

Abstract
Background
Medulloblastoma (MB) with metastases at diagnosis and recurrence correlates with poor prognosis. Unfortunately, the molecular mechanism underlying metastases growth has received less attention than primary therapy-naïve MB. Though astrocytes have been frequently detected in brain tumors, their roles in regulating the stemness properties of MB stem-like cells (MBSCs) in disseminated lesions remain elusive.
Methods
Effects of tumor-associated astrocyte (TAA)–secreted chemokine C-C ligand 2 (CCL2) on MBSC self-renewal was determined by immunostaining analysis. Necroptosis of TAA was examined by measuring necrosome activity. Alterations in Notch signaling were examined after inhibition of CCL2. Progression of MBSC-derived tumors was evaluated after pharmaceutical blockage of necroptosis.
Results
TAA, as the essential components of disseminated tumor, produced high levels of CCL2 to shape the inflammation microenvironment, which stimulated the enrichment of MBSCs in disseminated MB. In particular, CCL2 played a pivotal role in maintaining stem-like properties via Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3)–mediated activation of Notch signaling. Loss of CCL2/C-C chemokine receptor 2 (CCR2) function repressed the JAK2/STAT3-Notch pathway and impaired MBSC proliferation, leading to a dramatic reduction of stemness, tumorigenicity, and metastasizing capability. Furthermore, necroptosis-induced CCL2 release depended on activation of receptor-interacting protein 1 (RIP1)/RIP3/mixed lineage kinase domain-like pseudokinase (MLKL) in TAA, which promoted the oncogenic phenotype. Blockade of necroptosis resulted in CCL2 deprivation and compromised MBSC self-proliferation, indicating MBSCs outsourced CCL2 from necroptotic TAA. Finally, CCL2 was upregulated in high-risk stages of MB, further supporting its value as a prognostic indicator.
Conclusion
These findings highlighted the critical role of CCL2/CCR2 in Notch signaling activation in MBSCs and revealed a necroptosis-associated glial cytokine microenvironment driving stemness maintenance in disseminations.
Key Points
1. TAA-derived CCL2 promoted stemness in disseminated MBSCs through Notch signaling activation via the JAK2/STAT3 pathway.
2. TAA released CCL2 in a RIP1/RIP3/MLKL-dependent manner leading to necroptosis.
Keywords: medulloblastoma, cancer stem-like cells, tumor-associated astrocytes, CCL2, necroptosis
Importance of the Study.
Recurrent or disseminated medulloblastoma accounts for a majority of pediatric brain tumor–related mortality and points to a significant challenge for conventional treatment modalities. Although targeted therapies represent an important approach to improve prognosis, the molecular mechanisms regarding metastases and current treatments approved for disseminated medulloblastoma remain ambiguous, leading to poor outcomes. Herein, we demonstrate that TAA, the essential components of the microenvironment in recurrent medulloblastoma, secrete CCL2, which is required for maintaining MBSC stemness via JAK2/STAT3-mediated Notch signaling activation. Furthermore, TAA shape the cytokine microenvironment in a necroptosis-dependent manner. Our findings are the first to demonstrate that ablation of necroptosis can dramatically suppress TAA-derived CCL2 release and block disseminated MB progression when combined with other chemotherapeutics.
Medulloblastoma (MB) constitutes the most common malignant brain tumor in childhood, for which comprehensive management is operation followed by chemotherapy and radiation.1 Recurrences (local or disseminated lesions) are observed among nearly all MB, along the neuraxis including leptomeninges, the ventricular system, and cerebrospinal parenchyma, signifying a formidable treatment challenge.2,3 However, a majority of studies focus on the biological basis and therapeutic strategies of primary tumors, leaving a lack of complete understanding of molecular mechanisms regulating metastases and establishment of specific treatments approved for disseminated MB.
Previously, medulloblastoma stem-like cells (MBSCs) have been identified to drive tumorigenesis and recurrence with the potential of self-renewal and resistance to chemotherapy and radiation.4 MBSCs show restricted capacity to maintain stemness when undergoing metastasis, which requires the efficient cooperation of the MBSC niche to protect stem-like properties.5 In the tumor microenvironment (TME), astrocytes, the most abundant of glial cells, are reactivated to play a critical role in supporting tumor growth and inducing protection from chemotherapy.6 Recently, we reported that tumor-associated astrocytes (TAA) are distributed across the MB releasing cytokines to provide a supportive TME for tumor proliferation.7 Though gene functions associated with stemness are well studied in cancer, microenvironmental factors regulating stem-like properties remain poorly understood. Furthermore, because cancer stem-like cells exhibit extreme refractoriness to various therapies and are enriched in tumor relapses after treatment in the setting of chronic inflammation induced by chemoradiotherapy, we ask whether generation of the inflammatory microenvironment can prevent MBSCs from differentiating.
In this study, we found that MBSC enrichment was attributed to an increased level of C-C motif ligand 2 (CCL2) released by TAA. CCL2 derived from a wide range of cells plays a key role in inducing inflammation and promoting metastasis.8,9 Herein, we identified that a Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3)–dependent Notch signaling triggered MBSC stemness activated by TAA-derived CCL2. As cancer cells disrupt the apoptotic process to metastasize,10 we investigated necroptosis, a type of programmed necrosis, which was activated by receptor-interacting protein 1/3 (RIP1/3) and acted to shape the cytokine microenvironment and promote oncogenesis.11 Our study provides insight into how stemness is regulated in MB metastasis, and explains how the principle necrosome leads to inflammatory TME.
Materials and Methods
Human MB Samples
A total of 74 patients with MB recurrence (local or disseminated) were included in this study from January 2013 to January 2019. Clinicopathological information was provided in Supplementary Tables 1 and 2. In addition, a total of 136 cases with primary MB were collected in our hospital from January 2010 to November 2018. Tissue collection information is provided in the Supplementary Material. All procedures were approved by the research ethics committee of our institution, and written informed consent was taken from each patient included in the study. The following methods have detailed descriptions in the Supplementary profiles.
Cell Isolation and Culture
To isolate MBSCs, human MB samples were minced and digested in a solution containing papain (20 U/mL) and DNase (4%). Cluster of differentiation (CD)133+/CD15+ (Abcam Biotechnology) cells were sorted by fluorescence activated cell sorting and cultured at 1 × 107/mL in Neurobasal medium supplemented with B27 (2%), basic fibroblast growth factor (20 ng/mL), epidermal growth factor (50 ng/mL), L-glutamine (1%), and penicillin/streptomycin (1%). The differentiated control cells were cultured in fetal bovine serum (FBS)–containing Dulbecco’s modified Eagle’s medium (DMEM). Functional studies were performed with not more than 3 passages. TAA were isolated from human MB and labeled “TAA + pathology #. . . .” Briefly, MB was digested using papain to obtain a single cell suspension, and then centrifuged through a 35‒65% Percoll gradient (Sigma). Cells from phosphate buffered saline (PBS) to 35% interface were cultured in full DMEM with 10% FBS.
Slice Culture
The 400 μm slices of human surgical MB samples were prepared using a VT1000S vibratome (Leica Microsystems) as previously described.7 Slices were transferred onto Millicell culture membrane inserts (Merck Millipore) in a 6-well plate with 1 mL DMEM containing 25 mmol/L HEPES, and cultured in a humid incubator with 5% CO2 at 37°C for the indicated time.
Histology and Immunohistochemistry
Surgical MB tissues were fixed in 10% formalin overnight. Samples were embedded in paraffin, and 5 µm sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry (IHC), paraffin-embedded sections were stained with indicated antibodies at 4°C overnight, and then washed and incubated with secondary antibodies for 1 hour. The number of positive stained cells was determined based on counting of 500 nuclei in 4 high-magnified (400×) representative fields and reported as a percentage.
Western Blot and Immunoprecipitation Analysis
Tissues and cells were lysed in splitting buffer (pH = 7.4) containing protease-inhibitor cocktail. Whole proteins were separated in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred on polyvinylidene difluoride membranes. Membranes were subsequently incubated with associated antibodies overnight at 4°C, and then probed with secondary antibodies for 1 hour at room temperature. Anti-JAK2 antibody or anti-RIP1 antibody cross-linked with protein A/G beads were incubated with 500 μg lysates overnight at 4°C, respectively. Then proteins were eluted from beads and added into 10% SDS-PAGE gel for electrophoresis. Immunoblotting for indicated antibodies was performed.
Quantitative PCR
RNA was isolated using TRIzol reagent according to the manufacturer’s procedures (Invitrogen). Complementary DNA was synthesized by using the iScript reverse transcription kit. Quantitative levels of mRNA were determined by using SYBR Green on the Mx3000P quantitative (q)PCR system and normalized for expression of glyceraldehyde 3-phosphate dehydrogenase mRNA.
Transwell Assay
MBSCs or TAA (1 × 103/100 μL) were seeded in the lower or upper chambers, respectively. The indicative reagents were supplemented in lower chambers. After 8 hours, the filters were fixed and stained with 0.1% crystal violet for 15 minutes.
Luciferase Reporter Assay
MBSCs transfected with CSL/pCI or Jag-1/pCl were treated with CCL2 or PBS, and then lysed with 1× passive lysis buffer as supplied in the luciferase reporter assay kit (Promega). Luciferase and Renilla luciferase activity were separately tested using a fluorescence spectrophotometer (Infinite M200).
In Situ Hybridization
Digoxigenin (DIG)–uridine triphosphate labeled probes for CCL2 were added to this mixture on frozen sections, and hybridization was performed at 60°C for 48 hours. After hybridization, sections were incubated overnight at 4°C with alkaline phosphatase–conjugated anti-DIG antibodies. Bound probe was visualized by incubating sections in NBT/BCIP (nitro blue tetrazolium / 5-bromo-4-chloro-3-indolyl phosphate) overnight in the dark.
Transplantation and Pharmaceutical Administration
All animal experiments were performed in accordance with the national guidelines at the Chinese PLA General Hospital. The MBSCs and TAA (1:5, totally 5 × 105 cells in 5 μL) were used for intracranial co-transplantation into the right hemisphere by utilizing a mouse stereotaxic apparatus as previously described.12 Engraftments were checked on the seventh day after transplantation and mice with equivalent volumes were randomized into several groups (n = 6/group) for gavage treatment: necrostatin-1 (Nec-1) plus methotrexate (MTX), medium chain triglycerides (MCT) plus MTX, and Nec-1 plus MCT.
Statistical Analysis
Data statistical handling was performed using SPSS 19.0 and GraphPad Prism 8.0 software. Values were shown as mean ± SD with error bars representing SEM. An unpaired t-test was utilized between 2 groups and comparison of mean values between multiple groups was evaluated by one-way ANOVA. For all statistical methods, P < 0.05 was considered significance.
Results
MBSCs Are Enriched in Disseminated MB
To observe the presence of MBSCs in disseminated MB, we examined expression of sex determining region Y–box 2 (Sox2) and paired box 6 (Pax6) by IHC in human MB samples. A large number of MBSCs (Sox2+ or Pax6+) were present among the MB cells (positive for zinc finger of the cerebellum protein [Zic+]; Fig. 1A and Supplementary Fig. 1A). Previously we identified the MBSCs in primary tumors.13 Herein, we compared the proportion of MBSCs in disseminated MB with the primary compartment, which displayed a higher percentage of Sox2+ cells in disseminations (Fig. 1B). To further confirm the enrichment of this population, we purified MBSCs from human surgical recurrent samples by harvesting CD133+/CD15+ cells via fluorescence activated cell sorting. Flow cytometry analysis revealed that about 5% of cells from disseminated MB were positive for CD133/CD15, which was higher than that (~2.5%) in primary tumors (Supplementary Fig. 1B). Strong expression of Nestin was observed in MBSCs. Moreover, a majority of Sox2+ cells expressed Ki-67 in both sorted cells and tumor sections from disseminated MB addressing their proliferative potential (Fig. 1C and Supplementary Fig. 1C). Thus, these data suggest that MBSC enrichment promotes the enhanced stemness in disseminations.
Fig. 1.

TAA were enriched by human MBSC-derived IL-6. (A) Representative images of Zic1 expression in sagittal section from SHH-MB9578 and CB and expression of Sox2 and Pax6 in disseminations. (B) The percentage of Sox2+ cells in disseminated (n = 8) and primary (n = 15) MB. (C) Representative images of Nestin and Sox2/Ki-67 expression in CD133+/CD15+ cells sorted from disseminated MB (n = 5). (D) Representative images of S100β/Zic1 or GFAP/Zic1 expression in disseminated MB (n = 9). (E) Representative images of S100β and GFAP expression in TAA (n = 7). (F) The GFAP expression in disseminated and primary MB (n = 10). (G) TAA migration with IL-6, IL-6 + MP5-20F3 and DMSO in the lower wells of Transwell system (n = 4). (H) Immunostaining for GFAP when MB slices were cultured with or without MP5-20F3 (n = 3). Scale bars, 10 μm.
TAA Are Reactivated by MBSC-Derived Interleukin-6
A previous study has reported that interleukin (IL)-6/IL-6 receptor (IL-6R) is directly responsible for astrocyte activation.14 Since a high level of IL-6 was observed in both primary and disseminated MBSCs in our study (Supplementary Fig. 2A and 2B), we next sought to investigate whether MBSC-derived IL-6 could activate astrocytes in disseminated MB. The presence of TAA which were positive for astrocyte activation markers glial fibrillary acidic protein (GFAP) and S100β were identified surrounding MB cells throughout the tumor (Fig. 1D). TAA were also abundantly detected in MBSC-derived intracranial xenografts (Supplementary Fig. 2C). Similarly, robust staining of GFAP and S100β was observed in TAA purified from metastases (Fig. 1E). Moreover, a greater number of TAA were detected in disseminated MB compared with primary therapy-naïve tumors (Fig. 1F). Then we tested the chemotactic properties of TAA using a Transwell system containing IL-6 in the lower wells, and challenged the chemotactic behavior by using IL-6 neutralizing antibody (MP5-20F3). More than 70% of TAA migrating were induced in the presence of IL-6 and this recruitment was suppressed by IL-6 blockage (Fig. 1G). Furthermore, we treated the Transwell co-culture of MBSCs (lower) and TAA (upper) with MP5-20F3. At 24 hours after treatment, approximately 30% of TAA were recruited through the membranes of upper wells compared with the absence of MP5-20F3 (Supplementary Fig. 2D). In order to determine the role of IL-6/IL-6R in TAA recruitment, we knocked down IL-6R expression in TAA with a lentiviral system producing short hairpin (sh)RNA against IL-6R (Supplementary Fig. 2E). As expected, when co-cultured with MBSCs, TAA recruitment was inhibited, suggesting that IL-6R deficiency impaired the migration of TAA (Supplementary Fig. 2F). We continued to use organotypic slice culture to examine the chemotactic function of IL-6. Tumors with adjacent normal cerebellum (CB) slices were prepared from surgical samples and treated with or without MP5-20F3 for 3 days. Following treatment, GFAP expression was examined showing the abrogating effects of IL-6 on TAA activation (Fig. 1H). Thus, these data suggest that within the TME of disseminated MB, TAA are recruited by MBSCs via IL-6.
TAA in Disseminated MB Secrete CCL2
Previously, astrocytes have been reported to secrete several cytokines supporting brain development or regulating inflammation progression.15,16 To assess cytokine production, we collected sera from 20 patients with metastases and screened cytokines by human cytokine array. CCL2 was the most commonly secreted cytokine (Supplementary Fig. 3A). To test whether CCL2 was derived from TAA, we harvested the supernatant of TAA to examine cytokines and found the remarkably increased level of CCL2 (Fig. 2A and B). Moreover, we found that the level of CCL2 in TAA media from disseminated MB was higher than that from primary MB, indicative of the specific function of CCL2 in MB dissemination (Fig. 2C). Abundant expression of CCL2 mRNA was obtained in TAA, but lower in MBSCs or MB tumor cells (Fig. 2D), which suggested the limited production of tumor cells and highlighted the cytokine support of TAA. To further confirm the presence of CCL2, we looked for it in tumor tissues finding high protein and mRNA levels (Fig. 2E and Supplementary Fig. 3B).
Fig. 2.

TAA secreted CCL2 in disseminated MB. (A) ELISA-detected cytokines in CM of TAA isolated from Group 4 MB7609, Group 3 MB7965, and Group 3 MB9678. (B) Concentration of CCL2 in CM of TAA (n = 4) and normal astrocytes (n = 3). (C) Concentration of CCL2 in supernatant of disseminated and primary TAA culture (n = 3). (D) Expression of CCL2 mRNA in TAA, MBSCs, and MB cells derived from MB (n = 6, 2 WNT, 2 sonic hedgehog and 2 Group 3 MB). (E) Immunoblotting analysis of CCL2 in disseminated MB and CB. (F) CCL2 mRNA expression in disseminated MB examined by in situ hybridization. (G) CCL2 mRNA expression in 9-month, 3-year, and 42-year CB examined by in situ hybridization. (H) CCL2 mRNA expression in isolated astrocytes from 9-month CB. Scale bars, 10 μm. Abbreviation: AS, astrocytes.
To identify the anatomical area that was positive for CCL2, we examined the presence of CCL2 mRNA in MB, developing and mature CB by in situ hybridization. CCL2 mRNA was found in disseminated MB (Fig. 2F), as well as in the Purkinje layer of 9-month pediatric CB, but not after 3 years old (Fig. 2F and G). The co-location of CCL2 and calbindin, the marker of Purkinje cells, was observed in molecular layers in the 9-month CB, but not in the 3-year CB (Supplementary Fig. 3C). We isolated astrocytes from developing CB and detected remarkable amplification of CCL2 mRNA (Fig. 2H). Additionally, CCL2 was stimulated by treatment of 50% granule neuron progenitor media (Supplementary Fig. 3D). Taken together, these data demonstrate that TAA efficiently secrete functional CCL2 into the TME.
CCL2 Is a Clinically Relevant Chemokine in Recurrent MB
The abundance of CCL2 prompted us to further explore the association with clinicopathological characters. High levels of CCL2 (>4500 pg/mL) were observed in more than two-thirds of cases, and about 75% of patients suffered from recurrence in less than 19.6 months (Supplementary Table 1). Clinically, operations were performed when recurrent patients suffered from severe intracranial hypertension or spinal cord compression, which resulted in a limited bank of samples. We collected 21 samples of recurrent MB during operations or biopsies to examine CCL2 expression by IHC. CCL2 was significantly elevated in large-cell/anaplastic MB and Group 3 MB (Fig. 3A and B), correlating with more aggressive tumor subtypes (Supplementary Table 1). We then analyzed the numbers of TAA among subgroups. More TAA were observed in the Group 3 MB subgroup, and CCL2 levels correlated with the numbers of TAA (Supplementary Fig. 4). Additionally, we collected 136 therapy-naïve MB from our hospital and analyzed the expression of CCL2, which presented a robust expression in the large-cell/anaplastic subgroup (Supplementary Fig. 5A), and cases in the higher risk stage were more likely to express CCL2 (Supplementary Fig. 5B). These data indicate that CCL2 correlates positively with frequent local recurrence or dissemination.
Fig. 3.

CCL2 correlated with poor prognosis of patients with recurrent MB. (A) CCL2 expression in classic MB, MB with extensive nodularity, and large-cell/anaplastic MB. (B) CCL2 expression in molecular subgroups of recurrent MB. (C) Representative enhanced T1-weighted images for one pair of matched primary and disseminated Group 3 MB7965 (red arrow), with H&E staining and IHC staining for Nestin, Ki-67, and MMP9. (D) Representative images of CCL2 expression in one pair of matched MB and the statistical graph showing CCL2 scores in 10 pairs. (E) The association of CCL2 with Nestin or Ki-67 estimated by Pearson analysis. (F) The overall survival Kaplan–Meier curve of recurrent MB patients. Scale bars, 10 μm.
To further compare the expression of CCL2 in primary and relapsed MB, 10 pairs of matched tumors were selected. Nestin, Ki-67, and matrix metalloproteinase (MMP)9 expressions were elevated in disseminated MB, and CCL2 was strongly expressed in relapsed MB (Fig. 3C and D). Moreover, CCL2 was positively associated with Nestin or Ki-67 estimated by Pearson analysis (Fig. 3E), supporting the evidence linking CCL2 with encouragement of MBSCs to proliferate. In the cohort of recurrent cases, the median follow-up time was 39.6 months (range, 2.0‒61.0 mo). The 21 patients were grouped by the average of CCL2 immunostaining intensity. Patients with lower levels showed a longer survival (Fig. 3F). Similar results were obtained among therapy-naïve patients (Supplementary Fig. 5C). Both logistic analysis and the Cox proportional model identified CCL2 as a valuable prognostic indicator (Supplementary Tables 1 and 3). To confirm the survival analysis, we analyzed data from pan-cancers in the database of The Cancer Genome Atlas, finding that patients with higher CCL2 expression had poorer prognosis (Supplementary Fig. 5D). Therefore, the clinical data highly suggest that CCL2 plays a critical role in MB recurrence, conferring poor prognosis.
CCL2 Maintains the Stem-like Properties of Disseminated MBSCs
Having identified that CCL2 correlated with MB recurrence, we next investigated the potential contribution of CCL2 to the stem-like properties of MB. MBSCs were cultured in Neurobasal media without any growth factors to detect the proliferation and differentiation at different timepoints. Most MBSCs remained viable and undifferentiated for 48 hours (Supplementary Fig. 6A). However, they started ceasing proliferation and differentiating after 48 hours. These data suggested that MBSCs couldn’t maintain their own properties of stemness. To test whether exogenous CCL2 contributed to sustaining stemness of MBSCs, cells were cultured in the presence of CCL2 for 72 hours. Approximately 40% of MBSCs were Ki-67+ compared with the 10% in control, and half of MBSCs were Sox2+ in the presence of CCL2 (Fig. 4A). Similarly, the percentage of CD133+/CD15+ cells was significantly elevated when MB cells dissociated from tumors were treated with CCL2 (Supplementary Fig. 6B). When cultured without poly-D-lysine coating for 10 days, both the size and number of MBSC spheres were increased by CCL2 stimulation (Supplementary Fig. 6C), indicating that exogenous CCL2 supports MBSC proliferation and maintains stemness properties.
Fig. 4.

TAA-derived CCL2 maintained MBSC stemness. (A) MBSCs treated with CCL2 or PBS for 72 hours and immunostained for Ki-67 and Sox2 (n = 3). (B) MBSCs cultured in the media mixed 1:1 with supernatant of FBS-free TAA culture or PBS and immunostained for Ki-67 (n = 3). (C) Up, expression of Sox2 in MBSCs expressing shCCR2#2 or scrambled shRNA in presence of CCL2 (n = 3). Down, expression of Sox2 in MBSCs co-cultured with CCL2-deficient or CCL2-normal TAA (n = 3). (D) Immunostaining for Sox2 and GFP in the co-culture of MBSCs and shCCL2-GFP expressing TAA after treatment with CCL2 (n = 4). (E) Sox2, Pax6, and Nestin mRNA levels in MBSCs cultured in the media mixed 1:1 with supernatant of FBS-free TAA culture or PBS (n = 3). (F) Immunostaining for Ki-67 and NeuN in disseminated MB slices cultured with or without PF6309 (n = 3). (G) Growth rate and tumor size of subcutaneous co-transplantation of MBSCs and TAA infected with shCCL2#3-GFP, shCCL2#5-GFP, or scrambled control (n = 6/group). (H) Immunostaining for Ki-67, NeuN, and GFP in subcutaneous xenografts (n = 6). Scale bars, 10 μm.
To examine the effects of TAA-derived CCL2 on stem-like features, FBS-free conditioned medium (CM) from TAA cultures was provided to these cells mixed 1:1 with MBSC cultures. We found that about 50% of cells were proliferating at 72 hours treatment (Fig. 4B). Similarly, we treated co-cultures of MBSCs and TAA with CCR2 inhibitor (PF6309) and examined the expression of Sox2 and Zic1. TAA-protected stemness features were significantly declined by PF6309 (Supplementary Fig. 6D), suggesting that TAA maintained MBSC stemness relying on CCL2. To further determine whether the CCL2/CCR2 axis promoted MBSC stemness, we manipulated these cells through a lentiviral approach (shRNA) to reduce the expression of CCR2 (Supplementary Fig. 6E). After CCR2-low cells were treated with CCL2 for 72 hours, Sox2 expression was found to be downregulated, demonstrating decreased stemness in MBSCs lacking CCR2 (Fig. 4C up). In turn, TAA were infected with lentiviruses carrying shCCL2–green fluorescent protein (GFP) and co-cultured with MBSCs for 3 days. The decreased percentage of Sox2+ cells indicated that MBSC stemness depended on TAA-secreted CCL2 (Fig. 4C down). As a rescue, exogenous CCL2 was added into co-cultures of MBSCs and CCL2-low TAA. As expected, the inhibited expression of Sox2 was significantly increased (Fig. 4D). To better corroborate our findings, we collected MBSCs cultured in media enriched with serum-free TAA CM (1:1) and evaluated them for stemness-associated genes expression by qPCR. The mRNA levels of Sox2, Pax6, and Nestin were elevated after TAA media stimulation (Fig. 4E). Collectively, these data suggest that TAA-derived CCL2 maintains stemness and stimulates proliferation of MBSCs.
To further investigate the supporting role of CCL2 in the TME, organotypic tumor slices were treated with or without PF6309 for 4 days. Immunohistochemistry revealed that proliferation was compromised and a significant proportion of tumor cells were under differentiation (NeuN+) following the treatment (Fig. 4F). Decreased levels of MMP2 and MMP9 in supernatant were also observed by enzyme-linked immunosorbent assay (ELISA), indicating that the markers of aggressive tumor cells were suppressed by CCL2/CCR2 blockade (Supplementary Fig. 6F). In addition, we subcutaneously co-transplanted MBSCs and TAA infected with shCCL2-GFP lentivirus. Following the transplantation, growth of xenografts after CCL2 deletion in TAA was significantly reduced (Fig. 4G). Proliferation was impaired and amounts of differentiated cells were enriched after CCL2 deletion (Fig. 4H). Collectively, the above data support that TAA-secreted CCL2 is required for MBSC stemness.
CCL2 Regulates Stemness through Notch Signaling Activation
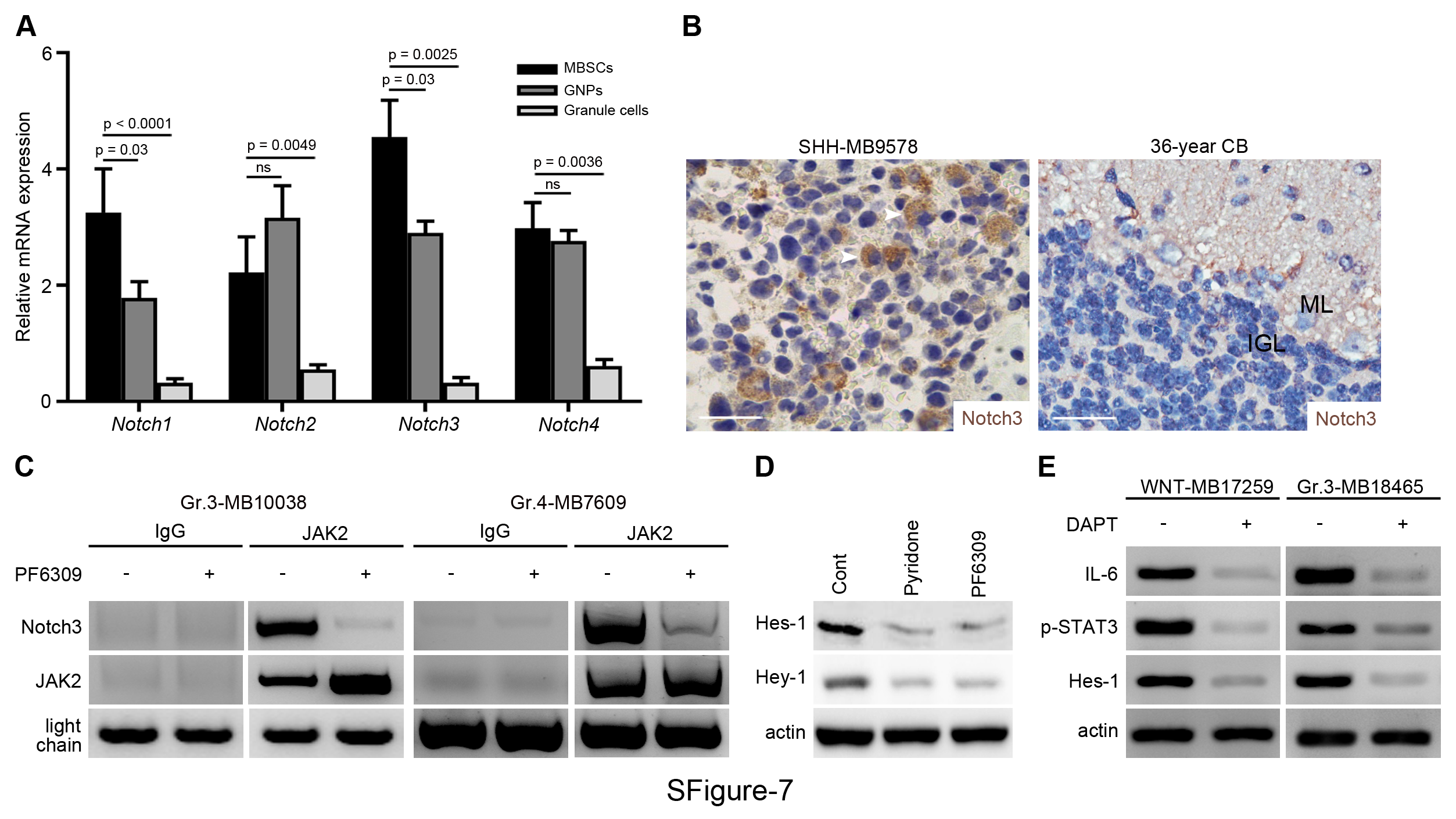
Having identified the indispensable role of CCL2 in stemness maintenance, we next sought to investigate the mechanism underlying our findings. Notch signaling has been reported to be implicated in brain cancer stem-like genesis.17 Recent studies have also revealed that the Notch pathway is activated by protein atonal homolog 1 in MB with leptomeningeal dissemination18; moreover an inflammatory TME has shown to activate Notch signaling.19 Thus, we asked whether the CCL2/CCR2 axis could induce Notch signaling activation to maintain stemness of MBSCs. To address this, we examined mRNA expression of Notch paralogs (Notch 1–4) in MBSCs, GNPs from developing CB, and granule cells from adult CB by qPCR, finding a significant difference of Notch3 expression in MBSCs (Fig. 5A and Supplementary Fig. 7A). Similarly, Notch3 expression was observed by IHC in disseminated MB (Supplementary Fig. 7B). To further confirm activation of Notch signaling, we examined Notch pathway targeted transcription factor expression in sorted MBSCs by immunofluorescence. Co-localizing of downstream members of Notch signaling cascade, Hey-1/Hes-1 and Sox2 were detected in a majority of MBSCs (Fig. 5B). In sum, our data demonstrate that Notch signaling is activated in MBSCs during MB dissemination.
Fig. 5.

CCL2-induced stemness required Notch signaling activation. (A) Notch1–4 mRNA expression in MBSCs and MB cells (n = 6). (B) Representative images of Hey-1, Hes-1, and Sox2 expression in MBSCs (n = 3). (C) Immunoblotting analysis of JAK2, STAT3, phosphorylated STAT3 (p-STAT3) and Notch3 expression in MBSCs treated with or without PF6309. (D) Lysate from MBSCs that had been treated with PF6309 or DMSO immunoprecipitated with anti-JAK2 antibody and tested for expression of JAK2 and Notch3. Input control was probed. (E) CSL/Notch promoter and Jag-1 promoter induced by CCL2 or PBS (n = 3). (F) Hey-1 and Hes-1 mRNA level in MBSCs treated with pyridone, PF6309, or DMSO (n = 4). (G) Representative images of Sox2 expression in slices from disseminated MB cultured with or without DAPT (n = 3). Scale bars, 10 μm.
Interplay of JAK2/STAT3 and Notch signaling has been studied in development and tumorigenesis.20 We next investigated the activation of the Notch pathway mediated by CCL2/CCR2 via JAK2/STAT3 signaling. For this purpose, immunoblotting was performed after MBSCs were cultured in the presence or absence of PF6309 demonstrating the suppression of JAK2, phosphorylated STAT3, and Notch3 with CCR2 inhibition (Fig. 5C). Furthermore, binding of endogenous Notch3 to JAK2 was detected by immunoprecipitation (IP) of the lysates from MBSCs using an anti-JAK2 antibody. As expected, similar results were replicated in the presence of PF6309 (Fig. 5D and Supplementary Fig. 7C). Additionally, luciferase reporter assays revealed that CCL2 stimulated activity of CBF-1/suppressor of hairless/Lag1 (CSL)/Notch and Jag-1 promoters (Fig. 5E), indicating that CCL2/CCR2 axis promoted the interaction between JAK2/STAT3 and Notch signaling. To elucidate the role of JAK2/STAT3 in mediating Notch signaling activation, we restrained the functions of JAK2 and CCR2 using a JAK2 inhibitor (pyridone) and PF6309, finding a significant repression of Hey-1 and Hes-1 (Fig. 5F and Supplementary Fig. 7D). To test whether Notch signaling promoted stemness in MB cells, slices from disseminated MB were treated with a known γ-secretase inhibitor (DAPT) which indirectly inhibits Notch3.21 After 72 hours of treatment, Notch inhibition dramatically impaired stemness, as evidenced by a decreased Sox2 expression (Fig. 5G). Previous work has reported a positive feedback loop between Notch signaling and STAT3 mediated by toll-like receptor–stimulated IL-6.20 In accord with those reports, we found reduced IL-6 secretion following Notch signaling inhibition (Supplementary Fig. 7E). Because MBSC-derived IL-6 induced astrocyte activation (Fig. 1G, H and Supplementary Fig. 2D), targeting Notch signaling in MBSCs impaired TAA recruitment. Collectively, the above data suggest that the CCL2/CCR2 complex confers stemness to disseminated MB by activating Notch signaling through the JAK2/STAT3 pathway.
Production of CCL2 Relies on RIP1/3-Dependent Necroptosis
The critical role of CCL2 prompted us to ask how TAA produced CCL2. When MB patients received chemoradiotherapy, progressive chronic inflammation occurred throughout the tumor. Given the slow proliferation of TAA and limited sensitivity to chemoradiotherapy, we performed staining by TUNEL (terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling) to detect cell death, which in turn shaped the inflammatory TME.22 We found more dead cells throughout disseminated lesions (Supplementary Fig. 8A). Recently necroptosis has been reported to be involved in oncogenesis and injury-associated astrocyte activation via necrosome (RIP1/RIP3/MLKL complex).23,24 Since distinct types of cell death (necrosis, apoptosis, or necroptosis) couldn’t be discriminated based on TUNEL results, we performed immunoblotting analysis to assess the modes of cell death, which revealed high levels of RIP3 and MLKL, but low expression of the apoptosis marker cleaved caspase-3 in TAA compared with MB cells (Fig. 6A), indicating that TAA underwent necroptosis in disseminated MB. To further confirm the occurrence of necroptosis, we examined RIP3 in matched primary and recurrent MB, finding the strong RIP3 immunoreactivity within metastases (Supplementary Fig. 8B). Both RIP1 and RIP3 co-localized with S100β (Fig. 6B). Furthermore, IP with anti-RIP1 antibodies displayed that RIP3 interacted with RIP1 only in disseminated TAA, but not in primary TAA (Fig. 6C and Supplementary Fig. 8C). We then infected TAA with a lentivirus carrying shRNA against RIP3. Downregulation of phosphorylated MLKL was identified following infection (Supplementary Fig. 8D). These data suggest that the necrosome is activated in TAA within disseminated MB.
Fig. 6.

Targeting TAA necroptosis suppressed MB stemness and dissemination. (A) Immunoblotting analysis of RIP3, MLKL, and cleaved caspase-3 expression in TAA and MB cells within disseminated MB. (B) Representative images of RIP1, RIP3, and S100β expression in TAA. (C) Lysate from primary TAA (pTAA) and disseminated TAA (dTAA) immunoprecipitated with anti-RIP1 antibody and tested for expression of RIP1 and RIP3. Input control was probed. (D) Concentration of CCL2 in media of MTX-treated primary TAA following pretreatment of Nec-1 or DMSO (n = 3). Immunoblotting analysis of RIP1/RIP3/MLKL and cleaved caspase-3 expression in these TAA. (E) MB disseminations (white arrow) examined by microPET/CT. Ratio of SUV showing radioactive absorption of metastases (n = 3/group). (F) Tumor volumes of xenografts (red arrow) derived from primary MBSCs following treatment with Nec-1 plus MTX, MCT plus MTX or Nec-1 plus MCT examined by microMRI (n = 6/group). (G) Survival curve of xenograft-bearing mice in above-mentioned groups. (H) Immunoblotting analysis of RIP1/RIP3/MLKL and CCL2 expression in xenografts following treatment with Nec-1 plus MTX or MCT plus MTX. (I) The schematic model showing necroptosis-associated TAA cytokine microenvironment underlying MB recurrence or dissemination contributes to maintaining MBSC stemness via Notch signaling activation. Scale bars, 10 μm.
Because necroptotic cells modulate the TME via cytokine release,25 we next tested CCL2 levels after necroptosis inhibition. For that, TAA were purified from primary MB and cultured in the presence of necroptosis inhibitor Nec-1.26 Two days following pretreatment with Nec-1, TAA were treated with MTX, carboplatin (CBP), or ifosfamide (IFO) for 2 days. Pretreatment with Nec-1 significantly compromised the concentration of CCL2 in supernatant and expression of RIP1/RIP3/MLKL, as well as promoted apoptosis in TAA as evidenced by increased caspase-3 expression (Fig. 6D and Supplementary Fig. 8E). Similarly, CCL2 was also dramatically decreased when TAA were treated with MTX or an IFO/CBP/etoposide (VP-16) scheme following infection with shRIP3 lentivirus (Supplementary Fig. 8F). These data suggest that inhibiting necroptosis efficiently prevents CCL2 production. Since TAA-derived CCL2 played a supportive role in MBSC survival, we next explored if necroptotic TAA regulated MBSC stemness. The serum-free CM was harvested from dimethyl sulfoxide (DMSO) plus MTX-treated or Nec-1 plus MTX-treated TAA to treat primary MBSCs. Twenty-four hours later, more Sox2+ cells were observed in the presence of CM of single MTX-treated TAA (Supplementary Fig. 8G). Taken together, these data indicate that MBSC stemness requires microenvironmental cues regulated by necroptosis in disseminations.
Targeting Necroptosis Presents Therapeutic Potential for Disseminated MB
The above-mentioned results have reflected that necroptotic TAA contributed to the progression of disseminated MB. We then generated cohorts of MBSC-bearing mice to investigate whether metastases could be attenuated by targeting necroptosis. MBSCs purified from primary MB were intracranially transplanted into the forebrains of severe combined immunodeficient mice in order to obtain disseminated lesions. After 1 week following transplantation, animals received pretreatment with Nec-1 for 1 week prior to MTX chemotherapy once every 2 days. Six weeks later, uptake of 18F-fluorodeoxyglucose on microPET/CT demonstrated that Nec-1 plus MTX powerfully inhibited metastases, as evidenced by the standard uptake value (SUV; Fig. 6E). Based on microMRI, MBSCs pretreated with Nec-1 gave rise to much smaller masses (Fig. 6F). The Nec-1–pretreated mice also had longer survival (Fig. 6G). Moreover, a majority of MB cells in the MCT group were highly proliferative and aggressive, expressing high levels of Ki-67 and MMP9 (Supplementary Fig. 9). Noticeably, the Nec-1–pretreated tumors displayed a remarkable increase of the differentiated neuron marker NeuN (Supplementary Fig. 9), suggesting that ablation of necroptosis significantly inhibited stemness and led to differentiation. To determine the TAA necroptosis, we examined caspase-8 expression,11 which showed reduction of caspase-8 in xenografts receiving Nec-1 plus MTX treatment (Supplementary Fig. 9). Furthermore, the levels of CCL2 in transplanted MB significantly declined following Nec-1 pretreatment, coupled with the decreased levels of RIP1/RIP3/MLKL (Fig. 6H), supporting that CCL2 release depended on chemotherapy-induced necroptosis. Collectively, these data demonstrate that the necroptosis-associated TAA inflammatory TME underlying MB metastases activates Notch signaling in disseminated MBSCs (Fig. 6I), providing a potential therapeutic opportunity.
Discussion
Given a lack of successful treatment modalities in MB recurrence or metastasis, current efforts to target disseminated lesions have become a focus for improving patient survival. The functions and cell populations within the TME vary dynamically throughout tumor progression, which requires a detailed understanding of these complex interactions for effective treatment. Chronic inflammation is considered as the seventh hallmark of cancer, which can be induced by chemoradiotherapy.27 However, the effects of microenvironmental inflammation on metastasis remain unknown. Herein, we demonstrate that, upon RIP1/RIP3/MLKL-mediated necroptosis, TAA release CCL2 to maintain MBSC stemness in disseminated lesions through Notch signaling activation. These findings, for the first time, reveal an indispensable role of TAA in promoting stem-like features of MBSCs, highlighting the importance of an inflammatory microenvironment in metastasis.
MBSCs with low response to existing therapies are further enriched in MB recurrence, which relies on TAA. The CCL2/CCR2 axis bridges the junction of 2 populations, TAA and MBSCs. Garzia et al discovered that CCL2/CCR2 drove metastases through circulating MB cells.28 Interestingly, by analyzing therapy-naïve models, the authors suggest that MB cells are a source of CCL2 as opposed to TAA in this study. We purify MBSCs and TAA from human recurrent or disseminated MB samples after chemoradiotherapy. Our findings demonstrate that TAA-derived CCL2 secretion relies on the RIP1/3-dependent necroptosis which is induced by chemoradiotherapy. We detected a higher level of CCL2 mRNA in TAA compared with MB cells (Fig. 2D). Furthermore, chemotherapy or radiation leads to a reduction of tumor cells and a relatively elevated proportion of TAA in recurrent lesions (Fig. 1F). The increased population of TAA secrete more CCL2.
CCL2 has been shown to mediate cancer stem-like properties via inducing Notch1 in breast cancers.29 The function of Notch signaling in brain development and tumors has been well established via influencing stemness and terminal differentiation. Currently, the anti-Notch1 negative regulatory region antibody is being used as therapy in MB to prevent a high frequency of spinal disseminations.30 Notch signaling is activated in recurrent MB, which is known to harbor cancer stem cell–like characters. Nevertheless, the function of CCL2-mediated JAK2/STAT3 in regulating Notch3 promoter activity may underlie the induction of the p38 mitogen-activated protein kinase axis.20
Having identified the role of CCL2-mediated Notch signaling in maintaining stemness during MB metastasis, we further detected the mechanism involved in TAA-derived CCL2 release. It is likely that MB inevitably progresses within a chronic inflammatory microenvironment induced by chemoradiotherapy, in which different types of cell death occur.11 Uncontrolled necrosis is traditionally deemed to account for acute cell death. However, a recent study about cell death has unveiled the molecular mechanism of necroptosis, the programmed necrosis, which is mediated by intracellular RIP1/RIP3/MLKL cascade.22 Previous studies have reported the tumor-promoting effects of necroptosis in many cancers, including liver and pancreatic cancer.23,31 Based on the research that astrocytes underwent necroptosis releasing cytokines to harm the recovery after spinal cord injury,24 we discovered that necroptotic TAA also release abundant cytokines. Although most studies examine tumor cell necroptosis, the pro-tumoral effects of necroptosis are also driven by host nontumoral cells within the TME, consistent with our data regarding the necroptotic TAA. Considering the impacts of inflammation on metastasis, the beneficial effects of targeting necroptosis can result in a restricted production of cytokines. However, necroptosis is considered a double-edged sword, exerting both tumor-promoting and antitumor effects.32 The anticancer roles include immunogenicity elicitation and therapy-driven immunosurveillance. Although accumulating evidence suggests necroptosis is the antitumor target, a deeper understanding of the mechanisms in tumorigenesis is critical to evaluate the clinical application.
Funding
This work was supported by the National Natural Science Foundation of China (81902975 and 81871087), the 65th China Postdoctoral Science Foundation (2019M653940), Medical Big Data Research Program of Chinese PLA General Hospital (2018MBD-20), and the Clinical Research Support Program of Chinese PLA General Hospital (2016FCCXYY2010).
Conflict of interest statement. The authors declare no conflict of interest.
Authorship statement. Drs H. Liu and X. Yu contributed to the whole conception and design of this project. Drs H. Liu and Y. Sun were responsible for the details of experimental performance. Drs X. Qi, J. Zhang, S. Feng, and W. Jin made many efforts to the pathological estimates. Drs H. Liu, Y. Sun and J. O’Brien contributed to the analysis and interpretation of data. Drs C. Yu, S. Feng, C. Gu, and Z. Zhao worked as the neurosurgeons to collect the cases and analyze the clinical data. Drs H. Liu and X. Yu completed the manuscript, Fig. s, and tables. Drs J. O’Brien and J. Franco-Barraza made valuable comments and promotion of the writing.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to acknowledge Drs P. Y. Wen (Dana-Farber Cancer Institute) and Y. Liu (Chinese Medical University of Guangdong) for providing valuable suggestions about experimental design. The authors thank Dr I. Nakano (University of Alabama at Birmingham) for sharing plasmids. The authors also thank Drs K. Yao and C. Li (China Capital Medical University) for interpretation of pathological and radiographic data, Q. Fan (Soochow University) for double checking the associated information, and J. Jiang (Clinical Data and Specimen Repositories, Chinese PLA General Hospital) for providing the information of samples. The authors especially would like to thank Dr Z. Yang (Fox Chase Cancer Center) for the thoughtful comments and research training.
References
- 1. Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nat Rev Neurol. 2012;8(6):340–351. [DOI] [PubMed] [Google Scholar]
- 2. Northcott PA, Robinson GW, Kratz CP, et al. Medulloblastoma. Nat Rev Dis Primers. 2019;5(1):11. [DOI] [PubMed] [Google Scholar]
- 3. Ramaswamy V, Taylor MD. Medulloblastoma: from myth to molecular. J Clin Oncol. 2017;35(21):2355–2363. [DOI] [PubMed] [Google Scholar]
- 4. Aref D, Croul S. Medulloblastoma: recurrence and metastasis. CNS Oncol. 2013;2(4):377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang GH, Xu QF, Cui YH, Li N, Bian XW, Lv SQ. Medulloblastoma stem cells: promising targets in medulloblastoma therapy. Cancer Sci. 2016;107(5):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Priego N, Zhu L, Monteiro C, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med. 2018;24(7):1024–1035. [DOI] [PubMed] [Google Scholar]
- 7. Liu Y, Yuelling LW, Wang Y, et al. Astrocytes promote medulloblastoma progression through hedgehog secretion. Cancer Res. 2017;77(23):6692–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang AL, Miska J, Wainwright DA, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol. 2019;16(5):282–295. [DOI] [PubMed] [Google Scholar]
- 10. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23(12):620–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wegner KW, Saleh D, Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci. 2017;38(3):202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H, Sun Q, Zhang M, et al. Differential expression of folate receptor 1 in medulloblastoma and the correlation with clinicopathological characters and target therapeutic potential. Oncotarget. 2017;8(14):23048–23060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu H, Sun Q, Sun Y, et al. MELK and EZH2 cooperate to regulate medulloblastoma cancer stem-like cell proliferation and differentiation. Mol Cancer Res. 2017;15(9):1275–1286. [DOI] [PubMed] [Google Scholar]
- 14. Brunello AG, Weissenberger J, Kappeler A, et al. Astrocytic alterations in interleukin-6/soluble interleukin-6 receptor alpha double-transgenic mice. Am J Pathol. 2000;157(5):1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Molofsky AV, Deneen B. Astrocyte development: a guide for the perplexed. Glia. 2015;63(8):1320–1329. [DOI] [PubMed] [Google Scholar]
- 16. Brandao M, Simon T, Critchley G, Giamas G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia. 2019;67(5):779–790. [DOI] [PubMed] [Google Scholar]
- 17. Teodorczyk M, Schmidt MHH. Notching on cancer’s door: Notch signaling in brain tumors. Front Oncol. 2014;4:341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grausam KB, Dooyema SDR, Bihannic L, et al. ATOH1 promotes leptomeningeal dissemination and metastasis of sonic hedgehog subgroup medulloblastomas. Cancer Res. 2017;77(14):3766–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin X, Kim SH, Jeon HM, et al. Interferon regulatory factor 7 regulates glioma stem cells via interleukin-6 and Notch signalling. Brain. 2012;135(Pt 4):1055–1069. [DOI] [PubMed] [Google Scholar]
- 20. Hildebrand D, Uhle F, Sahin D, Krauser U, Weigand MA, Heeg K. The interplay of notch signaling and STAT3 in TLR-activated human primary monocytes. Front Cell Infect Microbiol. 2018;8:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y, Xie X, Zhu Y, et al. Inhibition of Notch3 prevents monocrotaline-induced pulmonary arterial hypertension. Exp Lung Res. 2015;41(8):435–443. [DOI] [PubMed] [Google Scholar]
- 22. Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. [DOI] [PubMed] [Google Scholar]
- 23. Seifert L, Werba G, Tiwari S, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532(7598):245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fan H, Zhang K, Shan L, et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener. 2016;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. [DOI] [PubMed] [Google Scholar]
- 26. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. [DOI] [PubMed] [Google Scholar]
- 27. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. [DOI] [PubMed] [Google Scholar]
- 28. Garzia L, Kijima N, Morrissy AS, et al. A hematogenous route for medulloblastoma leptomeningeal metastases. Cell. 2018;173(6):1549. [DOI] [PubMed] [Google Scholar]
- 29. Tsuyada A, Chow A, Wu J, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kahn SA, Wang X, Nitta RT, et al. Notch1 regulates the initiation of metastasis and self-renewal of Group 3 medulloblastoma. Nat Commun. 2018;9(1):4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seehawer M, Heinzmann F, D’Artista L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562(7725):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qin X, Ma D, Tan YX, Wang HY, Cai Z. The role of necroptosis in cancer: a double-edged sword? Biochim Biophys Acta Rev Cancer. 2019;1871(2):259–266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.