

Abstract
Background & Aims
Colonic stem cells are essential for producing the mucosal lining, which in turn protects stem cells from insult by luminal factors. Discovery of genetic and biochemical events that control stem cell proliferation and differentiation can be leveraged to decipher the causal factors of ulcerative colitis and aid the development of more effective therapy.
Methods
We performed in vivo and in vitro studies from control (nuclear receptor corepressor 1 [NCoR1F/F]) and intestinal epithelial cell–specific NCoR1-deficient mice (NCoR1ΔIEC). Mice were challenged with dextran sodium sulfate to induce experimental ulcerative colitis, followed by colitis examination, barrier permeability analysis, cell proliferation immunostaining assays, and RNA sequencing analysis. By using crypt cultures, the organoid-forming efficiency, cell proliferation, apoptosis, and histone acetylation were analyzed after butyrate and/or tumor necrosis factor α treatments.
Results
NCoR1ΔIEC mice showed a dramatic increase in disease severity in this colitis model, with suppression of proliferative cells at the crypt base as an early event and a concomitant increase in barrier permeability. Genome expression patterns showed an important role for NCoR1 in colonic stem cell proliferation and secretory cell differentiation. Colonic organoids cultured from NCoR1ΔIEC mice were more sensitive to butyrate-induced cell growth inhibition and apoptosis, which were exaggerated further by tumor necrosis factor α co-treatment, which was accompanied by increased histone acetylation.
Conclusions
NCoR1 regulates colonic stem cell proliferation and secretory cell differentiation. When NCoR1 is disrupted, barrier protection is weakened, allowing luminal products such as butyrate to penetrate and synergistically damage the colonic crypt cells. Transcript profiling: RNA sequencing data have been deposited in the GEO database, accession number: GSE136153.
Keywords: NCoR1, Ulcerative Colitis, Colonic Crypt Cell, Butyrate
Abbreviations used in this paper: AMP, antimicrobial peptide; BrdU, bromodeoxyuridine; BW, body weight; Cldns, claudins; DCS, deep crypt secretory; DSS, dextran sodium sulfate; HDAC, histone deacetylase; HFD, high-fat diet; H3K, histone H3; IBD, inflammatory bowel disease; IEC, intestinal epithelial cell; FITC-d, fluorescein isothiocyanate–dextran; IL, interleukin; NCoR1, nuclear receptor co-repressor 1; RT-qPCR, quantitative reverse-transcription polymerase chain reaction; TNFα, tumor necrosis factor α; UC, ulcerative colitis; UCSD, University of California, San Diego; WRN, Wnt-3A, R-Spondin 3, and Noggin
Graphical abstract
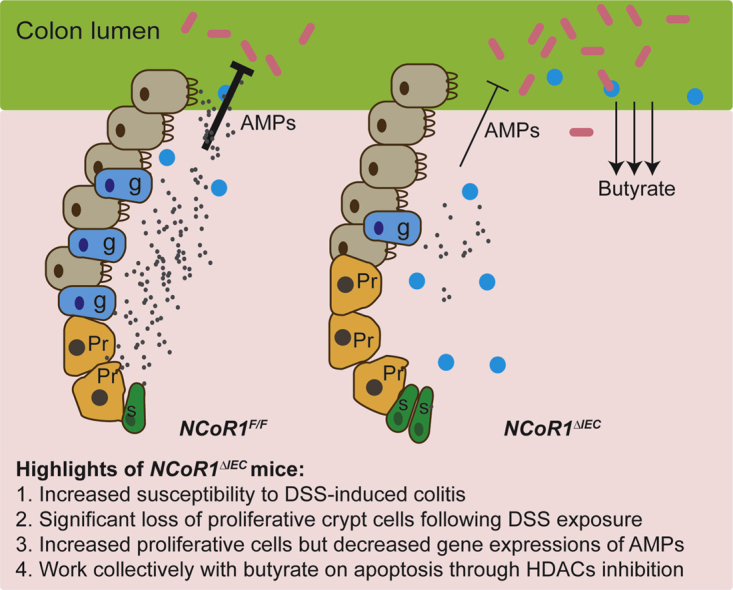
Summary.
Intestinal epithelial cell–specific nuclear receptor corepressor 1 (NCoR1)-deficient mice show increased susceptibility to chemical-induced ulcerative colitis. Colonic stem cell proliferation and secretory cell differentiation are regulated by NCoR1. Disruption of NCoR1 weakened barrier protection, allowing luminal products such as butyrate to penetrate and synergistically damage colonic crypt cells.
Ulcerative colitis (UC), one of the principal types of inflammatory bowel disease (IBD), is characterized as a long-term condition that results in colon and rectum inflammation. Often starting in the rectum with bloody diarrhea, UC can extend continuously to proximal segments and even the entire large intestine. Associated with society modernization and a Western lifestyle, UC has become an expanding global health concern with the highest incidence and prevalence in Northern Europe, Australia, and North America.1 After worldwide industrialization, a significant increase in the incidence and prevalence of UC was observed in Asia, especially South-East Asia and Japan.1,2 In China, the most populated country with the fastest growth of urbanization and a Westernized lifestyle, the prevalence for UC increased more than 3-fold from the 1980s to 1990s, and the incidence rate increased 6-fold between 1985 and 2006.3 Quality of life for individuals with UC is compromised substantially because of frequent flare-ups leading to additional complications, including an increased risk for colorectal cancer.4, 5, 6
The exact etiology of UC has not been identified, but the pathogenesis of UC clearly is multifactorial, involving genetic predisposition, epithelial barrier defects, dysregulated immune responses, and environmental factors.7 Most of the current therapeutic approaches target symptoms such as inflammation, using monoclonal antibodies targeting the inflammatory cytokine tumor necrosis factor α (TNFα) and the integrins.1,8 However, these approaches fail to change the relapsing-remitting course of UC or prevent additional clinical interventions such as colectomy.9,10 After the applications of genetically modified animal models and high-throughput sequencing, many key factors that are crucial in UC initiation and progression have been gradually identified and characterized. Emerging therapeutic targets provide opportunities to implement precision medicine designed to improve specific disease outcomes, such as mucosal healing,11,12 which has been confirmed to be associated with improved long-term outcome of UC treatment.13,14 Colonic stem cells, similar to other intestinal stem cells, reside at the bottom of the crypt base and are responsible for continuous mucosal regeneration to maintain normal intestinal homeostasis and to repair the mucosal layer after injury. Impaired function of intestinal stem cells results in mucosal barrier breakdown and the penetration of luminal contents. The importance of the colonic stem cells have been leveraged in clinical studies by experimenting with stem cell transplantation to relieve the inflammatory symptoms of UC.15, 16, 17 A thorough understanding of the disruptive events leading to colonic stem cell impairment is essential to identify those events leading to damage of the mucosal lining.
Nuclear receptor corepressor 1 (NCoR1), a major epigenetic regulator, interacts with a set of nuclear receptors and limits chromatin accessibility by recruiting chromatin-modifying enzymes, such as histone deacetylases (HDACs), resulting in transcriptional inhibition.18,19 In adipocytes and macrophages, the deletion of NCoR1 has been linked with anti-inflammatory effects through nuclear receptors peroxisome proliferator-activated receptor-γ and liver X receptor,20,21 but in bone marrow, the disruption of Bcl6-silencing mediator of retinoic acid and thyroid hormone receptor/NCoR1 interactions results in severe atherosclerosis.22 These findings indicate a prominent tissue- and cell-specific role for NCoR1 in regulating inflammation. The importance of intrinsic NCoR1 in intestinal homeostasis was discovered in our laboratory when we generated intestinal epithelial cell (IEC)-specific NCoR1 deletion mice (NCoR1ΔIEC) and identified the role of NCoR1 in regulating IEC proliferation and enterocyte maturation during neonatal development.23 However, the role of NCoR1 in the context of gastrointestinal disease remains unknown. By using dextran sodium sulfate (DSS), a standard agent used to induce experimental colitis in mouse models, the current work has focused on mechanistic studies of NCoR1 in colonic crypt cells and colitis development.
Results
Mice With Intestinal NCoR1 Deletion Are More Sensitive to DSS-Induced Colitis
We generated IEC-specific NCoR1 deletion mice (NCoR1ΔIEC) by cross-breeding floxed NCoR1 mice (NCoR1F/F) with mice carrying the Villin-Cre transgene (Figure 1A). The successful deletion of NCoR1 was shown at both transcriptional and protein levels by quantitative reverse-transcription polymerase chain reaction (RT-qPCR) of colon tissues and Western blot analysis of nuclear proteins prepared from ileum samples (Figure 1B). During development, NCoR1ΔIEC mice had no obvious abnormalities, both male and female mice developed into adulthood with regular reproductivity and normal body weight (BW) (Figure 1C). We then used DSS, a standard agent used to induce experimental colitis in murine models.24 Both NCoR1F/F and NCoR1ΔIEC mice were treated with 2.5% (w/v) DSS in their drinking water for 6 days and BW changes were monitored daily for up to 13 days. As shown in Figure 1D, NCoR1F/F mice were minimally affected, whereas NCoR1ΔIEC mice showed profound BW loss (P < .0001; 2-way analysis of variance; n = 10). The BW difference was observed initially at day 5 after DSS exposure. The greatest BW loss was observed on day 8 (DSS 6 days plus water 2 days) with a 17.7% ± 1.5% weight loss in NCoR1ΔIEC vs 8.1% ± 2.0% in NCoR1F/F mice (male mice). After day 8, BW began to recover in both groups, but NCoR1ΔIEC mice showed slower recovery compared with controls. No gender difference was observed in this experiment; both male and female mice showed a similar DSS-induced BW loss (Figure 1D). Compared with DSS-NCoR1F/F mice, DSS-NCoR1ΔIEC mice showed shrinkage of the cecum and signs of inflammation (Figure 1E). Although there was no difference in colon length at this time point between the 2 groups, the percentage of colon length decrease in DSS-NCoR1ΔIEC mice was much greater than in DSS-NCoR1F/F mice (Figure 1F and G).When H&E staining was performed in the drinking water group, NCoR1ΔIEC mice showed limited histologic difference from NCoR1F/F mice. However, DSS-treated NCoR1ΔIEC mice showed increased disease severity as quantitated by the histopathologic colitis score, which is based on the severity of ulcerative lesions, disrupted epithelial structure, and increased inflammatory cell infiltration (Figure 1H and I). Consistently, RT-qPCR also showed increased expression of immune cell–derived proinflammatory cytokines TNFα, interleukin (IL)1β, and IL6 in the colon tissues in DSS-NCoR1ΔIEC mice (Figure 1J–L). These results suggest that NCoR1 plays an important role in protecting the colon from DSS-induced colitis.
Figure 1.

Intestinal NCoR1 deletion mice are more sensitive to DSS-induced colitis. (A) An outline of the removal of exon 11 in the NCoR1 gene that leads to the creation of mice with an IEC-specific NCoR1 deletion (NCoR1ΔIEC). (B) RT-qPCR of NCoR1 concentrations in colon tissue from NCoR1F/F (F/F) and NCoR1ΔIEC (ΔIEC) and Western blot analysis using nuclear fractions isolated from ileum to examine NCoR1 and HDAC1 concentrations. (C) BW of adult mice at 8 weeks of age (n = 10 per group). (D) Kinetics of BW change (n = 10). Both male (♂) and female (♀) mice at 8 weeks of age were treated with either water (naïve) or 2.5% DSS dissolved in drinking water. After 6 days, mice were restored to regular drinking water. BW was monitored daily for 13 days (n = 10 per group). (E) Gross pictures of naïve (water) and DSS-treated mice of colons collected from mice (6 days of DSS treatment and 2 days of water). (F) Colon tissues were dissected on day 8 after 6 days of DSS treatment and 2 days of water, colon lengths were measured (n = 6). (G) The decrease of colon length of DSS-treated mice was described as the percentage change of colon lengths of DSS-treated over naïve mice (n = 6). (H) Pathologic colitis score. (I) Colon tissues were collected on DSS day 5. H&E staining showing damaged colon in NCoR1ΔIEC mice. Scale bar: 40 μm. Images were examined using a 20× Plan-Apochromat objective (numeric aperture, 0.8) on an upright Imager A2 microscope (Zeiss) with an Axiocam 506 color camera and ZEN2012 imaging software. (J–L) RT-qPCR of cytokine expression in colon tissues collected from both naïve and DSS-treated mice (DSS day 5, n = 6). Results were described as means ± SEM, Student t test analyses were performed, and P values smaller than .05 were considered statistically significant. *P < .05, **P < .01, and ***P < .001.
Suppression of Proliferative Cells at the Crypt Base Is an Early Event in DSS-Treated NCoR1ΔIEC Mice With Concomitant Increase of Barrier Permeability
To investigate if NCoR1 deletion compromises the epithelial barrier function, we tested the ability of fluorescein isothiocyanate–dextran (FITC-d), a 3- to 5-kilodalton marker, to pass through the colonic barrier. In addition to naïve mice, we examined 2 DSS exposure time points. An early time point on DSS day 3, which precedes any signs of BW loss or severe inflammation, and the other on DSS day 5 when mice have significant BW loss. Naïve NCoR1F/F and NCoR1ΔIEC mice showed similar permeability to FITC-d (Figure 2A). On day 3 after DSS exposure, NCoR1ΔIEC mice started to show a significant increase of the fluorescence in their sera (P < .05), but no changes were observed in NCoR1F/F serum samples. On day 5, increased FITC-d in serum samples were observed in both strains, with significantly increased permeability still observed in DSS-NCoR1ΔIEC mice (Figure 2A). Thus, compared with NCoR1F/F mice, NCoR1ΔIEC mice are more prone to the disruption of barrier integrity.
Figure 2.

NCoR1ΔIEC mice show increased epithelial permeability after DSS treatment and altered proliferative cells. (A) FITC-d permeability analysis. NCoR1F/F and NCoR1ΔIEC mice were treated with water or DSS for 3 or 5 days, respectively. On the last day, each mouse was administered 20 mg of FITC-d through oral gavage. After 4 hours, blood samples were collected for serum, and FITC-d concentrations were measured and calculated from a FITC-d standard curve. Data are described as FITC concentration (n = 6). (B) BrdU incorporation analysis. Mice were treated with BrdU at 100 mg/kg by intraperitoneal injection on the last day of DSS exposure. Slides were examined under an upright Imager A2 microscope (Zeiss), at least 10 photographs of different areas were taken of each slide. Scale bar: 40 μm. BrdU+ cells were enumerated and described as BrdU+ cells per villous (n = 5). (C) On day 3 after DSS exposure, colon samples were dissected for immunostaining of Ki67. Photographs were taken under a Leica TCS SP5 X confocal microscope and LAS AF imaging software. Scale bar: 50 μm. Ki67+ cells were counted and described as average Ki67+ cells per field (n = 6). DAPI, 4′,6-diamidino-2-phenylindole. ∗P < .05, ∗∗P < .01.
To further investigate the role of NCoR1 toward cell proliferation, bromodeoxyuridine (BrdU) incorporation analysis was performed. Four hours after BrdU intraperitoneal injection, mouse tissues were collected for immunostaining of BrdU-positive (BrdU+) cells. We showed that in naïve NCoR1ΔIEC mice BrdU+ cells had increased by approximately 70% (n = 5; P < .05) (Figure 2B). On day 2 after DSS exposure, no significant changes were observed in DSS-NCoR1F/F mice; however, BrdU+ cells were decreased significantly more than 35% in DSS-NCoR1ΔIEC mice (n = 5; P < .01). On day 4, a decrease of BrdU+ cells also was observed in DSS-NCoR1F/F mice, but DSS-NCoR1ΔIEC mice showed more severe damage (P < .05). The decrease of proliferative cells likewise was observed through the loss of proliferative marker Ki67 (Figure 2C). These results showed that NCoR1 plays a biphasic role in regulating colonic cell proliferation. Under physiological conditions, IEC-NCoR1 deletion stimulates colonic cell proliferation, but in colitic mice cell proliferation was decreased sharply in NCoR1ΔIEC mice.
DSS Exposure Induces Differential Gene Expression in NCoR1ΔIEC Vs NCoR1F/F Mice, Highlighted by Altered Inflammatory Response and Attenuated Cell Division
To investigate the underlying mechanisms of NCoR1 in DSS-induced colitis development, we performed RNA sequencing analysis. NCoR1F/F and NCoR1ΔIEC mice were treated with water (naïve) or 2.5% DSS. On day 3 after DSS treatment, RNA was prepared from colon tissue. This experiment consisted of 4 groups, as follows: (1) water only for NCoR1F/F mice (F/F_naïve); (2) DSS-treated NCoR1F/F mice (F/F_DSS); (3) water only for NCoR1ΔIEC mice (ΔIEC_naïve); and (4) DSS-treated NCoR1ΔIEC mice (ΔIEC_DSS). RNA from 3 mice per group were combined as 1 sample, with 4 samples from each group subjected for RNA sequencing as previously described.23 Sequencing reads were aligned to the mouse reference genome (mm10) produced by the University of California, Santa Cruz (UCSC). Results showed that DSS treatment was associated with alterations in transcript abundance for more than 2000 genes in NCoR1F/F mice (F/F_naïve vs F/F_DSS) and more than 3000 genes in NCoR1ΔIEC mice (ΔIEC_naïve vs ΔIEC_DSS) (Figure 3A). We then further compared ΔIEC_DSS vs F/F_DSS samples and identified 633 genes with a fold change greater than 0.75 (up or down) and a P value less than .05. Gene Ontology Enrichment Pathway analysis showed that in the up-regulated genes, the increased inflammatory response was hallmarked (Figure 3B and C). This is consistent with the increased transcription of IL1β, TNFα, and other cytokines shown in Figure 1J–L. In those genes that were down-regulated, pathway analysis targeted organelle division and cell-cycle control as the most impacted group of genes (Figure 3B), represented by many cyclins, cyclin-dependent kinases, kinesins, and many other cell division–associated genes (Figure 3C). In particular, we identified that stem cell markers, including leucine-rich repeat-containing G-protein–coupled receptor 5 (Lgr5),25 and achaete-scute family basic helix-loop-helix transcription factor 2 (Ascl2),26 which are expressed mitotically in active intestinal stem cells and contribute robustly to intestinal homeostatic regeneration, were decreased more significantly in DSS-NCoR1ΔIEC mice (Figure 3D and E). These results indicate that NCoR1 plays an important role in protecting the proliferative cells in the colon.
Figure 3.

RNA sequencing and pathway analysis.NCoR1F/F and NCoR1ΔIEC mice were treated with water or DSS. On day 3, colon RNA was prepared. Samples from 3 mice were combined as 1 sample, with 4 samples from each group subjected for RNA sequencing analysis. Total RNA was used for preparation of the sequencing libraries using the Illumina Trueseq RNA Sample Prep Kit, and the sequencing was performed on a Hiseq4000 (Illumina, San Diego, CA). RNA-seq reads were aligned to the mouse mm10 genome. (A) Heat map comparing gene expression differences between NCoR1F/F and NCoR1ΔIEC mice treated with either water or DSS. (B) Pathway analysis of DSS-NCoR1F/F vs DSS-NCoR1ΔIEC mice. (C) Representative markers of down-regulated (in blue) and up-regulated (in red) genes between DSS treated NCoR1F/F and NCoR1ΔIEC mice. (D and E) RNA sequencing reads of intestinal stem cell marker genes Lgr5 and Ascl2. The data are presented as the percentage change when compared with naïve NCoR1F/F mice. (F) Transcriptional alteration of DCS cell marker Reg4 and AMP genes. (G) Expression of the Cldns. The data are described as reads in fragments per kilobase of exon per million mapped reads. Ctrol, control; PLD, Phospholipase D; PLK, polo-like kinase-1. *P < .05, **P < .01, ***P < .001, and ****P < .0001.
In RNA sequencing data analysis, we noticed that 2 groups of genes, associated with cell differentiation and barrier integrity, were regulated significantly (Figure 3F and G). Intestinal stem cells arise and differentiate into 2 lineages: absorptive and secretory. The absorptive cells, enterocytes in the small intestine or colonocytes in the colon, constitute most of the epithelium needed to absorb water and electrolytes. The secretory cells, including the goblet cells, enteroendocrine cells, and Paneth cells, secret mucins, hormones, and antimicrobial peptides (AMPs), respectively, to form the epithelial barrier that facilitates intestinal protection.27, 28, 29, 30 Paneth cells reside at the base of the crypts in the small intestine, protecting the adjacent stem cells by secreting high concentrations of AMPs.29,30 Colons lack Paneth cells, but deep crypt secretory (DCS) cells serve as Paneth equivalents in the colon crypt.31 Single-cell messenger RNA sequencing identified DCS cells as a subpopulation of goblet cells,32,33 marked by regenerating islet-derived family member 4 (Reg4) and cKit+.31, 32, 33 Ablation of Reg4+ DCS cells results in loss of stem cells from colonic crypts and disrupts gut homeostasis.31 In our RNA sequencing study, we found that Reg4 largely was inhibited in naïve NCoR1ΔIEC mice without DSS exposure (Figure 3F). A group of AMPs, such as Ang4, Itln1, Retnlb, and Wfdc2, produced mainly by secretory cells including Paneth, DCS, and goblet cells,28,34,35 also were inhibited dramatically (Figure 3F). The altered secretory cells may compromise the innate protection in NCoR1ΔIEC mice and predispose mice to the initiation and development of colitis. Another group of genes are the claudins (Cldns). Cldns are the main determinants of barrier properties of tight junctions.36 From our preliminary RNA sequencing data, several of the Cldn genes were induced after DSS treatment, with an accelerated induction in NCoR1ΔIEC colons, whereas others were reduced (Figure 3G). Many Cldn genes show differential regulation in naïve mice after NCoR1 deletion, similar to Reg4 and AMPs. Because these proteins maintain tight junction properties, this indicates a disruption of colon barrier integrity. Cldn2 and Cldn15 both were down-regulated in DSS treatment, and to a greater extent in DSS-NCoR1ΔIEC mice. This is of interest because their expressions are localized to the deep crypt cells.37,38 In brief, these data strongly suggest that NCoR1 plays an important role in maintaining colonic epithelial cell homeostasis and the barrier integrity, and the absence of NCoR1 leads to the compromise of epithelial barrier protection, increasing mice vulnerability to various disease factors.
NCoR1ΔIEC Crypts Were More Sensitive to Butyrate-Induced Suppression of Proliferative Cells
The faster and stronger barrier disruption in DSS-treated NCoR1ΔIEC mice will lead to the exposure of deep colonic stem cells to luminal contents, resulting in damage to cell proliferation. Butyrate is one of the short-chain fatty acids that is the end product of dietary fibers fermented by anaerobic intestinal microbiota. Recent reports have shown that butyrate, but not other short-chain fatty acids, is a potent suppressor of colonic epithelial stem/progenitor proliferation.39 We have shown that NCoR1ΔIEC mice show greater barrier permeability, providing the possibility that colonic crypt cells can be exposed to luminal factors, including butyrate. Therefore, we examined if NCoR1 protects cells from butyrate-induced growth suppression. Based on our experience isolating intestinal organoids40 and published protocols,41 we have established a protocol for colonic crypt culture. The role of NCoR1 in cultured crypt cells isolated from NCoR1F/F and NCoR1ΔIEC colons was determined (Figure 4A and B). Colony-forming efficiency assays were performed from single cells isolated from spheroids. Cells were incubated in culture medium in the absence or presence of butyrate. On day 5, colonies were quantified, and the size of the spheroids was measured via microscopy. Both reduced numbers of colonies and decreased spheroid size were observed in NCoR1ΔIEC in the presence of 1 mmol/L butyrate (Figure 4C and D). Cells in active proliferation also were analyzed by using the Click-iT EdU Alexa Fluor 488 Imaging Kit (Thermo Fisher, Waltham, MA). Cells positive in EdU fluorescence were largely decreased after butyrate exposure, with a more significant change in NCoR1ΔIEC cells (Figure 4E). When NCoR1F/F and NCoR1ΔIEC crypts were treated with different concentrations of butyrate ranging from 0.1 to 30 mmol/L, apoptosis marker cleaved caspase 3 was increased at 3 mmol/L of butyrate exposure in NCoR1ΔIEC but not in NCoR1F/F mice (Figure 4F). Butyrate is a HDAC inhibitor that has been linked to its suppression of colonic stem/progenitor cell proliferation.39 We found that NCoR1ΔIEC crypts showed higher induction of histone H3 lysine 9 acetylation (H3K9ac) and H3K27ac after butyrate exposure (Figure 4F). NCoR1 may increase the resistance of crypt cells to the thresholds of antiproliferative stimuli during injury.
Figure 4.

NCoR1ΔIEC crypts were more sensitive to butyrate-induced suppression of proliferative cells. (A) Image of colonic crypt cells. Scale bar: 200 μm. Images were acquired using a 10× Ph1 ADL (numeric aperture, 0.25) phase-contrast objective on an inverted Nikon Ts2 microscope with a Nikon DS-Fi3 camera and Nikon NIS elements imaging software (Nikon Instruments, Inc, Melville, NY). (B) RT-qPCR of NCoR1 in colonic crypt cells after a few time passages (n = 3). (C) Colony-forming efficiency. Crypt cells were counted and plated at 10 cells/5 μL Matrigel and cultured in media supplemented with 1 mmol/L butyrate. After 5 days, spheroids developed from crypt cells and were enumerated and described as the percentage of control cells with no butyrate treatment. (D) The size of spheroids (diameter) was measured under a microscope, and grouped as large (>200 μm), medium (100–200 μm), and small (<100 μm). The results were described as the percentage of total cell numbers. (E) EdU incorporation assay. Two days after plating, cells were exposed to 1 mmol/L butyrate. Twenty-four hours later, cells were incubated with EdU at final concentration of 10 μmol/L. One hour later, cells were fixed and permeated for immunostaining with EdU Click-iT reaction cocktail containing Alexa Fluor–labeled primary antibody, followed by 4′,6-diamidino-2-phenylindole staining. Images were acquired with a Leica TCS SP5 X confocal microscope and LAS AF imaging software. Scale bar: 50 μm. EdU+ cells were counted and described as the percentage of total cell numbers. (F) NCoR1F/F and NCoR1ΔIEC spheroids were treated with vehicle or 0.1, 1, 3, 10, and 30 mmol/L butyrate for 24 hours. Whole-cell extracts were prepared for electrophoresis and Western blot. Band density of targets and tubulin were quantified using Image Lab 5.2.1 software, and the results are described as the ratio of band density of targets over tubulin. ∗P < .05, ∗∗∗P < .001. BUT, butyrate; C-Casp, cleaved caspase.
Synergetic Effects of Butyrate and TNFα in Inducing Crypt Cell Apoptosis, Especially in NCoR1ΔIEC Cells
To study the potential interrelationships among inflammatory signaling, butyrate, and NCoR1, we treated the cells with butyrate in the presence of TNFα. Results showed that cotreatment with TNFα largely increased cell apoptosis in both NCoR1F/F and NCoR1ΔIEC spheroids, as examined by the induced apoptotic marker cleaved caspase 3 (Figure 5A). Histone markers H3K9ac and H3K27ac also were increased dramatically. We suspected that the increased apoptosis of the cotreatment likely was a result of HDAC inhibition. We then used another HDAC inhibitor trichostatin A to verify this possibility. We found that trichostatin A, similar to butyrate, shows similar effects in promoting cell death in combination with TNFα treatment (Figure 5B). It is worth noting that TNFα also can minimize histone deacetylation by depleting HDAC1.42 Therefore, these results show that NCoR1 plays an important role in maintaining the status of histone deacetylation. When interrupted, histones are acetylated more easily by factors including TNFα and butyrate, resulting in up-regulation of genes that are associated with apoptosis. To study this further, we used the histone acetyltransferase inhibitor anacardic acid and cotreated with butyrate. The presence of a histone acetyltransferase inhibitor successfully counteracted the accumulation of H3K9ac and H3K27ac and minimized butyrate-induced apoptosis in both NCoR1F/F and NCoR1ΔIEC spheroids (Figure 5C), results that further confirmed the importance of epigenetic modification in butyrate-induced apoptosis.
Figure 5.

Synergetic role of butyrate, TNFα, and NCoR1 deletion in inducing crypt cell apoptosis. (A) Cells were treated with different concentrations of butyrate in the presence or absence of TNFα (20 ng/mL). Twenty-four hours after treatment, cells were collected and whole-cell extracts were prepared for Western blot analysis. (B) Cells were treated with different concentrations of trichostatin (TSA) in the presence or absence of TNFα (20 ng/mL) for 24 hours and followed by the preparation of whole-cell extracts that were used in Western blot analysis. (C) Cells were treated with butyrate (5 mmol/L) in the presence or absence of anacardic acid (ANA, 10 μmol/L, MedChemExpress), a histone acetyltransferase inhibitor. After 24 hours, whole-cell extracts were prepared for electrophoresis and Western blot. The band densities of targets and tubulin were quantified using Image Lab 5.2.1 software, and the results were described as the ratio of band density of targets over tubulin. BUT, butyrate; C-Casp, cleaved caspase.
Discussion
Our findings define a critical function of NCoR1 in fighting against UC by guarding proliferative cells in the colon tissue. In our studies, we have shown that mice with IEC-NCoR1 deletion show more severe colitis when exposed to DSS. DSS itself does not cause intestinal inflammation directly. Instead, DSS-elicited chemical injury to the epithelium, leading to exposure of the lamina propria and submucosal compartment to luminal antigens and enteric bacteria, triggering inflammation.43 By using the FITC-d permeability analysis, we determined that FITC-d was detected earlier and at a higher concentration in serum samples in DSS-treated knockout mice, indicating that IEC-NCoR1 deletion mice showed increased fragility of the epithelial barrier toward DSS-induced chemical injury. In analyzing the potential cause, we discovered that in IEC-NCoR1 colons, the expression of AMP genes, produced mainly by secretory cells including goblet and DCS cells, were down-regulated significantly. The changes were noticed significantly in mice without DSS treatment. This suggests the possibility that secretory cell lineage is compromised after IEC-NCoR1 deletion. Secretory cell lineage originates from colonic stem cells. Activation of Notch signaling in Lgr5+ cells maintains self-renewal and proliferation of stem cells, but it negatively regulates the transcription factor Math1, which is necessary for differentiation of the secretory cell lineages. Notch thus biases cell fate toward the absorptive lineage.44 Because we have observed that BrdU+ cells are increased after IEC-NCoR1 deletion in concert with AMP inhibition, it suggests that NCoR1 might be important in balancing Notch signaling and maintaining the secretory cell differentiation. It has been reported that the decrease of histone deacetylation, for example, by using HDAC inhibitor valproic acid, synergistically can maintain self-renewal of mouse Lgr5+ intestinal stem cells, resulting in nearly homogeneous cultures of Lgr5+ cells, stimulating cell proliferation but minimizing cell differentiation.45 Valproic acid has been shown for its role in activating Notch signaling.46 We suspect that NCoR1 may show this function through regulating histone deacetylation. NCoR1 is known as a nuclear-receptor corepressor that intermediates HDAC with a host of nuclear receptors in regulating downstream target genes. Thus, the deletion of IEC-NCoR1 leads to a decrease in HDAC, which may activate Notch signaling and minimize secretory cell differentiation. The decreased production of secretory products, such as AMPs, predispose mice toward various insults, such as DSS-induced chemical insult, resulting in increased colitis severity.
We showed that NCoR1 deletion leads colonic crypt cells to be more sensitive to butyrate-induced cytotoxicity. The colon has high concentrations of butyrate-producing bacteria. In the lumen of the colon, butyrate concentrations can reach 10 mmol/L in mice, which is used primarily by mature colonocytes as an energy source through fatty acid oxidation.47,48 This process titrates butyrate concentrations to 0.05–1 mmol/L when it reaches the crypt base where colonic stem cells reside.47, 48, 49 This is important because the concentration appears to be an important factor in determining the outcome of butyrate treatment. In our experiments, we found that IEC-NCoR1 deletion cells have a higher sensitivity toward butyrate-induced cell growth and apoptosis, as determined by organoid-forming efficiency and the cleavage of caspase 3, and these effects are exaggerated in the presence of TNFα. This is of great clinical significance. Because UC patients have deteriorated barriers and inflamed tissue, butyrate likely is penetrating to the base area of the crypts, in which it synergizes with inflammatory molecules, such as TNFα, resulting in severe damage of colonic stem cells. Many studies have emphasized the beneficial role of butyrate for gut health in conditions that include IBD, for which probiotics are popularly used to promote the good bacteria producing butyrate. Thus, the finding of the potential deteriorating effects of butyrate is of great significance in human health, especially in individuals with UC. This finding is concordant with recent publications in which butyrate or butyrate-producing fermentable fiber are found to exacerbate the severity of experimental colitis.39,50, 51, 52, 53 These observations also coincide with current practices to exclude fermentable oligosaccharides, disaccharides, monosaccharides, and polyols from the diet of IBD patients.54,55
Caloric intake regulates tissue homeostasis and health.56 Diets with a high intake of fat, common in the Western world and countries with similar lifestyles, have been identified as risk factors for the development of UC and IBD.1,57 In animal models, a high-fat diet (HFD) has been associated with factors leading to the deterioration of intestinal homeostasis, including gut dysbiosis, increased barrier permeability, and amplified chronic inflammation.58,59 Yamamoto et al60 confirmed that a HFD impairs NCoR1-specific enhanced exercise endurance, a property in NCoR1 deleted muscle tissue that induces the capacity to metabolize oxygen after activation of peroxisome proliferator-activated receptor-δ and/or estrogen-related receptor alpha.60,61 This study provided convincing evidence that the regulation of NCoR1 was linked directly to metabolic homeostasis. The transcriptional co-repressor NCoR1 activity also can be interrupted post-translationally by triggering the removal of NCoR1 from the nucleus, an event that is induced by phosphorylation and ubiquitination.19,62 These events can be impacted directly by a HFD because caloric overload is tied to the development of chronic inflammation59 and the production of cytokines TNFα and IL1β, both of which are signaling mechanisms for induction of phosphorylation cascades. For example, mitogen-activated protein kinase kinase kinase 1 phosphorylates NCoR1 after IL1β,63 and c-Jun phosphorylation removed NCoR1 from promoter binding sites after the stress of liposaccharide treatment.64 Therefore, we suspect that NCoR1 activity can be inhibited by HFD factors, such as excessive fatty acids and/or induced cytokines, and the decrease of NCoR1-repressive activity may function as a causal factor in colitis development. Taken together, in this study we have shown in vivo and in vitro the protecting role of NCoR1 in the development of experimental colitis in mice. The environmental triggers–NCoR1–colonic epithelium–butyrate axis may serve as a causal factor in driving colitis initiation and development and could be exploited as therapeutic targets to reduce disease progression.
Methods
Animal Studies and Tissue Collections
We crossed NCoR1F/F mice23 with Villin-Cre transgenic mice (Jackson Laboratory, Bar Harbor, ME)65 to generate NCoR1F/F (F/F) and NCoR1ΔIEC (ΔIEC) mice. All mouse strains were housed in a pathogen-free University of California, San Diego (UCSD) vivarium. All animal protocols were reviewed and approved by the UCSD Animal Care and Use Committee. Both male and female mice were included, with siblings preferred for all the experiments. Colon tissues were dissected from mice, washed with ice-cold phosphate-buffered saline, snap-frozen in liquid nitrogen, and stored at -80°C. Frozen tissues were pulverized for further RNA and protein extraction. For histology studies, colons were removed, opened longitudinally, and cleaned. Tissues were prepared in a Swiss roll style and then either fixed in 10% phosphate-buffered formalin for histologic examination or embedded into optimal cutting temperature compound for frozen sectioning.
Total RNA Preparation, RT-qPCR, and RNA Sequencing Analysis
Total RNA from tissue and spheroid samples was extracted using TRIzol reagent (Thermo Fisher Scientific). For RT-qPCR analysis, the iScript complementary DNA synthesis kit (Bio-Rad, Hercules, CA) was used for reverse transcription, and real-time quantitative PCR experiments were performed on a CFX96 qPCR system (Bio-Rad) using Ssoadvanced SYBR Green reagent (Bio-Rad). For RNA sequencing studies, each RNA sample consisted of RNA from 3 mice, and 4 RNA samples (a total of 12 mice) per group were analyzed as previously performed in our laboratory.23 The sequencing library was prepared using the Illumina TruSeq RNA Sample Prep Kit (FC-122-1001; Illumina, San Diego, CA) with 1 ug of total RNA. The sequencing was performed on an Illumina HiSeq4000 sequencer in the IGM Genomics Center at UCSD. RNA sequencing data analysis was performed by Dr Christopher Benner (UCSD). Image deconvolution, quality value calculation, and the mapping of exon reads and exon junctions were performed by Dr Chris Benner (UCSD). Base calling was performed using bcl2fastq (v2.17.1.14; Illumina). RNA sequencing reads were aligned to the mouse genome (mm10) with STAR (v2.2.0c) with default parameters, only uniquely alignable reads were used for downstream analysis. Gene expression values were calculated using HOMER by quantifying strand-specific reads across annotated gene exons (RefSeq) and reported as fragments per kilobase of exon per million mapped reads. Sequencing reads were aligned to the Mus musculus (UCSC mm10) genome.
Histology, Immunohistochemistry, and Immunofluorescence
Paraffin-embedded sections were used for routine H&E staining, which were performed at the UCSD Cancer Center Histology Core. The histopathologic colitis score was quantitated based on the presence and severity of ulcerative lesions, disrupted epithelial structure, damaged crypt architecture, and increased inflammatory cell infiltration, with each parameter scored on a scale of 0–3 (where none = 0; mild = 1; moderate = 2; and severe = 3), which were summed to provide an overall score.66 BrdU immunohistochemistry was performed according to the manufacturer’s instructions (8800-6599-45; eBioscience Inc, San Diego, CA). Briefly, mice were treated with BrdU at 100 mg/kg by intraperitoneal injection. Paraffin-embedded colon sections were applied for immunohistochemistry staining and detected by the ABC detection system (eBioscience, Inc. San Diego, CA). Slides were examined using a 20× Plan-Apochromat objective (numeric aperture, 0.8) on an upright Imager A2 microscope (Carl Zeiss Microscopy, LLC, White Plains, NY) with an Axiocam 506 color camera and ZEN2012 imaging software. Ten photographs at different areas were taken of each slide. BrdU+ cells were enumerated and described as BrdU+ cells per villous. Frozen sections were prepared in optimal cutting temperature compound for immunofluorescence staining. After the overnight incubation with primary antibodies, slides then were washed and stained with Alexa-488–conjugated secondary antibodies (Life Technologies, Carlsbad, CA), alongside 4′,6-diamidino-2-phenylindole counterstaining. The following antibodies were used: anti-Ki67 (GTX16667; GeneTex, Irvine, CA), anti–β-catenin (sc-1496; Santa Cruz Biotechnology, Dallas, TX). Mounted slides were examined under a Leica TCS SP5 X confocal microscope (Leica Microsystems Inc, Buffalo Grove, IL) and LAS AF imaging software. Ten photographs at different areas were taken of each slide. Ki67-positive (Ki67+) cells were enumerated and described as Ki67+ cells per field.
FITC-d Permeability Analysis
Both NCoR1F/F and NCoR1ΔIEC mice were exposed to either water or challenged with 2.5% DSS (40–50 kilodaltons; Affymetrix, Santa Clara, CA) in their drinking water. We examined 2 exposure time points after DSS treatment on day 3, which preceded any signs of BW loss or severe inflammation, and DSS day 5, when mice had significant BW loss. On the last day of DSS exposure, both naïve and DSS-treated groups were administered 20 mg FITC-d dissolved in injectable phosphate-buffered saline by oral gavage. After 4 hours, blood samples were collected and prepared for serum. FITC-d levels of diluted sera were measured at an excitation wavelength of 485 nm and an emission wavelength of 528 nm (FilterMax F5 Multi-Mode microplate reader; Molecular Devices, San Jose, CA) and calculated from a FITC-d standard curve. Data are described as the concentration of FITC (ng/mL).
Crypt Isolation, Culture, and Passage
Colonic organoids (spheroids) were cultured as described previously.40,41 Briefly, colon tissue isolated from NCoR1F/F and NCoR1ΔIEC mice were incubated in 2 mg/mL collagenase I solution (17018029; Thermo Fisher Scientific). After incubation, the tissue mixture was filtered through a 70-μm cell strainer. Crypts were plated in Matrigel (BD 356231; BD Bioscience, San Diego, CA) and cells were cultured in advanced Dulbecco’s modified Eagle medium/F12 (12634028; Thermo Fisher Scientific) with 50% Wnt-3A, R-spondin 3, and Noggin (WRN) conditioned medium, supplemented with 20% fetal bovine serum, 10 μmol/L of A83-01 (a transforming growth factor β inhibitor), and 10 μmol/L Y-27632 (a ROCK inhibitor that enhances the survival and cloning efficiency of dissociated stem cells without affecting their pluripotency). The WRN conditioned medium containing Wnt-3A, R-spondin 3, and Noggin is produced by the L-WRN cell line, a generous gift from Dr Thaddeus S Stappenbeck (Washington University, St. Louis, MO). When passaging for maintenance, spheroids were incubated with 0.25% Trypsin–EDTA and cells were dissociated by vigorous pipetting.
Colony Forming Efficiency Assay
Colonic spheroids were removed from Matrigel and then trypsinized for 10 minutes in 0.25% Trypsin–EDTA with vigorous pipetting to achieve single cells. Cells were filtered through a 40-μm cell strainer and then collected by centrifugation. Cells were resuspended in Matrigel at 10 cells/5 μL. Cells were cultured, and media were changed every other day in the presence or the absence of 1 mmol/L butyrate. On day 5, single cells that formed spheroid colonies were quantified. Colony-forming efficiency was described as a percentage of colonies formed in butyrate vs control media (as a percentage). Spheroid size was measured under microscopy and grouped as large (>200 μm), medium (100–200 μm), and small (<100 μm). The cell counts of each group was compared with the total cell counts and described as a percentage of the total.
EdU Click-iT Cell-Cycle Imaging Assay
The EdU Click-iT cell cycle imaging assay was performed according to the manufacturer’s manual with slight modification (C10639; Thermo Fisher Scientific). Briefly, 2 days after cells were plated, half of the media was replaced with fresh media containing 20 μmol/L EdU (5-ethynyl-2'-deoxyuridine). One hour later, cells were fixed in freshly made 4% paraformaldehyde, washed with 3% bovine serum albumin, and then blocked with phosphate-buffered saline containing 0.5% Triton X-100 (Sigma-Aldrich Corp, St. Louis, MO). Cells then were immunostained with Click-iT reaction cocktail containing Alexa Fluor–labeled primary antibody, followed by 4′,6-diamidino-2-phenylindole staining. Cells were mounted gently on glass slides and imaged under a Leica TCS SP5 X confocal microscope with LAS AF imaging software. The percentage of EdU-positive cells (S-phase cells) was enumerated.
Protein Preparation and Western Blots
Tissue or cell lysates were prepared in a RIPA buffer supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail. Electrophoresis was performed by using BOLT 4%–12% gradient gel (Thermo Fisher Scientific). Western blots were developed and imaged using a ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA), band density was quantified by using Image Lab 5.2.1 software. The following antibodies were used: cleaved caspase 3 (9661; Cell Signaling), cleaved caspase 8 (8592; Cell Signaling, Danvers, MA), H3K9ac (39137; Active Motif, Carlsbad, CA), H3K27ac (ab4729; Abcam, Cambridge, MA), and anti–α-tubulin (T9026; Sigma-Aldrich, St. Louis, MO).
Statistics
All results were subjected to statistical analysis. Student t test analyses (nonparametric Mann–Whitney test) were performed for most of the studies unless specified otherwise. All statistics and graphs were generated using GraphPad Prism software (GraphPad Software, San Diego, CA). Data are expressed as means ± SEM, and P values smaller than .05 were considered statistically significant.
Acknowledgments
The authors would like to thank Dr Robert H. Tukey at UCSD for providing great support for this study; Dr Ricard Garcia-Carbonell, Dr Monica Guma, and Dr Michael Karin at UCSD for advice regarding crypt culture; Dr Thaddeus S. Stappenbeck at Washington University, St. Louis, MO, for his generosity in providing the L-WRN cell line; and Dr Nissi Varki at the UCSD Cancer Center Histology Core in reviewing sectioning and staining of intestinal tissue.
Footnotes
Conflicts of interest The authors disclose no conflicts.
CRedit Authorship Contributions Elvira Mennillo, Ph.D (Investigation: Lead; Writing – original draft: Equal) Xiaojing Yang, Ph.D (Investigation: Supporting; Methodology: Supporting; Writing – original draft: Supporting) Miles Paszek, Graduate candidate (Investigation: Supporting; Writing – original draft: Supporting) Johan Auwerx, Ph.D (Resources: Supporting) Christopher W. Benner, Ph.D (Formal analysis: Equal; Writing – original draft: Supporting) Shujuan Chen, Ph.D. (Conceptualization: Lead; Data curation: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Lead; Project administration: Lead; Resources: Lead; Supervision: Lead; Writing – original draft: Lead)
Funding This work was supported by US Public Health Service Grants from the National Institutes of Environmental Health Sciences [ES010337], General Medical Sciences [GM100481], and Allergy and Infectious Disease [AI135677], and the UCSD Graduate Training Program in Cellular and Molecular Pharmacology [GM007752].
References
- 1.Ungaro R., Mehandru S., Allen P.B., Peyrin-Biroulet L., Colombel J.F. Ulcerative colitis. Lancet. 2017;389:1756–1770. doi: 10.1016/S0140-6736(16)32126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahuja V., Tandon R.K. Inflammatory bowel disease in the Asia-Pacific area: a comparison with developed countries and regional differences. J Dig Dis. 2010;11:134–147. doi: 10.1111/j.1751-2980.2010.00429.x. [DOI] [PubMed] [Google Scholar]
- 3.Chow D.K., Leong R.W., Tsoi K.K., Ng S.S., Leung W.K., Wu J.C., Wong V.W., Chan F.K., Sung J.J. Long-term follow-up of ulcerative colitis in the Chinese population. Am J Gastroenterol. 2009;104:647–654. doi: 10.1038/ajg.2008.74. [DOI] [PubMed] [Google Scholar]
- 4.Choi J.K., Kim D.W., Shin S.Y., Park E.C., Kang J.G. Effect of ulcerative colitis on incidence of colorectal cancer: results from the nationwide population-based cohort study (2003-2013) J Cancer. 2016;7:681–686. doi: 10.7150/jca.14264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romano M., De Francesco F., Zarantonello L., Ruffolo C., Ferraro G.A., Zanus G., Giordano A., Bassi N., Cillo U. From inflammation to cancer in inflammatory bowel disease: molecular perspectives. Anticancer Res. 2016;36:1447–1460. [PubMed] [Google Scholar]
- 6.Grivennikov S., Karin E., Terzic J., Mucida D., Yu G.Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pizarro T.T., Stappenbeck T.S., Rieder F., Rosen M.J., Colombel J.F., Donowitz M., Towne J., Mazmanian S.K., Faith J.J., Hodin R.A., Garrett W.S., Fichera A., Poritz L.S., Cortes C.J., Shtraizent N., Honig G., Snapper S.B., Hurtado-Lorenzo A., Salzman N.H., Chang E.B. Challenges in IBD research: preclinical human IBD mechanisms. Inflamm Bowel Dis. 2019;25:S5–S12. doi: 10.1093/ibd/izz075. [DOI] [PubMed] [Google Scholar]
- 8.Strober W., Fuss I., Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biondi A., Zoccali M., Costa S., Troci A., Contessini-Avesani E., Fichera A. Surgical treatment of ulcerative colitis in the biologic therapy era. World J Gastroenterol. 2012;18:1861–1870. doi: 10.3748/wjg.v18.i16.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannom R.R., Kaiser A.M., Ault G.T., Beart R.W., Jr., Etzioni D.A. Inflammatory bowel disease in the United States from 1998 to 2005: has infliximab affected surgical rates? Am Surg. 2009;75:976–980. [PubMed] [Google Scholar]
- 11.Im J.P., Ye B.D., Kim Y.S., Kim J.S. Changing treatment paradigms for the management of inflammatory bowel disease. Korean J Intern Med. 2018;33:28–35. doi: 10.3904/kjim.2017.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denson L.A., Curran M., McGovern D.P.B., Koltun W.A., Duerr R.H., Kim S.C., Sartor R.B., Sylvester F.A., Abraham C., de Zoeten E.F., Siegel C.A., Burns R.M., Dobes A.M., Shtraizent N., Honig G., Heller C.A., Hurtado-Lorenzo A., Cho J.H. Challenges in IBD research: precision medicine. Inflamm Bowel Dis. 2019;25:S31–S39. doi: 10.1093/ibd/izz078. [DOI] [PubMed] [Google Scholar]
- 13.Shah S.C., Colombel J.F., Sands B.E., Narula N. Mucosal healing is associated with improved long-term outcomes of patients with ulcerative colitis: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2016;14:1245–1255 e8. doi: 10.1016/j.cgh.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 14.Peyrin-Biroulet L., Ferrante M., Magro F., Campbell S., Franchimont D., Fidder H., Strid H., Ardizzone S., Veereman-Wauters G., Chevaux J.B., Allez M., Danese S., Sturm A. Scientific Committee of the European Crohns and Colitis Organization. Results from the 2nd Scientific Workshop of the ECCO. I: impact of mucosal healing on the course of inflammatory bowel disease. J Crohns Colitis. 2011;5:477–483. doi: 10.1016/j.crohns.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Yui S., Nakamura T., Sato T., Nemoto Y., Mizutani T., Zheng X., Ichinose S., Nagaishi T., Okamoto R., Tsuchiya K., Clevers H., Watanabe M. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat Med. 2012;18:618–623. doi: 10.1038/nm.2695. [DOI] [PubMed] [Google Scholar]
- 16.Fordham R.P., Yui S., Hannan N.R., Soendergaard C., Madgwick A., Schweiger P.J., Nielsen O.H., Vallier L., Pedersen R.A., Nakamura T., Watanabe M., Jensen K.B. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell. 2013;13:734–744. doi: 10.1016/j.stem.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuda M., Mizutani T., Mochizuki W., Matsumoto T., Nozaki K., Sakamaki Y., Ichinose S., Okada Y., Tanaka T., Watanabe M., Nakamura T. Small intestinal stem cell identity is maintained with functional Paneth cells in heterotopically grafted epithelium onto the colon. Genes Dev. 2014;28:1752–1757. doi: 10.1101/gad.245233.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishizuka T., Lazar M.A. The N-CoR/histone deacetylase 3 complex is required for repression by thyroid hormone receptor. Mol Cell Biol. 2003;23:5122–5131. doi: 10.1128/MCB.23.15.5122-5131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mottis A., Mouchiroud L., Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–835. doi: 10.1101/gad.214023.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li P., Spann N.J., Kaikkonen M.U., Lu M., Oh d.Y., Fox J.N., Bandyopadhyay G., Talukdar S., Xu J., Lagakos W.S., Patsouris D., Armando A., Quehenberger O., Dennis E.A., Watkins S.M., Auwerx J., Glass C.K., Olefsky J.M. NCoR repression of LXRs restricts macrophage biosynthesis of insulin-sensitizing omega 3 fatty acids. Cell. 2013;155:200–214. doi: 10.1016/j.cell.2013.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li P., Fan W., Xu J., Lu M., Yamamoto H., Auwerx J., Sears D.D., Talukdar S., Oh D., Chen A., Bandyopadhyay G., Scadeng M., Ofrecio J.M., Nalbandian S., Olefsky J.M. Adipocyte NCoR knockout decreases PPARgamma phosphorylation and enhances PPARgamma activity and insulin sensitivity. Cell. 2011;147:815–826. doi: 10.1016/j.cell.2011.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barish G.D., Yu R.T., Karunasiri M.S., Becerra D., Kim J., Tseng T.W., Tai L.J., Leblanc M., Diehl C., Cerchietti L., Miller Y.I., Witztum J.L., Melnick A.M., Dent A.L., Tangirala R.K., Evans R.M. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012;15:554–562. doi: 10.1016/j.cmet.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S., Lu W., Yueh M.F., Rettenmeier E., Liu M., Auwerx J., Yu R.T., Evans R.M., Wang K., Karin M., Tukey R.H. Intestinal NCoR1, a regulator of epithelial cell maturation, controls neonatal hyperbilirubinemia. Proc Natl Acad Sci U S A. 2017;114:E1432–E1440. doi: 10.1073/pnas.1700232114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perse M., Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barker N., van Es J.H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P.J., Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 26.van der Flier L.G., van Gijn M.E., Hatzis P., Kujala P., Haegebarth A., Stange D.E., Begthel H., van den Born M., Guryev V., Oving I., van Es J.H., Barker N., Peters P.J., van de Wetering M., Clevers H. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. doi: 10.1016/j.cell.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 27.Birchenough G.M., Johansson M.E., Gustafsson J.K., Bergstrom J.H., Hansson G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015;8:712–719. doi: 10.1038/mi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johansson M.E., Hansson G.C. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol. 2016;16:639–649. doi: 10.1038/nri.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bevins C.L., Salzman N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 30.Sato T., van Es J.H., Snippert H.J., Stange D.E., Vries R.G., van den Born M., Barker N., Shroyer N.F., van de Wetering M., Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sasaki N., Sachs N., Wiebrands K., Ellenbroek S.I., Fumagalli A., Lyubimova A., Begthel H., van den Born M., van Es J.H., Karthaus W.R., Li V.S., Lopez-Iglesias C., Peters P.J., van Rheenen J., van Oudenaarden A., Clevers H. Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc Natl Acad Sci U S A. 2016;113:E5399–E5407. doi: 10.1073/pnas.1607327113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rothenberg M.E., Nusse Y., Kalisky T., Lee J.J., Dalerba P., Scheeren F., Lobo N., Kulkarni S., Sim S., Qian D., Beachy P.A., Pasricha P.J., Quake S.R., Clarke M.F. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology. 2012;142:1195–1205 e6. doi: 10.1053/j.gastro.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grun D., Lyubimova A., Kester L., Wiebrands K., Basak O., Sasaki N., Clevers H., van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 34.Hooper L.V., Stappenbeck T.S., Hong C.V., Gordon J.I. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee S., Hooper L.V. Antimicrobial defense of the intestine. Immunity. 2015;42:28–39. doi: 10.1016/j.immuni.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 36.Tsukita S., Tanaka H., Tamura A. The claudins: from tight junctions to biological systems. Trends Biochem Sci. 2019;44:141–152. doi: 10.1016/j.tibs.2018.09.008. [DOI] [PubMed] [Google Scholar]
- 37.Fujita H., Chiba H., Yokozaki H., Sakai N., Sugimoto K., Wada T., Kojima T., Yamashita T., Sawada N. Differential expression and subcellular localization of claudin-7, -8, -12, -13, and -15 along the mouse intestine. J Histochem Cytochem. 2006;54:933–944. doi: 10.1369/jhc.6A6944.2006. [DOI] [PubMed] [Google Scholar]
- 38.Lili L.N., Farkas A.E., Gerner-Smidt C., Overgaard C.E., Moreno C.S., Parkos C.A., Capaldo C.T., Nusrat A. Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4-alpha cascades. Tissue Barriers. 2016;4 doi: 10.1080/21688370.2016.1214038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiko G.E., Ryu S.H., Koues O.I., Collins P.L., Solnica-Krezel L., Pearce E.J., Pearce E.L., Oltz E.M., Stappenbeck T.S. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016;165:1708–1720. doi: 10.1016/j.cell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu W., Rettenmeier E., Paszek M., Yueh M.F., Tukey R.H., Trottier J., Barbier O., Chen S. Crypt organoid culture as an in vitro model in drug metabolism and cytotoxicity studies. Drug Metab Dispos. 2017;45:748–754. doi: 10.1124/dmd.117.075945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyoshi H., Stappenbeck T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vashisht Gopal Y.N., Arora T.S., Van Dyke M.W. Tumour necrosis factor-alpha depletes histone deacetylase 1 protein through IKK2. EMBO Rep. 2006;7:291–296. doi: 10.1038/sj.embor.7400613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Y., Kolachala V., Dalmasso G., Nguyen H., Laroui H., Sitaraman S.V., Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch U., Lehal R., Radtke F. Stem cells living with a Notch. Development. 2013;140:689–704. doi: 10.1242/dev.080614. [DOI] [PubMed] [Google Scholar]
- 45.Yin X., Farin H.F., van Es J.H., Clevers H., Langer R., Karp J.M. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods. 2014;11:106–112. doi: 10.1038/nmeth.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenblatt D.Y., Vaccaro A.M., Jaskula-Sztul R., Ning L., Haymart M., Kunnimalaiyaan M., Chen H. Valproic acid activates notch-1 signaling and regulates the neuroendocrine phenotype in carcinoid cancer cells. Oncologist. 2007;12:942–951. doi: 10.1634/theoncologist.12-8-942. [DOI] [PubMed] [Google Scholar]
- 47.Fleming S.E., Fitch M.D., DeVries S., Liu M.L., Kight C. Nutrient utilization by cells isolated from rat jejunum, cecum and colon. J Nutr. 1991;121:869–878. doi: 10.1093/jn/121.6.869. [DOI] [PubMed] [Google Scholar]
- 48.Roediger W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology. 1982;83:424–429. [PubMed] [Google Scholar]
- 49.Donohoe D.R., Collins L.B., Wali A., Bigler R., Sun W., Bultman S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48:612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miles J.P., Zou J., Kumar M.V., Pellizzon M., Ulman E., Ricci M., Gewirtz A.T., Chassaing B. Supplementation of low- and high-fat diets with fermentable fiber exacerbates severity of DSS-induced acute colitis. Inflamm Bowel Dis. 2017;23:1133–1143. doi: 10.1097/MIB.0000000000001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Q., Wu Y., Wang J., Wu G., Long W., Xue Z., Wang L., Zhang X., Pang X., Zhao Y., Zhao L., Zhang C. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci Rep. 2016;6:27572. doi: 10.1038/srep27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuo S.M., Chan W.C., Hu Z. Wild-type and IL10-null mice have differential colonic epithelial gene expression responses to dietary supplementation with synbiotic Bifidobacterium animalis subspecies lactis and inulin. J Nutr. 2014;144:245–251. doi: 10.3945/jn.113.185249. [DOI] [PubMed] [Google Scholar]
- 53.Vancamelbeke M., Laeremans T., Vanhove W., Arnauts K., Ramalho A.S., Farre R., Cleynen I., Ferrante M., Vermeire S. Butyrate does not protect against inflammation-induced loss of epithelial barrier function and cytokine production in primary cell monolayers from patients with ulcerative colitis. J Crohns Colitis. 2019;13:1351–1361. doi: 10.1093/ecco-jcc/jjz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrett J.S., Gibson P.R. Fermentable oligosaccharides, disaccharides, monosaccharides and polyols (FODMAPs) and nonallergic food intolerance: FODMAPs or food chemicals? Therap Adv Gastroenterol. 2012;5:261–268. doi: 10.1177/1756283X11436241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrett J.S. How to institute the low-FODMAP diet. J Gastroenterol Hepatol. 2017;32(Suppl 1):8–10. doi: 10.1111/jgh.13686. [DOI] [PubMed] [Google Scholar]
- 56.Rafalski V.A., Mancini E., Brunet A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J Cell Sci. 2012;125:5597–5608. doi: 10.1242/jcs.114827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ananthakrishnan A.N., Khalili H., Konijeti G.G., Higuchi L.M., de Silva P., Fuchs C.S., Willett W.C., Richter J.M., Chan A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn's disease. Gut. 2014;63:776–784. doi: 10.1136/gutjnl-2013-305304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugihara K., Morhardt T.L., Kamada N. The role of dietary nutrients in inflammatory bowel disease. Front Immunol. 2018;9:3183. doi: 10.3389/fimmu.2018.03183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duan Y., Zeng L., Zheng C., Song B., Li F., Kong X., Xu K. Inflammatory links between high fat diets and diseases. Front Immunol. 2018;9:2649. doi: 10.3389/fimmu.2018.02649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamamoto H., Williams E.G., Mouchiroud L., Canto C., Fan W., Downes M., Heligon C., Barish G.D., Desvergne B., Evans R.M., Schoonjans K., Auwerx J. NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez-Schindler J., Summermatter S., Salatino S., Zorzato F., Beer M., Balwierz P.J., van Nimwegen E., Feige J.N., Auwerx J., Handschin C. The corepressor NCoR1 antagonizes PGC-1alpha and estrogen-related receptor alpha in the regulation of skeletal muscle function and oxidative metabolism. Mol Cell Biol. 2012;32:4913–4924. doi: 10.1128/MCB.00877-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perissi V., Jepsen K., Glass C.K., Rosenfeld M.G. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 63.Baek S.H., Ohgi K.A., Rose D.W., Koo E.H., Glass C.K., Rosenfeld M.G. Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 64.Huang W., Ghisletti S., Perissi V., Rosenfeld M.G., Glass C.K. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madison B.B., Dunbar L., Qiao X.T., Braunstein K., Braunstein E., Gumucio D.L. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 66.Kimball E.S., Wallace N.H., Schneider C.R., D'Andrea M.R., Hornby P.J. Vanilloid receptor 1 antagonists attenuate disease severity in dextran sulphate sodium-induced colitis in mice. Neurogastroenterol Motil. 2004;16:811–818. doi: 10.1111/j.1365-2982.2004.00549.x. [DOI] [PubMed] [Google Scholar]