Orphanet J Rare Dis. 2018;13(1):104. doi: 10.1186/s13023-018-0851-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adams D, Samuel D, Goulon-Goeau C, et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(7):1495–1504. doi: 10.1093/brain/123.7.1495. [DOI] [PubMed] [Google Scholar]
- 20.Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 21.Algalarrondo V, Antonini T, Theaudin M, et al. Prediction of long-term survival after liver transplantation for familial transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(19):2154–2156. doi: 10.1016/j.jacc.2015.08.870. [DOI] [PubMed] [Google Scholar]
- 22.Arruda-Olson AM, Zeldenrust SR, Dispenzieri A, et al. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid. 2013;20(4):263–268. doi: 10.3109/13506129.2013.845745. [DOI] [PubMed] [Google Scholar]
- 23.Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24(3):194–204. doi: 10.1080/13506129.2017.1357545. [DOI] [PubMed] [Google Scholar]
- 24.Bittencourt PL, Couto CA, Farias AQ, Marchiori P, Bosco Massarollo PC, Mies S. Results of liver transplantation for familial amyloid polyneuropathy type I in Brazil. Liver Transpl. 2002;8(1):34–39. doi: 10.1053/jlts.2002.29764. [DOI] [PubMed] [Google Scholar]
- 25.de Carvalho M, Conceicao I, Bentes C, Luis MLS. Long-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M) Amyloid. 2002;9(2):126–133. doi: 10.3109/13506120208995245. [DOI] [PubMed] [Google Scholar]
- 26.Coelho T, Inês M, Conceição I, Soares M, de Carvalho M, Costa J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):e1999. doi: 10.1212/wnl.0000000000006543. [DOI] [PubMed] [Google Scholar]
- 27.Coutinho MCA, Cortez-Dias N, Cantinho G, et al. Reduced myocardial 123-iodine metaiodobenzylguanidine uptake: a prognostic marker in familial amyloid polyneuropathy. Circ Cardiovasc Imaging. 2013;6(5):627–636. doi: 10.1161/CIRCIMAGING.112.000367. [DOI] [PubMed] [Google Scholar]
- 28.Ericzon B-G, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. doi: 10.1097/TP.0000000000000574. [DOI] [PubMed] [Google Scholar]
- 29.Franz C, Hoffmann K, Hinz U, et al. Modified body mass index and time interval between diagnosis and operation affect survival after liver transplantation for hereditary amyloidosis: a single-center analysis. Clin Transplant. 2013;27(Suppl 2):40–48. doi: 10.1111/ctr.12193. [DOI] [PubMed] [Google Scholar]
- 30.González-López E, Laura L, Marín Gómez LM, et al. Liver transplantation for hereditary transthyretin amyloidosis (hATTR) in Spain—25-year experience. In: Paper presented at the 16th international symposium on Amyloidosis. Kumamoto, Japan. 2018.
- 31.Inês M, Coelho T, Conceição I, Soares M, Carvalho M, Costa J. Clinical paradigms in treatment effectiveness for hereditary transthyretin amyloidosis. In: Paper presented at the 16th international symposium on Amyloidosis. Kumamoto, Japan. 2018.
- 32.Kawaji T, Inoue T, Hara R, Eiki D, Ando Y, Tanihara H. Long-term outcomes and complications of trabeculectomy for secondary glaucoma in patients with familial amyloidotic polyneuropathy. PLoS One. 2014;9(5):e96324. doi: 10.1371/journal.pone.0096324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagarto F, Gomes B, Couto PS, et al. Perioperative predictors of survival after liver transplantation for familial amyloid polyneuropathy in a Portuguese Center. Transplant Proc. 2016;48(6):2098–2101. doi: 10.1016/j.transproceed.2016.04.020. [DOI] [PubMed] [Google Scholar]
- 34.Loavenbruck AJ, Singer W, Mauermann ML, et al. Transthyretin amyloid neuropathy has earlier neural involvement but better prognosis than primary amyloid counterpart: an answer to the paradox? Ann Neurol. 2016;80(3):401–411. doi: 10.1002/ana.24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lobato L, Beirao I, Silva M, et al. End-stage renal disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factors. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis. 2004;11(1):27–37. doi: 10.1080/13506120410001673884. [DOI] [PubMed] [Google Scholar]
- 36.Mariani L-L, Lozeron P, Theaudin M, et al. Genotype–phenotype correlation and course of transthyretin familial amyloid polyneuropathies in France. Ann Neurol. 2015;78(6):901–916. doi: 10.1002/ana.24519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munar-Ques M, Saraiva MJM, Viader-Farre C, Zabay-Becerril JM, Mulet-Ferrer J. Genetic epidemiology of familial amyloid polyneuropathy in the Balearic Islands (Spain) Amyloid. 2005;12(1):54–61. doi: 10.1080/13506120500032741. [DOI] [PubMed] [Google Scholar]
- 38.Ohya Y, Okamoto S, Tasaki M, et al. Manifestations of transthyretin-related familial amyloidotic polyneuropathy: long-term follow-up of Japanese patients after liver transplantation. Surg Today. 2011;41(9):1211–1218. doi: 10.1007/s00595-010-4488-5. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto S, Hornsten R, Obayashi K, Wijayatunga P, Suhr OB. Continuous development of arrhythmia is observed in Swedish transplant patients with familial amyloidotic polyneuropathy (amyloidogenic transthyretin Val30Met variant) Liver Transpl. 2011;17(2):122–128. doi: 10.1002/lt.22184. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl. 2009;15(10):1229–1235. doi: 10.1002/lt.21817. [DOI] [PubMed] [Google Scholar]
- 41.Okumura K, Yamashita T, Masuda T, et al. Long-term outcome of patients with hereditary transthyretin V30M amyloidosis with polyneuropathy after liver transplantation. Amyloid. 2016;23(1):39–45. doi: 10.3109/13506129.2015.1123149. [DOI] [PubMed] [Google Scholar]
- 42.Plante-Bordeneuve V, Lalu T, Misrahi M, et al. Genotypic-phenotypic variations in a series of 65 patients with familial amyloid polyneuropathy. Neurology. 1998;51(3):708–714. doi: 10.1212/WNL.51.3.708. [DOI] [PubMed] [Google Scholar]
- 43.Suhr OB, Ericzon B-G, Friman S. Long-term follow-up of survival of liver transplant recipients with familial amyloid polyneuropathy (Portuguese type) Liver Transpl. 2002;8(9):787–794. doi: 10.1053/jlts.2002.34386. [DOI] [PubMed] [Google Scholar]
- 44.Swiecicki PL, Zhen DB, Mauermann ML, et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid. 2015;22(2):123–131. doi: 10.3109/13506129.2015.1019610. [DOI] [PubMed] [Google Scholar]
- 45.Takei Y-I, Ikeda S-I, Ikegami T, Hashikura Y, Miyagawa S-I, Ando Y. Ten years of experience with liver transplantation for familial amyloid polyneuropathy in Japan: outcomes of living donor liver transplantations. Intern Med. 2005;44(11):1151–1156. doi: 10.2169/internalmedicine.44.1151. [DOI] [PubMed] [Google Scholar]
- 46.Tashima K, Suhr OB, Ando Y, et al. Gastrointestinal dysfunction in familial amyloidotic polyneuropathy (ATTR Val30Met)–comparison of Swedish and Japanese patients. Amyloid. 1999;6(2):124–129. doi: 10.3109/13506129909007313. [DOI] [PubMed] [Google Scholar]
- 47.Wixner J, Sundstrom T, Karling P, Anan I, Suhr OB. Outcome of gastric emptying and gastrointestinal symptoms after liver transplantation for hereditary transthyretin amyloidosis. BMC Gastroenterol. 2015;15:51. doi: 10.1186/s12876-015-0284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamamoto K, Ikeda S, Hanyu N, Takeda S, Yanagisawa N. A pedigree analysis with minimised ascertainment bias shows anticipation in Met30-transthyretin related familial amyloid polyneuropathy. J Med Genet. 1998;35(1):23–30. doi: 10.1136/jmg.35.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto S, Wilczek HE, Nowak G, et al. Liver transplantation for familial amyloidotic polyneuropathy (FAP): a single-center experience over 16 years. Am J Transplant. 2007;7(11):2597–2604. doi: 10.1111/j.1600-6143.2007.01969.x. [DOI] [PubMed] [Google Scholar]
- 50.Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78(9):637–643. doi: 10.1212/WNL.0b013e318248df18. [DOI] [PubMed] [Google Scholar]
- 51.Yamashita T, Ueda M, Misumi Y, et al. Genetic and clinical characteristics of hereditary transthyretin amyloidosis in endemic and non-endemic areas: experience from a single-referral center in Japan. J Neurol. 2018;265(1):134–140. doi: 10.1007/s00415-017-8640-7. [DOI] [PubMed] [Google Scholar]
- 52.Hellman U, Alarcon F, Lundgren H-E, Suhr OB, Bonaiti-Pellie C, Plante-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15(3):181–186. doi: 10.1080/13506120802193720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transpl. 2015;21(3):282–292. doi: 10.1002/lt.24058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.