

Summary
Understanding microbial communities' roles in human health and disease requires methods that accurately characterize the microbial composition and their activity and effects within human biological samples. We present sMETASeq (small RNA Metagenomics by Sequencing), a novel method that uses sequencing of small RNAs to jointly measure host small RNA expression and create metagenomic profiles and detect small bacterial RNAs. We evaluated the performance of sMETASeq on a mock bacterial community and demonstrated its use on different human samples, including colon cancer, oral leukoplakia, cervix cancer, and a panel of human biofluids. In all datasets, the detected microbes reflected the biology of the different sample types.
Subject Areas: Genomics, Microbiology, Bioinformatics
Graphical Abstract
Highlights
-
•
Our method “sMETASeq” generates metagenomic profiles from small RNA sequencing data
-
•
sMETASeq jointly profiles host and microbe small RNAs in human samples
-
•
sMETASeq measures and detects changes in abundance of microbes at species level
-
•
sMETASeq is available as open source scripts
Genomics; Microbiology; Bioinformatics
Introduction
Small RNA sequencing (sRNA-seq) has traditionally been a sequencing method for quantifying microRNAs (miRNAs), but increased understanding of small RNAs and improved databases have enabled identification of other small RNA classes such as transfer RNAs (tRNAs), small nucleolar RNAs (snoRNAs), small nuclear RNAs (snRNAs), and other small RNAs. Current sRNA-seq protocols require 3′ hydroxyl- and 5′ phosphate groups on the RNA for adapter ligation, whereas subsequent size selection usually enriches for RNAs approximately 22 nucleotides (nts) in length (Pritchard et al., 2012). Several classes of RNA meet these criteria and will therefore be part of the final sequencing library.
Detection and quantification of microbes currently rely on either 16S rDNA-seq, utilizing variable regions within the 16S ribosomal RNA (rRNA) gene (Hamady and Knight, 2009), or shotgun DNA-seq in which the DNA is randomly fragmented and sequenced (Venter et al., 2004). The 16S rDNA-seq method has been the gold-standard for metagenomics owing to its good sensitivity and specificity and relatively low cost. However, 16S rDNA-seq has some limitations including underrepresentation of species owing to primer mismatches (Schulz et al., 2017) and low phylogenetic power due to high DNA sequence similarity of the 16S rRNA genes (Janda and Abbott, 2007). Fungi are usually detected by sequencing the Internal Transcribed Spacer (ITS) (Pankaj, 2013, Schoch et al., 2012) and require a separate primer set than that for 16S rDNA-seq. Viruses are commonly detected using customized oligonucleotide capture probes (O'Flaherty et al., 2018) or ribo-depleted total RNA-seq (Visser et al., 2016), but they can also be detected using small RNA-seq (Massart et al., 2019). Shotgun DNA-seq has the advantage over 16S rDNA-seq in that it can detect other microbes than bacteria, has a higher species specificity than 16S rDNA-seq, and can assemble whole genes and infer gene function (Quince et al., 2017).
Small RNAs have been identified in bacteria and shown to play regulatory roles (Majdalani et al., 2005). Bacterial sRNAs are between 50 and 500 nts long and can be detected using total RNA-seq protocols, which have a bias against RNAs shorter than 50 nts. Bacterial sRNAs resemble eukaryotic miRNAs in their ability to base pair with target RNAs; however, they do not undergo a biogenesis pathway similar to that of miRNAs (Gottesman and Storz, 2011). Moreover, the base pairing usually occurs at the 5′end of the target RNA. The number of bacterial sRNAs varies between species, but the number is likely much smaller than in eukaryotes (Gottesman and Storz, 2011), although the identification has lagged that of eukaryotes because fewer sequencing studies have been performed. Some bacteria express tRNA-derived RNA fragments (tRFs) (Kumar et al., 2014) and yRNAs (Chen et al., 2014), two types of sRNAs frequently found in humans.
Here we present a novel metagenomic method, sMETASeq (small RNA Metagenomics by Sequencing), that can jointly measure host small RNAs and generate a metagenomics profile from the same sample. We evaluated the performance of the method together with 16S rDNA-seq on a mock bacterial community and showed that sMETASeq has high sensitivity, specificity, and quantitative performance. We further show that sMETASeq detects differentially expressed microbes in colon cancer and oral leukoplakia and characterizes bacteria and other microbes in human biofluids and cervix samples that reflect the sample type of origin.
Results
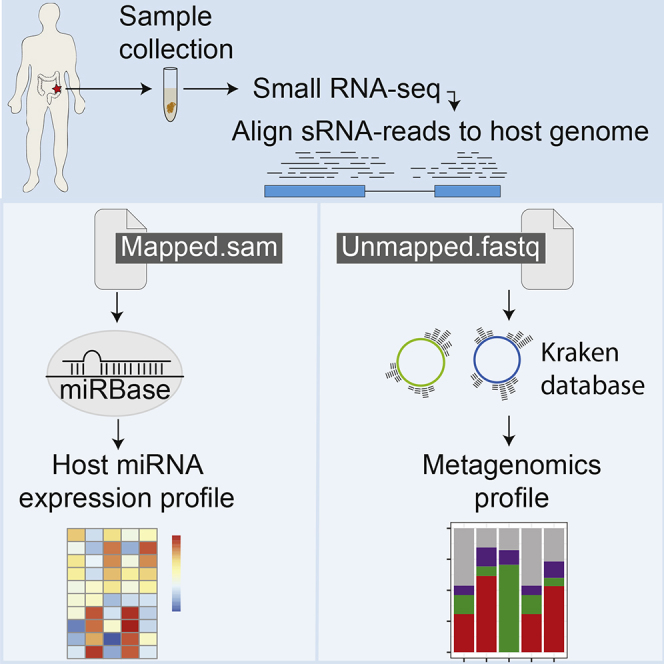
Overview of the sMETASeq Pipeline
The method sMETASeq was developed to enable microbiome characterization using sRNA-seq from samples containing both host small RNAs (e.g., microRNAs) and microbes, for instance, human gut biopsies or biofluids. The data for sMETASeq are generated using a standard sRNA-seq wet-lab protocol and can therefore be applied to already generated publicly available sRNA data. First, adapter-trimmed and collapsed sRNA sequencing reads are mapped to the human genome to identify mapped and unmapped human reads. The mapped reads are then compared with available database annotations of human miRNAs, for instance, miRBase (Griffiths-Jones et al., 2006), and used to generate expression profiles for miRNAs, and potentially other small RNAs, by counting the number of uncollapsed reads that map to each gene. The unique unmapped reads are further aligned to the kraken microbiome reference database (Wood and Salzberg, 2014). The microbiome alignment results are then used to generate metagenomics profiles and estimates of relative and absolute expression of microbes and microbial sRNAs.
Experimental Setup and Sequencing Statistics
To assess the performance of sMETASeq in identifying and quantifying microbes, we applied the method on a mock microbial community that had undergone serial dilution and compared the results with the widely used 16S rDNA-seq method. The mock community comprised 20 known bacterial species with a 5% contribution from each species. To better mimic a host-microbe environment, microbial DNA/RNA from the mock community was mixed with DNA/RNA from the human plasma cell line INA-6 at different concentrations (Table S1 and Figure S1). Since the species of the mock community is known, this approach allowed us to evaluate the sensitivity, specificity, and quantitative abilities of sMETASeq across the different dilutions. The experiment consisted of 15 dilutions for which samples D1–D6 contained increasing amounts of human DNA/RNA and samples D8–D15 contained decreasing amounts of bacterial DNA/RNA (Table S1). The amounts of human and bacterial DNA/RNA were fixed for samples D8–D15 and D1–D6, respectively; sample D7 contained equal amounts of human and bacterial DNA/RNA.
For sMETASeq, an average of 20 million reads per sample were generated (Figure S2A) of which on average 8 million reads mapped to the human genome and 10.5 million did not map to the human genome (Figure S2B). For 16S rDNA-seq, 135,980 reads were generated per sample on average, none of which mapped to the human genome (Figure S2C). About 1% of the reads were remove after quality filtering (see Methods) (Figures S2D and S2E). As expected, both methods showed a decrease in the number of non-human reads in samples with decreasing bacterial DNA/RNA (Figures S2B and S2C). The filtered 16S rDNA data were run through kraken to enable a direct comparison between the two methods. We also ran the 16S rDNA data through Qiime2 (Bolyen et al., 2019), to evaluate its performance on a well-established microbiome platform using a different reference database.
Effect of Bacterial RNA on miRNA Expression
We investigated the effect on miRNA expression of having bacterial RNA in the sample. As expected, the number of human miRNA reads increased as the amount of input human RNA increased (Figure S3A). Similarly, the detected number of unique miRNAs increased with increased input human RNA, from 86 unique miRNAs in sample D2 to 535 in sample D14 (Figure S3B). A principal component analysis of the miRNA showed clear separation between the samples with high and low bacterial biomass (Figure S3C). Interestingly, small amounts of bacterial RNA (<10%, sample D8-D15) did not alter the miRNA distribution significantly, indicating the sRNA-seq protocol is robust to low levels of non-human contaminants. Indeed, the miRNA expression profiles of the samples with low bacterial biomass (D8–D15) were highly correlated (r ≥ 0.99; Figure S3D). In contrast, the samples with high bacterial biomass showed more variation in their miRNA expression profiles and this variation was mainly related to the number of detected miRNAs.
Evaluation of Specificity, Sensitivity, and Quantitative Abilities of sMETASeq on a Mock Bacterial Community
We evaluated the ability of sMETASeq to correctly identify the expected species in a mock community of 20 bacteria. First, we investigated how many of the mock species sMETASeq and 16S rDNA-seq were able to identify. We found that sMETASeq identified 19 of the 20 species, whereas 16S rDNA-seq identified 18 of the 20 species (Figures 1A and 1B). Neither sMETASeq nor 16S rDNA-seq was able to identify Actinomyces odontolyticus and, additionally, 16S rDNA-seq failed to identify Lactobacillus gasseri. For both methods, the abundance of the bacteria decreased with decreasing input bacteria material (Figures 1A and 1B). For sMETASeq, the gram-negative Rhodobacter sphaeroides was the most abundant bacteria, and we also observed high expression of the gram-positive Deinococcus radiodurans.
Figure 1.

Bacterial Composition in Mock Community
(A) Relative abundance of the 20 mock bacteria across the dilutions as identified by sMETASeq. Contaminants are indicated in light and dark gray.
(B) Similar as in (A) for 16S rDNA-seq.
(C) Correlation of mock bacteria abundance between sMETASeq and 16S rDNA-seq for pooled samples. The Pearson's correlation values for each individual bacterium across dilutions are indicated with color. p = 0.00057. See also Table S2.
(D) Number of correctly assigned mock species for sMETASeq (light blue) and 16S rDNA-seq (dark blue) across dilutions. Dilutions D1–D8 have the same number of correct assignments and are therefore pooled.
(E) Comparison of diversity measurements between sMETASeq and 16S rDNA-seq across the dilutions.
Potential contaminant operational taxonomic units (OTUs) were identified using the decontam package in R (Davis et al., 2018). Consistent with the decreasing amounts of bacterial relative to human DNA/RNA, reads from the domain Eukaryota and the kingdom Metazoa were identified as the main contaminants in 16S rDNA-seq and sMETASeq, respectively, and the levels correlated well with the dilution series (Figures 1A and 1B). Contaminants from other OTUs were generally lowly expressed and likely represent cross-mapping of the sequencing reads or sequencing errors (Table S2). Importantly, none of the mock species were identified as a contaminant in either of the methods using the default decontam threshold parameter of 0.1.
To investigate the ability of sMETASeq to quantify the mock species we compared the normalized number of reads within and between the species. First, when grouping all the dilutions into one average estimate of species abundance we observed high correlation (r = 0.7) between sMETASeq and 16S rDNA-seq, indicating that sMETASeq is able to quantify the mock species with good accuracy (Figure 1C). Second, the within-species correlation across the dilutions was also high, indicating that sMETASeq can quantify bacteria at different biomass levels (Figure S4A). When the 16S rDNA data were analyzed using Qiime2 and the GTDB database (Parks et al., 2018), we also observed high correlation between the two methods, although four species were not detected by Qiime2 (Figure S5A). Next, we correlated the abundance of the 20 mock OTUs against the amount of input DNA/RNA to evaluate the ability of sMETASeq to directly quantify bacteria with respect to input bacterial biomass. When performing linear regression against all 15 dilutions, sMETASeq showed higher correlation than 16S rDNA-seq (p = 0.0001, Wilcoxon rank-sum test; Figure S4B). However, when limiting the correlation analysis to the most diluted samples (D8–D15), 16S rDNA-seq correlated better with input bacterial biomass than did sMETASeq (p = 0.0003, Wilcoxon rank-sum test) (Figure S4C). This indicated that sMETASeq has higher sensitivity at high bacterial biomass, whereas 16S rDNA-seq has higher sensitivity at low bacterial biomass.
The specificity of sMETASeq was investigated by measuring how many of the mock species were correctly assigned. A mock species was defined as correctly assigned if the species had the highest number of assigned reads within its corresponding genus. The correct predictions for sMETASeq ranged between 16 and 19 for the different dilutions, and for 16S rDNA-seq the correct predictions ranged between 13 and 15 (Figure 1D; p = 6 × 10−6, sign test). sMETASeq was particularly good at predicting the correct species for samples with high bacterial biomass. When performing the same analysis using Qiime2 and the GTDB database, we observed on average 13 correct predictions for the highest expressed species (Figure S5B). This slightly reduced performance by Qiime2 is partly due to the fact that four species were not detected by Qiime2. Together, these results indicate that sMETASeq is good at discriminating between species in diverse bacterial communities and that 16S rDNA-seq is more comparable with sMETASeq at low bacterial biomass and that sMETASeq is superior at high bacterial biomass.
Diversity Metrics of sMETASeq
Next, we measured the alpha and beta diversity of sMETASeq across the dilutions. We observed a distinct difference in diversity between the non-diluted and the diluted samples. For samples with high bacterial biomass (D1–D6), 16S rDNA-seq generally overestimate diversity (Shannon and Simpson index) and species richness (p = 0.007, p = 0.01, and p = 0.03, comparing “Simpson,” “Shannon,” and “Species richness,” respectively for D1–D6; paired Wilcoxon rank-sum test) (Figure 1E). When bacterial biomass decreased, the estimated diversity tended to increase more rapidly for sMETASeq followed by and increase for 16S rDNA-seq, but the estimated diversity was comparable for the two most diluted samples with lowest bacterial biomass (D14–D15; “Simpson,” “Shannon,” and “Species richness”). When comparing the diversity for all diluted samples where bacteria RNA/DNA was higher than human RNA/DNA (D8-D15), there was a significant difference in Shannon diversity and species richness between sMETASeq and 16S rDNA-seq but not in Simpson diversity (p = 0.04, p = 0.55, and p = 0.04 for “Shannon,” “Simpson,” and “Species richness,” respectively, paired Wilcoxon rank-sum test).
Reads from Protein Coding Genes Are Enriched at Species Level
Having shown that sRNAs can be used to measure bacteria in a mock community we wanted to investigate the genomic origins of the bacterial sRNAs. All sequencing reads assigned to the bacterial strains in the mock community or to their genus were therefore further mapped to the corresponding strain-specific genomes of the 20 mock species. The bacterial sRNAs overlapped different classes of RNAs, the most common being protein-coding RNAs, followed by rRNAs and tRNAs (Figure 2). Reads assigned to the strains were enriched for protein-coding genes, whereas reads assigned at the genus level were enriched for rRNAs and tRNAs. This difference is consistent with rRNA and tRNAs being more evolutionarily conserved than protein-coding genes. Furthermore, when analyzing the relative abundance of RNA types across dilutions at species level, we observed a relative enrichment of rRNAs and tRNAs in samples with low bacterial biomass (D8–D15) compared with samples with high or equal bacterial biomass (D1–D7) and opposite for protein-coding RNAs (Figure S4D). The relative increase for tRNAs in samples with low bacterial biomass was also present for reads mapping at the genus level. Together, these findings illustrate that species-specific identification of bacteria is a result of reads overlapping the protein coding part of the genome of the bacteria and can explain some of the differences in specificity between 16S rDNA-seq and sMETASeq.
Figure 2.

Relative Abundance of RNA Types in Mock Community
Shown are the relative abundances of the different gene classes for the reads assigned to the species (bottom) and genus (top) of the bacteria in the mock community. The dilutions are ordered along the x axis, and the annotation “D8” illustrates the shift from high bacterial biomass samples to low bacterial biomass samples.
Bacterial Identification by sMETASeq in Colon Tissue Reflects the Gut Microbiota
Our group previously performed sRNA-seq on tumor and adjacent normal samples from 48 patients with colon cancer (96 samples) (Mjelle et al., 2019). Bacteria are known to play important roles in the carcinogenesis of colon cancer (Dahmus et al., 2018), and we therefore wanted to investigate if sMETASeq could be used to detect bacteria in these samples. We detected high amounts of bacteria from the genus Bacteroides in most samples, and the most abundant species was Bacteroides fragilis and Bacteroides vulgatus, both naturally occurring in the colon microbiota (Figure 3A). We observed consistent abundance of other gut-associated bacteria, including the orders Clostridiales and Enterobacteriales, the species Enterococcus faecium and Faecalibacterium prausnitzii, as well as the colon-cancer-associated Fusobacterium (Figure 3A).
Figure 3.

OTUs in Colon Tissue as Identified by sMETASeq and 16S rDNA-seq
(A) Detected OTUs by sMETASeq in 96 colon tissue samples. The samples are ordered from left to right with respect to the number of bacteria reads identified. OTUs within the same taxonomic lineage are indicated with different saturation of the same color. The taxonomic level for each OTU is indicated as “d,” domain; “p,” phylum; “c,” class; “o,” order; “g,” genus; “s,” species, and bacteria are indicated as “Bac” and virus as “Vir.” Reads aligned to other OTUs than those listed are indicated as “other.” Samples are indicated with patient number and “T” and “N” for tumor and normal, respectively.
(B) Detected OTUs by 16S rDNA-seq in 48 colon tissue samples. See (A) for a description of the plot.
(C) Correlation plot showing the mean expression of OTUs across samples as identified by sMETASeq (y axis) and 16S rDNA-seq (x axis). The color of the points indicates the correlation coefficient (Pearson's) and the axis shows the expression. p < 2.2e-16.
Bacterial Identification by sMETASeq Correlates with that of 16S rDNA-Seq
Having identified bacteria using sMETASeq, we performed 16S rDNA-seq on a subset of the same samples (48 samples, owing to lack of DNA from all 96) and compared the abundance of the OTUs detected by the two methods. The 16S data also showed high amounts of bacteria from Bacteroides, Clostridiales, and Enterobacteriales, similar to that identified by sMETASeq (Figure 3B). We compared the abundance of the bacterial species that were identified by both 16S rDNA-seq and sMETASeq and observed a high correlation between the two methods (r = 0.82) (Figure 3C). When focusing on the individual species, most species were positively correlated at the sample level between the two methods and more than 60% of the species had a correlation value greater than 0.5. The species B. ovatus and F. prausnitzii showed the highest correlations between the two methods (Figure S6). When analyzing the 16S rDNA data using Qiime2 and the GTDB database, we observed good correlation between genera that were detected by both methods (Figure S5C).
Bacteria are shown to play important roles in colon cancer, and we therefore wanted to analyze differences in bacterial composition between tumor and normal samples and compare the differences across the two methods. We observed differential expression of several bacteria in tumor and normal samples, the clearest being Fusobacteria, Bacteroidetes, and Proteobacteria (Figure S7A). The differential expression observed by sMETASeq was highly reproducible in the 16S data, although to a lesser extent and partly at different taxonomic levels (Figure S7A, right panel). Furthermore, comparing the logFC values between tumor and normal samples for the two methods showed that most OTUs were changing in the same direction (Figure S7B and Table S3).
sMETASeq Detects Microorganisms in Human Biofluids, Cervicovaginal Self-Samples, and Oral Leukoplakia
Having established that sMETASeq is comparable with 16S rDNA-seq in identifying and quantifying bacteria, we went on applying sMETASeq on panels of different sample types. First, we analyzed a publicly available dataset comprising sRNA-seq data from nine different human body fluids (Seashols-Williams et al., 2016). The highest relative amounts of bacterial reads were found in vaginal secretion, menstrual secretion, feces, and saliva, whereas urine showed low amounts of bacterial reads (Figure S8A). The detected bacteria were representative of the respective biofluids, with Lactobacillus being enriched in vaginal and semen samples, Bacteroides in feces, Cutibacterium acnes in perspiration, and Prevotella in saliva (Figure 4A). Samples were generally consistent within the biological replicates but also indicated individual differences, such as Gardnerella vaginalis infections in two of the samples (Figure 4A). Next, we analyzed a second publicly available dataset comprising samples from human saliva, urine, serum, plasma, blood, and lymphocytes (El-Mogy et al., 2018). Similar as in the dataset of Seashols-Williams et al., saliva had the highest number of bacterial reads (Figure S8B) and were enriched with Prevotella and Fusobacterium, both common bacteria of the oral microbiota (Figure 4B).
Figure 4.

Microbes in Human Biofluids, Cervix and Oral Leukoplakia Samples
(A) Distribution of bacteria in nine different human biofluids (Seashols-Williams et al., 2016) as identified by sMETASeq. See Figure 3A for a description of the plot.
(B) Distribution of bacteria from eight different human biofluids (Seashols-Williams et al., 2016) as identified by sMETASeq. See Figure 3A for a description of the plot.
(C) Expression of fungi and parasites in cervix samples from persons with CIN1 (normal) cervix samples and persons with CIN3 (severe dysplasia) (Snoek et al., 2018). The samples are grouped into three quantiles (Q1–3) based on the number of reads aligning to the specific OTUs. The plots marked with Q3 show the samples with the highest number of reads for the specific OTU, and the plots marked with Q1 show the samples with the lowest number of reads for the specific OTU.
(D) Differentially expressed bacteria in progressive oral leukoplakia versus non-progressive oral leukoplakia. The x axis shows the logFC between progressive and non-progressive oral leukoplakia. The y axis shows the inverse (-log10) Benjamini-Hochberg-adjusted p values. Significant bacteria are indicated in red. The dotted gray lines indicated absolute logFC of 1 and the gray horizontal line indicates the significant threshold of 0.05.
To further investigate the bacterial composition in vaginal samples, and to show that sMETASeq also detects viruses, fungi, and other eukaryotes, we analyzed a dataset containing 56 HPV-positive cervicovaginal self-samples (Snoek et al., 2018). Twenty-four of the samples were histologically diagnosed with CIN3, characterized by dysplasia in the cervix, and 32 samples were CIN1 and HPV positive. Viral miRNAs can be detected by sMETASeq using miRBase; however, several viruses do not have miRNAs and PCR-based methods are used for detection. We analyzed the HPV-infected cervical samples and detected Alphapapillomavirus in 12 of the 24 CIN3 samples and one of the normal samples (p = 2.87 × 10−5, Chi-square test for the difference between CIN3 and normal), indicating that the level of Alphapapillomavirus increases with increasing dysplasia (Table S4). Next, we searched the literature for known fungi and parasites reported to be present in the female genital tract and focused on Candida and Trichomonas vaginalis (Bradford and Ravel, 2017). We detected two Candida species, tropicalis and albicans, of which Candida albicans was the highest expressed and found in many of the samples (Figure 4C). The protozoan parasite Trichomonas vaginalis was also detected in several samples.
Finally, we applied sMETASeq on 20 samples of oral leukoplakia, a potentially malignant disorder affecting the oral mucosa (Philipone et al., 2016). Leukoplakia is clinically important owing to its association with the development of oral squamous cell carcinoma (OSCC), a disease with high morbidity and mortality (Bewley and Farwell, 2017). The dataset consisted of 20 subjects divided into two groups; group 1 (n = 10) “progressive group” (patients with leukoplakia that progressed to OSCC within 5 years) and group 2 (n = 10) “non-progressive group” (patients with leukoplakia that did not progress to OSCC within 5 years). We compared bacterial expression between the two groups and detected one differentially expressed genus, Neisseria, which showed significantly higher expression in the progressive group compared with the non-progressive group (Figure 4D) (p = 0.005, Benjamini Hochberg-corrected). When investigating the most abundant microbes across all 20 samples, we detected high abundance of several microbes associated with the oral cavity (Figure S8C). Interestingly, one of the patients in the progressive group showed very high levels of Epstein-Barr virus (EBV, human gammaherpesvirus 4) (Figure S8C). EBV has been linked to both oral carcinomas and oral leukoplakia, although the sample size is too small to draw conclusions from this dataset (Guidry et al., 2018). Together, these results indicate that bacterial small RNAs can be used to differentiate progressive and non-progressive oral leukoplakia samples and potentially serve as biomarkers for OSCC development.
Bacterial Detection in Infected Cell Lines
To further validate that sMETASeq can indeed detect bacteria known to be present in a sample, we performed sRNA-seq of a mycoplasma-infected JJN-3 myeloma cell line and a matched non-infected cell line and compared the level of mycoplasma with a luciferase-based mycoplasma test assay. In the sRNA-seq data, the infected cell line showed between 100 and 200 times more mycoplasma than the non-infected cell line across replicates (Figure S8D). The same cell lines were tested using the MycoAlert Mycoplasma Detection Kit (Lonza), which showed no detectable levels of mycoplasma in the non-infected cell line (readout of 0.6 and 0.4) and high levels in the infected cell line (readout of 70 and 24) (Figure S8D). A readout above 1.2 indicates a positive test.
Discussion
In this article, we present sMETASeq for combined metagenomics and host small RNA profiling. The method provides high-quality metagenomic profiling, is an alternative to current DNA-based methods, and can be applied to various sample material from tissue biopsies to biofluids. The method is particularly suited for research questions where both host small RNAs, for instance, human miRNAs, and the host microbiome are of interest. The method can, for instance, be used to study associations between human miRNAs and microbial composition in one single experiment. sMETASeq displays the versatility of small RNA-based sequencing and shows that bacterial sRNAs are more widespread and consistently expressed than previously anticipated. This could indicate that bacterial sRNAs are protected from degradation in many sample types, either by binding to proteins or being contained in vesicles. Indeed, studies have shown that prokaryotic vesicles contain different RNA types that could be delivered and interact with eukaryotic cells Dauros-Singorenko et al. (2018). Given the stability of bacterial sRNA, they could also function as biomarker for disease.
In addition to describing sMETASeq, this is the first study to perform a comprehensive analysis of small RNA metagenomics and to compare it with the widely used 16S rDNA method. It was recently shown that a modified sRNA-seq protocol focusing on bacterial tRNAs (tRNA-seq) can be used to characterize the microbiome (Schwartz et al., 2018). In contrast to sMETASeq, tRNA-seq will not provide information on other RNA types, for instance, miRNAs, and is a more specific protocol to study tRNA modifications. The strengths of sMETASeq lie in its ability to investigate multiple types of sRNAs and that the data can be used to study both metagenomics and other biological questions. As shown in this paper, sMETASeq tends to perform better at higher bacterial concentration and to some degree lack the sensitivity of 16S rDNA-seq at very low bacterial concentrations. For instance, sMETASeq tended to slightly overestimate sample diversity at low concentrations, and fewer reads mapped to microbes when bacteria concentration decreased. Our results show that, for sample types with high microbial biomass such as fecal, colon, oral, and vaginal samples, sMETASeq would be a good method for identification and quantification of the microbiome. However, in samples with low microbial biomass, such as blood or different human tissues, sMETASeq would likely lack the sensitivity to detect lowly expressed microbes. However, future developments of sMETASeq, with, for instance, a microbial enrichment step, would make the method more sensitive toward microbes; however, this has to be optimized not to compromise the host sRNA profiles.
RNA is generally more prone to degradation compared with DNA, which makes sMETASeq more sensitive to samples with degraded RNA. However, since sMETASeq aims at small RNAs, even degraded RNA molecules contain valuable information that can be used to identify microbes in the sample. Using a k-mer-based analysis method as applied by sMETASeq in the kraken pipeline, short fragments will be suited for taxonomic classification even if they are degradation products.
To compare the sensitivity and the specificity of sMETASeq and 16S rDNA-seq we chose to perform an RNA- and DNA-based sequencing experiment of a mock bacterial community comprising 20 known bacterial species. In terms of specificity, sMETASeq showed good performance, both in samples with high and low bacterial biomass, although the specificity was slightly compromised in samples with low bacterial biomass. These differences in specificity between sMETASeq and 16S rDNA-seq could be attributed to the fact that sMETASeq is not limited to the 16S region for bacteria identification but utilizes reads mapping to the whole bacterial genome. For species with highly similar genomes, sMETASeq is likely to improve the discrimination. Indeed, when investigating the abundance of reads mapping to different RNA types for sMETASeq, we observed that species-specific identification is a result of reads mapping to protein-coding RNAs. The enrichment of protein-coding RNAs for reads that map to the specific bacterial strains shows that the protein-coding region contains valuable strain-specific information that can be utilized when discriminating between closely related species within the same genus. The reduced specificity and increased diversity measures in samples with low bacterial biomass could be explained by the reduction in detected protein-coding RNAs for these samples. Regarding sensitivity and quantitative abilities, the two methods correlated well with each other in measuring the abundance of the mock species across the dilution. All correlation values were above 0.9, and several correlation values were close to 1. When focusing on the samples with low bacterial biomass, 16S rDNA-seq correlated better with input bacterial biomass than did sMETASeq. In contrast, when including the high bacterial biomass samples, sMETASeq correlated better with biomass than did 16S rDNA-seq.
We applied sMETASeq on a previously published in-house sRNA-seq dataset from colon tissue and publicly available data from human biofluids and showed that the bacteria identified largely reflected the sample of origin, supporting that the findings are biologically relevant and not a result of sample contamination. Analyzing the colon dataset, we observed high levels of bacteria commonly found as part of the gut microbiota, including Faecalibacterium, Enterobacteriaceae, Bacteroides, and Fusobacterium (Garrett et al., 2010, Miquel et al., 2013). The latter has been identified as a potential player in colon cancer (Shang and Liu, 2018). Two patients showed high levels of the genus Brachyspira, both in the normal and tumor samples. Brachyspira has been associated with diarrhea and colitis in several animals and is the cause of spirochetosis in human, an infection of the colonic mucosa (Amat Villegas et al., 2004).
Human biofluids were analyzed using two publicly available datasets and were shown to contain a wide range of bacteria down to genus and species level. The bacteria identified largely reflected the known microbiota of the different biofluids. The saliva samples showed high levels of the oral bacterium Fusobacterium. Fusobacterium can also be isolated from the vaginal microbiome (Hillier et al., 1993) and one of the vaginal secretion sample showed very high levels of Fusobacterium, indicating potential vaginosis. The saliva and vaginal secretion samples also showed high levels of the bacteria Prevotella, which have previously been associated to the oral, vaginal, and gut microbiota (Gholizadeh et al., 2016, Ley, 2016, Si et al., 2017). Prevotella was highly expressed in the saliva samples in both datasets. The oral bacterium Veillonella parvula was detected in the saliva samples in the dataset of El-Mogy et al., and in the dataset of Seashols-Williams et al. we detected the phylum Firmicutes to which Veillonella parvula belongs. Stool samples are known to express high levels of bacteria, and sMETASeq showed that the feces samples had the highest proportion of bacterial reads. We found that these bacteria are mainly within the order Bacteroides, as expected from previous 16S studies (Eggerth and Gagnon, 1933). The perspiration samples showed high amounts of the skin-specific bacterium C. acnes and Moraxella osloensis, which is also frequently found in skin (Alkhatib et al., 2017, Dreno et al., 2018). In one of the vaginal secretion samples high amounts of the bacterium Gardernella vaginalis was detected. This bacterium is involved in bacterial vaginosis, a vaginal condition caused by abnormal bacterial composition in the vagina (Schwebke et al., 2014). In the cervix samples we detected many highly expressed bacteria at the species levels, all previously associated with the female genital tract. Interestingly, these samples also contained high levels of other eukaryotes, in particular two fungi species within the Candida genus, Candida albicans and tropicalis, and overgrowth of these fungi is shown to cause vaginal candidiasis, an infection in the vagina. Another interesting finding is the parasite Trichomonas vaginalis, which was detected in a subset of the samples. T. vaginalis has been isolated from samples of the vagina and has been associated with bacterial vaginosis (Franklin and Monif, 2000, Moodley et al., 2002). In the same dataset, we detected viral RNA fragments from the HPV virus Alphapapillomavirus 9. This HPV species has several subtypes, including HPV-16, which is one of the subtypes that can lead to cervix cancer (Holl et al., 2015).
Oral leukoplakia are white patches on the oral mucosa and has malignant potential as these patches can be precursor lesions of OSCC. The likelihood of malignant transformation of oral leukoplakia is observed to lie between 0.2% and 3% and varies between studies and population groups (Bewley and Farwell, 2017). Early detection of OSCC increases the survival rate from 20% to 30% to approximately 80%, which highlights the importance of developing new good diagnostic biomarkers (Noone et al., 2018). Different microorganisms have been linked to oral cancer; however, there are still discussions regarding the role and importance of these microorganisms (Gholizadeh et al., 2016, Healy and Moran, 2019, Pushalkar et al., 2011). Using sMETASeq we were able to find high abundance of Neisseria, a genus of gram-negative species belonging to the phylum Proteobacteria. Most Neisseria species are non-pathogenic; however, at least two species are regarded as pathogenic, Neisseria meningitidis and Neisseria gonorrhoeae. Neisseria species are highly abundant in the human oral cavity and have been detected in, for instance, saliva, plaque, mucosal surfaces in the mouth, and teeth, and Neisseria has been regarded as part of the “core microbiome” of the healthy human oral cavity (Keijser et al., 2008, Zaura et al., 2009). It has previously been shown that Neisseria is able to produce the carcinogenic organic compound acetaldehyde; however, it is not clear if in vivo production of acetaldehyde by Neisseria is related to carcinogenesis (Muto et al., 2000, Yokoyama et al., 2018). The other microorganisms detected by sMETASeq in the oral leukoplakia samples generally reflected the oral sample type. For instance, Capnocytophaga, Fusobacterium, Pasteurellaceae, Rothia, Gemella, and gammaherpesvirus are all related to the oral cavity, and some are implicated in oral cancer or oral leukoplakia.
In summary, applying sMETASeq to different human biofluids, cervical self-samples, and oral leukoplakia, we showed that bacteria, fungi, parasites, and viruses can be identified and quantified between groups. We show that the identification is comparable for similar sample types across datasets and that the microorganisms reflect the biology of the sample in which they are detected.
The establishment of sMETASeq for bacterial identification enables researchers to analyze publicly available datasets and to plan new experiments in which both human small RNAs and other organisms can be identified. sMETASeq also identifies viruses, independent of whether they encode viral miRNAs or not, as well as fungi and other eukaryotes and parasites, and is therefore one of the most versatile protocols for metagenomics. Moreover, by adjusting the gel-purification step during sRNA library preparation the ratio between long or short fragments can be changed to favor different RNA species. Another advantage of using sRNAs in metagenomics is that sRNAs provide information about transcription. If bacterial sRNAs are detected in the samples, it is possible that the bacterium has active transcription since sRNAs from latent or dead bacteria would be rapidly degraded; however, RNA from latent or dead bacteria could also be present in samples if the RNA is protected by proteins, such as for Hfq-associated sRNAs (De Lay et al., 2013). 16S DNA, on the other hand, would be stable for a longer period of time and using 16S metagenomics would be better in case of dormant cells and non-dividing bacteria.
Thousands of sRNA-seq datasets have been submitted to the NCBI sequence read archive. These datasets might contain valuable information on sample microbiomes that researchers could access and analyze through sMETASeq. Several large consortium projects include sRNA as part of the pipeline. For instance, the FANTOM consortium (de Rie et al., 2017) contains sRNA-seq from every major human organ as well as primary cell lines, and The Cancer Genome Atlas project (TCGA) (Chu et al., 2016) contains sRNA-seq from the most common human cancers and includes both cancer and normal samples. We expect that researchers with an interest in metagenomics and microbes will apply sMETASeq to gain new insights into the role of microorganisms in human health.
Limitations of the Study
We here present a method for metagenomics profiling using small RNAs. Although the method performs well at characterizing and quantifying microbes in samples with high bacterial biomass, the sensitivity might be a limiting factor in samples with low bacterial microbes. Furthermore, in samples with low sequencing depth, the number of bacterial reads will often be low, compromising both the sensitivity and the specificity of the method. Further developments of sMETASeq addressing both the library preparation and the data analysis could potentially improve the method.
Resource Availability
Lead Contact
Further information and requests for resources, code, and scripts should be directed to and will be fulfilled by the Lead Contact, Robin Mjelle (robin.mjelle@ntnu.no).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
sMETASeq is available through github (https://github.com/MjelleLab/sMETASeq).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank St. Olavs Hospital for providing colon tissue material for sRNA-seq and 16 rDNA-seq and for funding the sequencing. We thank the Genomics Core Facility at The Norwegian University of Science and Technology for performing sequencing.
Authors Contributions
R.M. performed 16S rDNA-seq, sRNA-seq, and data analysis and prepared the manuscript; K.R.A. provided cell lines and performed mycoplasma tests; E.H. and W.S. provided biobank material for colon samples and E.H. was responsible for acquiring funding; P.S. wrote the bioinformatics pipeline, generated figures, and helped preparing the manuscript. All authors commented on the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101131.
Supplemental Information
References
- Alkhatib N.J., Younis M.H., Alobaidi A.S., Shaath N.M. An unusual osteomyelitis caused by Moraxella osloensis: a case report. Int. J. Surg. Case Rep. 2017;41:146–149. doi: 10.1016/j.ijscr.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amat Villegas I., Borobio Aguilar E., Beloqui Perez R., de Llano Varela P., Oquinena Legaz S., Martinez-Penuela Virseda J.M. [Colonic spirochetes: an infrequent cause of adult diarrhea] Gastroenterol. Hepatol. 2004;27:21–23. doi: 10.1016/s0210-5705(03)70440-3. [DOI] [PubMed] [Google Scholar]
- Bewley A.F., Farwell D.G. Oral leukoplakia and oral cavity squamous cell carcinoma. Clin. Dermatol. 2017;35:461–467. doi: 10.1016/j.clindermatol.2017.06.008. [DOI] [PubMed] [Google Scholar]
- Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford L.L., Ravel J. The vaginal mycobiome: a contemporary perspective on fungi in women's health and diseases. Virulence. 2017;8:342–351. doi: 10.1080/21505594.2016.1237332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Sim S., Wurtmann E.J., Feke A., Wolin S.L. Bacterial noncoding Y RNAs are widespread and mimic tRNAs. RNA. 2014;20:1715–1724. doi: 10.1261/rna.047241.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A., Robertson G., Brooks D., Mungall A.J., Birol I., Coope R., Ma Y., Jones S., Marra M.A. Large-scale profiling of microRNAs for the cancer genome atlas. Nucleic Acids Res. 2016;44:e3. doi: 10.1093/nar/gkv808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmus J.D., Kotler D.L., Kastenberg D.M., Kistler C.A. The gut microbiome and colorectal cancer: a review of bacterial pathogenesis. J. Gastrointest. Oncol. 2018;9:769–777. doi: 10.21037/jgo.2018.04.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauros-Singorenko P., Blenkiron C., Phillips A., Swift S. The functional RNA cargo of bacterial membrane vesicles. FEMS Microbiol. Lett. 2018;365 doi: 10.1093/femsle/fny023. [DOI] [PubMed] [Google Scholar]
- Davis N.M., Proctor D.M., Holmes S.P., Relman D.A., Callahan B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome. 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lay N., Schu D.J., Gottesman S. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 2013;288:7996–8003. doi: 10.1074/jbc.R112.441386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rie D., Abugessaisa I., Alam T., Arner E., Arner P., Ashoor H., Astrom G., Babina M., Bertin N., Burroughs A.M. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017;35:872–878. doi: 10.1038/nbt.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreno B., Pecastaings S., Corvec S., Veraldi S., Khammari A., Roques C. Cutibacterium acnes (Propionibacterium acnes) and acne vulgaris: a brief look at the latest updates. J. Eur. Acad. Dermatol. Venereol. 2018;32(Suppl 2):5–14. doi: 10.1111/jdv.15043. [DOI] [PubMed] [Google Scholar]
- Eggerth A.H., Gagnon B.H. The Bacteroides of human feces. J. Bacteriol. 1933;25:389–413. doi: 10.1128/jb.25.4.389-413.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mogy M., Lam B., Haj-Ahmad T.A., McGowan S., Yu D., Nosal L., Rghei N., Roberts P., Haj-Ahmad Y. Diversity and signature of small RNA in different bodily fluids using next generation sequencing. BMC Genomics. 2018;19:408. doi: 10.1186/s12864-018-4785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin T.L., Monif G.R. Trichomonas vaginalis and bacterial vaginosis. Coexistence in vaginal wet mount preparations from pregnant women. J. Reprod. Med. 2000;45:131–134. [PubMed] [Google Scholar]
- Garrett W.S., Gallini C.A., Yatsunenko T., Michaud M., DuBois A., Delaney M.L., Punit S., Karlsson M., Bry L., Glickman J.N. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholizadeh P., Eslami H., Yousefi M., Asgharzadeh M., Aghazadeh M., Kafil H.S. Role of oral microbiome on oral cancers, a review. Biomed. Pharmacother. 2016;84:552–558. doi: 10.1016/j.biopha.2016.09.082. [DOI] [PubMed] [Google Scholar]
- Gottesman S., Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidry J.T., Birdwell C.E., Scott R.S. Epstein-Barr virus in the pathogenesis of oral cancers. Oral Dis. 2018;24:497–508. doi: 10.1111/odi.12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M., Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19:1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy C.M., Moran G.P. The microbiome and oral cancer: more questions than answers. Oral Oncol. 2019;89:30–33. doi: 10.1016/j.oraloncology.2018.12.003. [DOI] [PubMed] [Google Scholar]
- Hillier S.L., Krohn M.A., Rabe L.K., Klebanoff S.J., Eschenbach D.A. The normal vaginal flora, H2O2-producing lactobacilli, and bacterial vaginosis in pregnant women. Clin. Infect. Dis. 1993;16(Suppl 4):S273–S281. doi: 10.1093/clinids/16.supplement_4.s273. [DOI] [PubMed] [Google Scholar]
- Holl K., Nowakowski A.M., Powell N., McCluggage W.G., Pirog E.C., Collas De Souza S., Tjalma W.A., Rosenlund M., Fiander A., Castro Sanchez M. Human papillomavirus prevalence and type-distribution in cervical glandular neoplasias: results from a European multinational epidemiological study. Int. J. Cancer. 2015;137:2858–2868. doi: 10.1002/ijc.29651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda J.M., Abbott S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 2007;45:2761–2764. doi: 10.1128/JCM.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keijser B.J., Zaura E., Huse S.M., van der Vossen J.M., Schuren F.H., Montijn R.C., ten Cate J.M., Crielaard W. Pyrosequencing analysis of the oral microflora of healthy adults. J. Dent. Res. 2008;87:1016–1020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- Kumar P., Anaya J., Mudunuri S.B., Dutta A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014;12:78. doi: 10.1186/s12915-014-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R.E. Gut microbiota in 2015: Prevotella in the gut: choose carefully. Nat. Rev. Gastroenterol. Hepatol. 2016;13:69–70. doi: 10.1038/nrgastro.2016.4. [DOI] [PubMed] [Google Scholar]
- Majdalani N., Vanderpool C.K., Gottesman S. Bacterial small RNA regulators. Crit. Rev. Biochem. Mol. Biol. 2005;40:93–113. doi: 10.1080/10409230590918702. [DOI] [PubMed] [Google Scholar]
- Massart S., Chiumenti M., De Jonghe K., Glover R., Haegeman A., Koloniuk I., Kominek P., Kreuze J., Kutnjak D., Lotos L. Virus detection by high-throughput sequencing of small RNAs: large-scale performance testing of sequence analysis strategies. Phytopathology. 2019;109:488–497. doi: 10.1094/PHYTO-02-18-0067-R. [DOI] [PubMed] [Google Scholar]
- Miquel S., Martin R., Rossi O., Bermudez-Humaran L.G., Chatel J.M., Sokol H., Thomas M., Wells J.M., Langella P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013;16:255–261. doi: 10.1016/j.mib.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Mjelle R., Sjursen W., Thommesen L., Saetrom P., Hofsli E. Small RNA expression from viruses, bacteria and human miRNAs in colon cancer tissue and its association with microsatellite instability and tumor location. BMC Cancer. 2019;19:161. doi: 10.1186/s12885-019-5330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodley P., Wilkinson D., Connolly C., Moodley J., Sturm A.W. Trichomonas vaginalis is associated with pelvic inflammatory disease in women infected with human immunodeficiency virus. Clin. Infect. Dis. 2002;34:519–522. doi: 10.1086/338399. [DOI] [PubMed] [Google Scholar]
- Muto M., Hitomi Y., Ohtsu A., Shimada H., Kashiwase Y., Sasaki H., Yoshida S., Esumi H. Acetaldehyde production by non-pathogenic Neisseria in human oral microflora: implications for carcinogenesis in upper aerodigestive tract. Int. J. Cancer. 2000;88:342–350. [PubMed] [Google Scholar]
- Noone A.M., Howlader N., Krapcho M., Miller D., Brest A., Yu M., Ruhl J., Tatalovich Z., Mariotto A., Lewis D.R., editors. SEER Cancer Statistics Review, 1975-2015. National Cancer Institute; 2018. https://seer.cancer.gov/csr/1975_2015/ [Google Scholar]
- O'Flaherty B.M., Li Y., Tao Y., Paden C.R., Queen K., Zhang J., Dinwiddie D.L., Gross S.M., Schroth G.P., Tong S. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 2018;28:869–877. doi: 10.1101/gr.226316.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankaj K. Methods for rapid virus identification and quantification. Mater. Methods. 2013;3 [Google Scholar]
- Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- Philipone E., Yoon A.J., Wang S., Shen J., Ko Y.C., Sink J.M., Rockafellow A., Shammay N.A., Santella R.M. MicroRNAs-208b-3p, 204-5p, 129-2-3p and 3065-5p as predictive markers of oral leukoplakia that progress to cancer. Am. J. Cancer Res. 2016;6:1537–1546. [PMC free article] [PubMed] [Google Scholar]
- Pritchard C.C., Cheng H.H., Tewari M. MicroRNA profiling: approaches and considerations. Nat. Rev. Genet. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushalkar S., Mane S.P., Ji X., Li Y., Evans C., Crasta O.R., Morse D., Meagher R., Singh A., Saxena D. Microbial diversity in saliva of oral squamous cell carcinoma. FEMS Immunol. Med. Microbiol. 2011;61:269–277. doi: 10.1111/j.1574-695X.2010.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N. Corrigendum: shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017;35:1211. doi: 10.1038/nbt1217-1211b. [DOI] [PubMed] [Google Scholar]
- Schoch C.L., Seifert K.A., Huhndorf S., Robert V., Spouge J.L., Levesque C.A., Chen W., Fungal Barcoding Consortium. Fungal Barcoding Consortium Author List Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U S A. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz F., Eloe-Fadrosh E.A., Bowers R.M., Jarett J., Nielsen T., Ivanova N.N., Kyrpides N.C., Woyke T. Towards a balanced view of the bacterial tree of life. Microbiome. 2017;5:140. doi: 10.1186/s40168-017-0360-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.H., Wang H., Pan J.N., Clark W.C., Cui S., Eckwahl M.J., Pan D.W., Parisien M., Owens S.M., Cheng B.L. Microbiome characterization by high-throughput transfer RNA sequencing and modification analysis. Nat. Commun. 2018;9:5353. doi: 10.1038/s41467-018-07675-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwebke J.R., Muzny C.A., Josey W.E. Role of Gardnerella vaginalis in the pathogenesis of bacterial vaginosis: a conceptual model. J. Infect. Dis. 2014;210:338–343. doi: 10.1093/infdis/jiu089. [DOI] [PubMed] [Google Scholar]
- Seashols-Williams S., Lewis C., Calloway C., Peace N., Harrison A., Hayes-Nash C., Fleming S., Wu Q., Zehner Z.E. High-throughput miRNA sequencing and identification of biomarkers for forensically relevant biological fluids. Electrophoresis. 2016;37:2780–2788. doi: 10.1002/elps.201600258. [DOI] [PubMed] [Google Scholar]
- Shang F.M., Liu H.L. Fusobacterium nucleatum and colorectal cancer: a review. World J. Gastrointest. Oncol. 2018;10:71–81. doi: 10.4251/wjgo.v10.i3.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si J., You H.J., Yu J., Sung J., Ko G. Prevotella as a hub for vaginal microbiota under the influence of host genetics and their association with obesity. Cell Host Microbe. 2017;21:97–105. doi: 10.1016/j.chom.2016.11.010. [DOI] [PubMed] [Google Scholar]
- Snoek B.C., Verlaat W., Babion I., Novianti P.W., van de Wiel M.A., Wilting S.M., van Trommel N.E., Bleeker M.C.G., Massuger L., Melchers W.J.G. Genome-wide microRNA analysis of HPV-positive self-samples yields novel triage markers for early detection of cervical cancer. Int. J. Cancer. 2018;144:372–379. doi: 10.1002/ijc.31855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter J.C., Remington K., Heidelberg J.F., Halpern A.L., Rusch D., Eisen J.A., Wu D., Paulsen I., Nelson K.E., Nelson W. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Visser M., Bester R., Burger J.T., Maree H.J. Next-generation sequencing for virus detection: covering all the bases. Virol. J. 2016;13:85. doi: 10.1186/s12985-016-0539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S., Takeuchi K., Shibata Y., Kageyama S., Matsumi R., Takeshita T., Yamashita Y. Characterization of oral microbiota and acetaldehyde production. J. Oral Microbiol. 2018;10:1492316. doi: 10.1080/20002297.2018.1492316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaura E., Keijser B.J., Huse S.M., Crielaard W. Defining the healthy "core microbiome" of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
sMETASeq is available through github (https://github.com/MjelleLab/sMETASeq).