

Abstract
Background
Drug resistance and chemotherapy-induced peripheral neuropathy continue to be significant problems in the successful treatment of acute lymphoblastic leukemia (ALL). 5,7-Dibromo-N-alkylisatins, a class of potent microtubule destabilizers, are a promising alternative to traditionally used antimitotics with previous demonstrated efficacy against solid tumours in vivo and ability to overcome P-glycoprotein (P-gp) mediated drug resistance in lymphoma and sarcoma cell lines in vitro. In this study, three di-brominated N-alkylisatins were assessed for their ability to retain potency in vincristine (VCR) and 2-methoxyestradiol (2ME2) resistant ALL cell lines. For the first time, in vitro neurotoxicity was also investigated in order to establish their suitability as candidate drugs for future use in ALL treatment.
Methods
Vincristine resistant (CEM-VCR R) and 2-methoxyestradiol resistant (CEM/2ME2-28.8R) ALL cell lines were used to investigate the ability of N-alkylisatins to overcome chemoresistance. Interaction of N-alkylisatins with tubulin at the the colchicine-binding site was studied by competitive assay using the fluorescent colchicine analogue MTC. Human neuroblastoma SH-SY5Y cells differentiated into a morphological and functional dopaminergic-like neurotransmitter phenotype were used for neurotoxicity and neurofunctional assays. Two-way ANOVA followed by a Tukey’s post hoc test or a two-tailed paired t test was used to determine statistical significance.
Results
CEM-VCR R and CEM/2ME2-28.8R cells displayed resistance indices of > 100 to VCR and 2-ME2, respectively. CEM-VCR R cells additionally displayed a multi-drug resistant phenotype with significant cross resistance to vinblastine, 2ME2, colchicine and paclitaxel consistent with P-gp overexpression. Despite differences in resistance mechanisms observed between the two cell lines, the N-alkylisatins displayed bioequivalent dose-dependent cytotoxicity to that of the parental control cell line. The N-alkylisatins proved to be significantly less neurotoxic towards differentiated SH-SY5Y cells than VCR and vinblastine, evidenced by increased neurite length and number of neurite branch points. Neuronal cells treated with 5,7-dibromo-N-(p-hydroxymethylbenzyl)isatin showed significantly higher voltage-gated sodium channel function than those treated with Vinca alkaloids, strongly supportive of continued action potential firing.
Conclusions
The N-alkylisatins are able to retain cytotoxicity towards ALL cell lines with functionally distinct drug resistance mechanisms and show potential for reduced neurotoxicity. As such they pose as promising candidates for future implementation into anticancer regimes for ALL. Further in vivo studies are therefore warranted.
Keywords: Chemotherapy, Multi-drug resistance, N-alkylisatin, P-glycoprotein, Neurotoxicity, Acute lymphoblastic leukemia
Background
Acute lymphoblastic leukemia (ALL) is a cancer of the blood and bone marrow. It is the most common childhood cancer, and its incidence is expected to increase worldwide [1, 2]. Over the last 40 years, advances in the development of novel anti-microtubule agents, refinements in multi-agent chemotherapy regimens and precision medicine approaches have resulted in a dramatic improvement in survival rates of children with ALL [3]. However, the emergence of drug resistance remains a pertinent obstacle to successful treatment and accounts for ~ 20% of ALL treatment failures [4]. ALL is a polyclonal disease and mutations that confer a survival advantage in cellular subclones may be selected by chemotherapy, thereby promoting treatment resistance and subsequent patient relapse [5]. There has been minimal progress in treating relapsed disease for the last two decades highlighting the need for new approaches to improve the chances for cure for patients with relapsed and refractory ALL.
The current approach to treating relapsed ALL involves many of the same drugs used for newly diagnosed patients; however they are frequently delivered at increased doses or using alternative schedules. The Vinca alkaloid vincristine (VCR) is a standard component in every combination chemotherapy regimen used to treat ALL [5–7]. VCR is an antimitotic agent that targets the β-tubulin subunit of αβ-tubulin heterodimers, effectively destroying mitotic spindles and inhibiting cancer cell division through microtubule depolymerization. Though VCR is a potent antineoplastic agent, its clinical use is limited by a number of factors related to the development of resistance [8, 9] and off-target neurotoxicity [10–12]. Resistance to microtubule-targeted drugs, such as VCR, can be mediated by several mechanisms including the overexpression of transmembrane P-glycoprotein (P-gp), a member of the ATP-binding cassette (ABC) family [13, 14]. P-gp acts as a broad-spectrum drug efflux transporter which reduces the ability of cytotoxic agents to accumulate to therapeutic concentrations in the intracellular environment. Other ABC transporters such as multi-drug resistance-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP) can also play a role [15]. Additionally, conformational changes in the drug target, such as mutations in the tubulin binding site, alterations in tubulin isotypes and/or altered microtubule polymer levels are key contributors to VCR drug resistance [16, 17]. Current evidence suggests that these cellular changes favour more stable microtubules or affect the microtubule dynamics such that VCR effectiveness is decreased [18]. Altogether, this calls for the development of novel drugs that can circumvent drug-efflux mechanisms and/or changes in the tubulin target as an alternative therapeutic approach.
Molecules targeting the colchicine binding site on tubulin may provide an opportunity to overcome such ABC transporter-mediated drug resistance [19, 20]. In fact, mounting evidence indicates that compounds interacting with the colchicine binding site are also less sensitive to other clinically-relevant mechanisms of resistance, including overexpression of the βIII-tubulin isotype [21]. Colchicine binds at the interface between the α- and β-tubulin monomers and prevents α-tubulin from stacking onto the β-tubulin β-sheet, thereby inhibiting microtubule assembly [22]. While colchicine itself is not used as an anticancer agent due to off-target toxicities, a large number of structurally diverse tubulin inhibitors targeting the colchicine site are currently being evaluated in preclinical and clinical trials for cancer with promising outcomes [23]. Interestingly, compounds bearing an indole ring form part of a growing number of antimitotic compounds that bind to the colchicine site on tubulin [24]. A related class of compounds that show great potential in mitigating multi-drug resistance (MDR), possibly through binding to the colchicine site or other sites [25] are those based on the isatin (1H-indole-2,3-dione) scaffold [26, 27]. Synthesis and subsequent cytotoxicity evaluation of a library of 5,7-dibominated N-alkylated isatins by our group found them to elicit potent cytotoxic activity against a range of human cancer cell lines in the sub-micromolar range [28, 29]. Tubulin binding experiments demonstrated that they destabilize microtubule growth, similar to the Vinca alkaloids and colchicine, by inhibiting microtubule polymerization [28, 29]. Interestingly, these N-alkylisatins retained potency against P-gp-overexpressing cell lines including both doxorubicin resistant uterine sarcoma and vinblastine resistant lymphoma cell lines [30]. While preliminary investigations into the potential binding site of the N-alkylisatins on tubulin have been performed, it remains unknown whether this class of molecules bind at a known site, or exploit a novel site on tubulin.
In order to investigate the ability of N-alkylisatins (Fig. 1) to overcome chemoresistance in ALL and to further elucidate molecular interactions with tubulin, we conducted cytotoxicity assays using VCR and 2-methoxyestradiol (2ME2) resistant ALL cell lines, the latter harbouring single-base mutations in the colchicine and paclitaxel binding sites of class I β-tubulin. We further studied the interaction of N-alkylisatins with tubulin at the the colchicine-binding site using competition assays. Finally, given that vincristine-induced peripheral neuropathy continues to be a dose-limiting factor in ALL chemotherapy, the neurotoxicity of three N-alkylisatins was studied in vitro using neurite outgrowth assays and neurofunctional electrophysiological experiments in order to further assess their suitability as candidate drugs for future use in ALL treatment.
Fig. 1.

Chemical Structures of the three N-alkylisatin derivatives. Derivation of the 5,7-dibromoisatin scaffold (blue) led to the development of potent antimitotic N-alkylated analogues: 5,7-dibromo-N-(p-hydroxymethylbenzyl)isatin (HMB-isatin), 5,7-dibromo-N-(1H-indol-4-ylmethyl)isatin (I4M-isatin) and 5,7-dibromo-N-(naphthalene-2-ylmethyl)isatin (N2M-isatin)
Materials and methods
Cell lines and culture conditions
The CCRF-CEM human T-cell acute lymphoblastic leukemia (T-ALL) cell line (CEM) [31] was originally supplied by P. Slowiaczek (Ludwig Institute, Sydney, Australia). The vincristine-resistant (CEM-VCR R) and 2-methoxyestradiol-resistant (CEM/2ME2-28.8R) sublines were prepared by continuous exposure to either VCR or 2ME2 respectively, as described in [13, 17]. ALL cell lines were routinely cultured in RPMI-1640 media containing 2 mM l-glutamine (Sigma-Aldrich, Sydney, Australia), supplemented with 10% (v/v) heat-inactivated fetal calf serum (FCS) (Sigma-Aldrich). The human neuroblastoma SH-SY5Y cell line (American Type Culture Collection, CRL-2266) was maintained in DMEM/F12 media (Sigma-Aldrich) supplemented with 10% (v/v) heat-inactivated FCS. All cell lines were confirmed negative for mycoplasma contamination and cultured at 37 °C in a 95% humidified air atmosphere, containing 5% CO2. Culturing of cells for experimental use did not exceed 20 passages.
Cytotoxins
The N-alkylisatins; 5,7-dibromo-N-(p-hydroxymethylbenzyl)isatin (HMB-isatin), 5,7-dibromo-N-(1H-indol-4-ylmethyl)isatin (I4M-isatin), 5,7-dibromo-N-(naphthalene-1-ylmethyl)isatin (N1M-isatin) and 5,7-dibromo-N-(naphthalene-2-ylmethyl)isatin (N2M-isatin) were synthesized by TCG Lifesciences Pty Ltd (Kolkata, India) following methodology described by our group previously [28, 29]. Colchicine, paclitaxel and vinblastine sulfate were purchased from Sigma-Aldrich (St. Louis, MO, USA). Vincristine sulfate and 2-methoxyestradiol were purchased from Abcam (Sydney, Australia) and Selleck Chem (Sydney, Australia), respectively. All compounds were solubilized in 100% dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA) and diluted in the appropriate media/buffer for downstream assays as described below.
Cell viability (MTS) assay
Cytotoxicity of drugs was determined using the CellTiter 96 Aqueous One Solution Cell Proliferation (MTS) Assay (Promega, USA) in flat-bottomed 96-well microplates. Cells were centrifuged at 300×g for 5 min at room temperature, resuspended in fresh growth media and seeded at 10,000 cells/well for CEM and CEM-VCR R cells, and 20,000 cells/well for CEM/2ME2-28.8R cells (90 μL) based on differences in cell growth properties (Additional file 1: Figure S1). Cells were then incubated for 24 h under standard culture conditions before the addition of cytotoxins. DMSO stocks of cytotoxins were serially diluted in media to give 10 × working stock (10% DMSO). This was further diluted to give a final drug concentration of cytotoxin in 1% DMSO. Following treatment, cells were further incubated at 37 °C for 48 h. At this time-point, 20 μL of MTS reagent was added to each well and incubated for 3 h at 37 °C, allowing for sufficient color change. Optical density (OD) was subsequently measured at 490 nm using the Spectrostar Nano Microplate Reader (BMG Labtech, Mornington, Australia). Cytotoxicity of drugs was calculated from dose response curves generated in GraphPad Prism v7.0 (Graphpad Software Inc., USA) and reported as cell viability (percent of DMSO control). IC50 values were calculated from sigmoidal dose response curves and defined as the concentration required to inhibit the metabolic activity of 50% of the cell population. The resistance index (RI) was calculated to evaluate the degree of acquired resistance of each cell line to the various drug treatments and was determined using the following equation: . Cellular morphological changes upon drug exposure were also tracked using the IncuCyte ZOOM (Essen Bioscience, Ann Arbor, MI, USA) and real-time quantitative live-cell analysis.
Rhodamine 123 efflux assay
The rhodamine 123 (R123) efflux assay was carried out as described in [30] with slight alterations to the protocol. Briefly, P-gp was probed on CEM-VCR R cells using the fluorescent P-gp substrate, R123, in the absence or presence of cyclosporine A (CSA), a specific P-gp inhibitor. Cells were seeded (10,000 cells/well) in 96-well microplates at a volume of 100 μL and incubated at 37 °C for 24 h. The microplate was then centrifuged at 300×g and supernatant was removed. Cells were then resuspended in media containing R123 (5 μM) in the absence or presence of CSA (20 μM) or DMSO vehicle control (final concentration 1% v/v). Cells were incubated at 37 °C for a further 20 min in the dark. Culture media was removed via centrifugation and the cells washed with ice-cold PBS. Cells were successively spun at 300×g, supernatant removed and the cell pellet lysed with 1% Triton-X (100 µL). An aliquot (80 μL) of the cell lysate was then added to wells of a 96-well microplate. Intracellular fluorescence was subsequently measured using a FLUOstar Optima Microplate Reader (BMG Labtech, Ortenburg, Germany) using an excitation wavelength of 485 nm, and emission wavelength of 530 nm.
Tubulin binding assays
The tubulin target site of the N-alkylisatins and their associated binding affinity constant (Kb) were determined by competitive assay using a fluorescent colchicine analogue 2-methoxy-5-(2,3,4-trimethoxyphenyl)-2,4,6-cycloheptatrien-1-one (MTC) that reversibly binds to the colchicine-binding site as described in [32]. Equal amounts of pure tubulin and MTC (10 µM) were mixed in a phosphate-based buffer (10 mM phosphate buffer pH 7, 1 mM EDTA, 1.5 mM MgCl2, 0.1 mM GTP), and incubated for 30 min at 25 °C with increasing concentrations up to 50 μM of the respective drug prior to the addition of 10 μM MTC. Afterwards, displacement of MTC bound to tubulin was monitored by changes in fluorescence intensity using a Varioskan® Flash plate reader (ThermoFisher Scientific) (excitation and emission wavelengths of 370 nm and 423 nm, respectively; 100 ms of exposure time with a bandwidth of 12 nm). Tubulin and MTC alone served as negative controls for fluorescence normalization. Fluorescence intensity data were converted to fractional saturation and binding constants were obtained using Equigra V5.0 as described in [33].
Neurite outgrowth assay
SH-SY5Y cells were seeded at a density of 5000 cells/well (100 µL) in 96-well plates and incubated for 24 h (37 °C, 95% humidity, 5% CO2). Media (DMEM/F12 + 10% FCS) was replaced with DMEM/F12 + 1% FCS + 10 μM retinoic acid (RA) (Sigma-Aldrich, Sydney, Australia) to induce differentiation into neuronal-like phenotype [34]. RA supplemented media (DMEM/F12 + 1% + FCS + 10 µM RA) was replaced every 48 h. Following sufficient neurite growth (approximately 96 h after initial addition of RA), various concentrations (1 nM, 10 nM and 100 nM) of vincristine, vinblastine, HMB-isatin, I4M-isatin or N2M-isatin were added to the cells and incubated for a further 48 h. Neurite outgrowth analysis was performed using the NeuroTrack analysis software on the IncuCyte ZOOM (Essen Bioscience). The algorithm masks and quantifies each image and returns neuronal cell metrics including neurite length and number of branch points per cell body [35].
Automated patch-clamp electrophysiology
SH-SY5Y cells were detached from 6 well dishes using TrypLE (ThermoFisher Scientific) and re-suspended in cold external recording solution as described in [36]. Cells were kept in suspension by automatic pipetting at 4 °C. Patch-clamp measurements were performed on an NPC-16 Patchliner (Nanion Technologies, Munich, Germany) using single and multi-hole medium resistance NPC-16 chips with an average resistance of 3.1 and 1.1 MΩ, respectively. Recordings were made in the perforated-patch configuration with internal solution consisting of (in mM): 60 CsF, 50 CsCl, 20 NaCl, 10 HEPES, 10 EGTA (285 ± 3 mOsm; pH7.2 with CsOH); or 60 KF, 50 KCl, 20 NaCl, 10 HEPES, 10 EGTA (285 ± 3 mOsm; pH7.2 with KOH) supplemented with the membrane perforating agent Escin (25 µg/mL; Sigma-Aldrich). The extracellular solution contained (in mM): 140 NaCl, 5 Glucose, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES (298 ± 3 mOsm; pH7.4 with NaOH). Seal formation was enhanced by brief treatment with SE solution (in mM): NaCl 80, KCl 3, MgCl2 10, CaCl2 35, HEPES 10 (298 ± 3 mOsm, pH = 7.3 adjusted with NaOH) until stable seals were obtained and replaced with standard external solution. Solutions were filtered through 0.2 μm membranes. Recording seal resistance was > 500 MΩ and access resistance was < 8 MΩ in the perforated patch configuration.
Chip and whole-cell capacitance were fully compensated, and series resistance compensation was applied via Auto Rs Comp function. Recordings were acquired at 50 kHz with the low-pass filter set to 10 kHz with Patchmaster (HEKA Elektronik, Lambrecht/Pfalz, Germany) and stored on a computer running PatchControlHT software (Nanion Technologies GmbH, Munich, Germany). Leak subtraction was performed automatically using a P/4 procedure following the test pulse. Offline analysis was performed using Igor Pro-6.37 (WaveMetrics Inc.), GraphPad Prism 7 (Molecular Devices) and Microsoft Excel. The pulse protocol used to evaluate voltage dependent sodium conductance consisted of a family of 25 ms test pulses from −60 to 60 mV (+10 mV increments) following a 100 ms pre-pulse to −120 mV. The holding potential (Vh) was −80 mV and stimulation was performed at 0.1 Hz. All recordings were conducted at room temperature. A 300 nM solution of tetrodotoxin, (TTX, Sigma Aldrich) was applied after IV recordings to determine the relative contribution of TTX-sensitive Nav channels.
To examine the voltage-dependent parameters of the sodium current, peak current amplitude from 25 ms test pulse potentials (100 ms pre-pulse to −120 mV, followed by step depolarization from −60 to 60 mV, ∆10 mV, Vh: −80 mV) were measured. Peak IV curves were fitted using: I(V) = (V − Vrev)*Gmax/[1 + exp(V0.5−Vslope)], where I is the macroscopic current, V is the command potential, Vrev is the reversal potential (mV), Gmax is the maximal conductance (S), V0.5 is the half-activation potential (mV), and Vslope is the slope factor (mV/e-fold).
Statistical analysis
Statistical analysis of IC50 values and neurite outgrowth assays were calculated using a two-way ANOVA followed by a Tukey’s post hoc test. A two-tailed, paired t-test was applied to compare intracellular levels of R123 between resistant and parental cells, as well as CSA-treated and non-treated cells. A two-tailed, paired t-test was also applied to compare current density between retinoic acid differentiated cells to all other experimental groups. All calculations were performed using GraphPad Prism v7.0 (Graphpad Software Inc., USA). Statistically significant results were represented by p < 0.05 (α-value of 0.05).
Results
Cell viability and efflux assays
We have previously demonstrated that 5,7-dibromo-N-alkylisatins with varied substituents at the isatin nitrogen retain potency in drug resistant human lymphoma (U937) and uterine sarcoma (MES-SA) MDR cancer cell lines in vitro [30]. In order to determine the ability of three 5,7-dibromo-N-alkylisatins (Fig. 1) to retain cytotoxicity against ALL cell lines with differing mechanisms of resistance, the cytotoxic effects of commercial antimitotic drugs: vincristine (VCR), vinblastine (VBL), 2-methoxyestradiol (2ME2), colchicine and paclitaxel and the N-alkylisatins: HMB-isatin, I4M-isatin and N2M-isatin were tested against the VCR-resistant (CEM-VCR R) and 2ME2-resistant (CEM/2ME2-28.8R) ALL cell lines, and compared to the chemosensitive parental ALL cell line (CEM) using the MTS cell proliferation assay.
CEM-VCR R cells displayed a 101.4-fold (t6= 4.869, p < 0.005) decrease in sensitivity to VCR and were significantly cross-resistant to VBL, 2ME2, colchicine and paclitaxel (Table 1). Treatment of CEM/2ME2-28.8R cells with 2ME2 revealed a 106.5-fold (t8 = 4.529 p < 0.05) decrease in drug sensitivity. Interestingly, these cells did not demonstrate cross-resistance to VCR, VBL, colchicine or paclitaxel. Notably, both CEM-VCR R and CEM/2ME2-28.8R resistant cell lines treated with HMB-isatin, I4M-isatin or N2M-isatin showed a negligible change in IC50 values (1.0–1.4-fold increase, p > 0.05) after 48 h compared to their respective parental cells (Fig. 2). A Two-Way Analysis of Variance (2-way ANOVA) found no significant difference in the viability of CEM, CEM-VCR R or CEM/2ME2-28.8R cells treated with increasing concentrations of the N-alkylisatins, irrespective of differences in their molecular weight, lipophilicity or N1 substitution patterns.
Table 1.
IC50 (µM) values and fold decrease in drug sensitivity (RI) of parental (CEM) versus resistant (CEM-VCR R and CEM/2ME2 28.8R) human acute lymphoblastic leukemia cell lines treated with microtubule targeting agents
| IC50 (µM) ± SDa | Resistance Index (RI)b | ||||
|---|---|---|---|---|---|
| Compound | CEM | CEM-VCR R | CEM/2ME2-28.8R | CEM-VCR R:CEM | CEM/2ME2-28.8 R:CEM |
| Vincristine | 0.08 (± 0.05) | 8.11 (± 1.62) | 0.41 (± 0.19) | 101.4 < 0.05 | 5.1 > 0.05 |
| Vinblastine | 0.13 (± 0.06) | 1.12 (± 0.16) | 0.39 (± 0.13) | 8.6 < 0.05 | 3.0 > 0.05 |
| 2-Methoxy-estradiol | 0.08 (± 0.04) | 4.07 (± 1.34) | 8.52 (± 1.38) | 50.9 < 0.05 | 106.5 < 0.05 |
| Colchicine | 0.32 (± 0.21) | 1.01(± 0.36) | 0.56 (± 0.19) | 3.2 < 0.05 | 1.8 > 0.05 |
| Paclitaxel | 0.15 (± 0.09) | 1.11 (± 0.36) | 0.25 (± 0.12) | 7.4 < 0.05 | 1.7 > 0.05 |
| HMB-isatin | 1.02 (± 0.32) | 1.07 (± 0.37) | 1.29 (± 0.41) | 1.0 > 0.05 | 1.3 > 0.05 |
| I4M-isatin | 1.07 (± 0.44) | 1.26 (± 0.19) | 1.16 (± 0.46) | 1.2 > 0.05 | 1.1> 0.05 |
| N2M-isatin | 0.36 (± 0.19) | 0.49 (± 0.36) | 0.46 (± 0.32) | 1.4 > 0.05 | 1.3 > 0.05 |
aValues are the mean of three independent experiments, performed in triplicate (n = 9) ± SD
bThe RI was calculated to determine the degree of acquired resistance of each cell line to the various drug treatments and was determined using the following equation:. P-values < 0.05 were considered significant
Fig. 2.

Dose-response curves for parental (CEM), vincristine-resistant (CEM-VCR R) and 2ME2-resistant (CEM/2ME2-28.8R) ALL cell lines treated with varying concentrations of commercial drugs (a–e) or N-alkylisatins (f–h). CEM (black), CEM-VCR R (solid) and CEM-2ME2/28.8R (dashed) cells were incubated with increasing concentrations of drug at 37 °C for 48 h. Cells were then assessed for metabolic activity by addition of MTS reagent and the OD measured at 490 nm. Dose–response curves were generated using GraphPad Prism v7.0. Values are the mean of triplicates (± SD)
Overexpression of the P-gp efflux transporter has previously been reported as one of the primary mechanisms of resistance in the CEM-VCR R cell line [13, 37, 38]. In order to confirm that P-gp was functional, we monitored the efflux of a fluorescent Rhodamine 123 (R123), a known P-gp substrate. Intracellular fluorescence was significantly increased by 3.5-fold in CEM cells compared to CEM-VCR R cells (t4 = 30.97, p < 0.05) (Fig. 3a). To further confirm that R123 extrusion from CEM-VCR R cells was the result of P-gp mediated efflux, cells were incubated with R123 in the absence or presence of cyclosporin A (CSA), a specific P-gp inhibitor. Here, CEM-VCR R cells incubated with R123 showed a twofold increase in intracellular fluorescence in the presence of CSA (t4 = 11.08, p < 0.05) (Fig. 3b).
Fig. 3.

Levels of intracellular rhodamine 123 (R123) in CEM and CEM-VCR R cell lines in the presence and absence of cyclosporine A (CSA). a Parental (CEM) and VCR-resistant cells (CEM-VCR R) were incubated with R123 for 20 min before being washed with PBS and lysed with 1% Triton-X. b CEM-VCR cells were incubated with R123 in the presence or absence of 20 µM Cyclosporin A (CSA), a specific P-gp inhibitor, for 20 min before undergoing subsequent washing and lysis. Intracellular fluorescence was subsequently measured using a FLUOstar Optima Microplate Reader using an excitation wavelength of 485 nm, and emission wavelength of 530 nm. Mean ± SD, n = 3. ***P < 0.001, ****P < 0.0001
Competition binding assays
Pioneering work by our group has revealed that 5,7-dibromo-N-alkylisatins inhibit tubulin polymerization, resulting in the accumulation of cells with atypical cell morphologies, fragmented microtubules and abnormal mitotic spindles [28, 29]. To obtain further insight into the molecular interaction of our N-alkylisatins with tubulin, and because the colchicine site is a major binding pocket for many indole, quinolone and thiophene-based microtubule destabilizers [23], we investigated the ability of HMB-isatin, I4M-isatin and N1M-isatin (where the naphthalene is instead attached via the C1 position instead of the C2 position) to bind to this site using a reversible fluorescent colchicine analogue MTC [32]. All compounds were shown to compete with similar affinities (within the experimental error) to the colchicine analog, displacing it from its binding site and reducing its fractional saturation, consistent with binding to the colchicine site (Fig. 4 and Table 2).
Fig. 4.
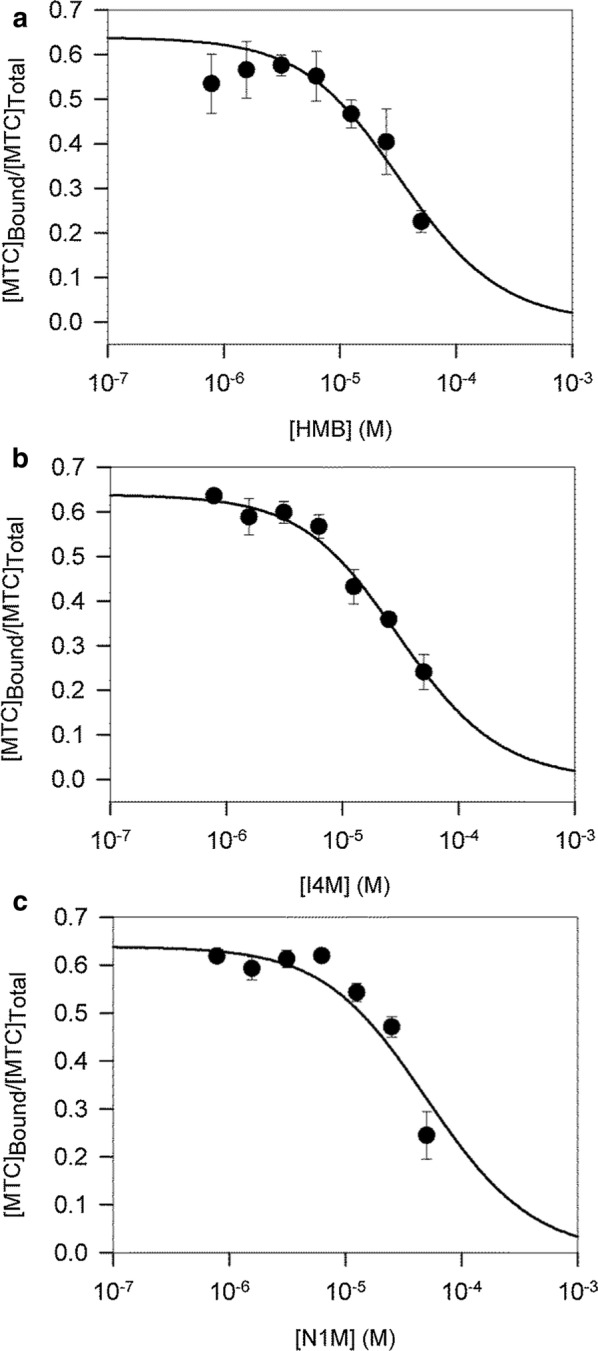
Displacement of MTC from the colchicine-binding site by a HMB-isatin, b I4M-isatin and c N1M-isatin. MTC fractional saturation values were calculated based on the concentration of MTC binding sites (0.8 per tubulin dimer) and the MTC binding constant (4.7 × 105 M−1). Solid line is the best fit to a single site competition model as calculated by EQUIGRA5. Values are the mean of triplicates ± SEM
Table 2.
Binding affinity values for HMB-isatin, I4M-isatin and N1M-isatin compared with reference compounds of the colchicine site
| Compound | Binding affinity (M−1) ± SEMa |
|---|---|
| Colchicine | 1.16 × 107 (37 °C) [70] |
| Podophyllotoxin | 1.8 × 106 [71] |
| MTC | 4.7 × 105 [72] |
| HMB-isatin | 2.7 ± 1.1 × 105 |
| I4M-isatin | 3.4 ± 1.5 × 105 |
| N1M-isatin | 1.4 ± 0.2 × 105(× 104) |
aValues (Kb) are the mean of three independent experiments performed in triplicate (n = 9) ± SEM
Neurite outgrowth assay
Peripheral neuropathy is the principal toxicity associated with the use of the Vinca alkaloids, especially in younger children with ALL [39]. Immortalized cell lines derived from CNS tissue are powerful tools for elucidating the mechanism of action of neurotoxic chemicals [40] as they recapitulate key aspects of neuronal function and overcome some of the limitations associated with the use of ex vivo and in vivo models such as heterogeneity (genetic variability) which obscures interpretation of mechanistic data. To compare the potential neurotoxic effect of our N-alkylisatins to VCR and VBL we performed neurite outgrowth assays using retinoic acid (RA) differentiated SH-SY5Y cells and measured alterations in neurite length and number of branch points (Figs. 5, 6 and Additional file 1: Figure S3). RA differentiated SH-SY5Y cells were treated with sub-IC50 concentrations of the Vinca alkaloids or N-alkylisatins and compared to vehicle control treated cells. No significant differences in neurite length were observed between any of the drugs and the control group at 1 nM (p > 0.05) (Fig. 5a). Cells treated with VCR and VBL at 10 nM and 100 nM however, showed a significant (p < 0.05) reduction in neurite length (56% and 45% for 100 nM VCR and VBL, respectively) compared to those vehicle treated. Conversely, treatment with HMB-isatin, I4M-isatin and N2M-isatin at 10 nM demonstrated 7% –15% reduction in neurite length and thus appeared significantly less neurotoxic than VCR and VBL (p < 0.05) at the same concentration. Similarly, SH-SY5Y cells treated with 100 nM HMB-isatin, I4M-isatin or N2M-isatin displayed 25%–35% reduction in neurite length, compared to the no drug control, but significantly less than VCR and VBL (P < 0.05).
Fig. 5.
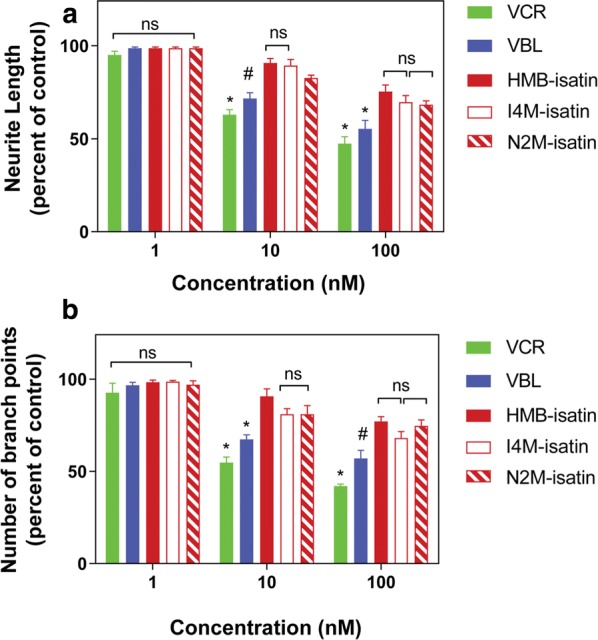
Effect of N-alkylisatins and Vinca alkaloids on SH-SY5Y neurite outgrowth and number of neurite branch points. SH-SY5Y neuroblastoma cells were incubated with 10 μM retinoic acid for 96 h to induce differentiation of cells into a mature dopaminergic-like neurotransmitter phenotype. Cells were then treated with increasing concentrations (1–100 nM) of N-alkylisatin (HMB-isatin, I4M-isatin or N2M-isatin) or Vinca alkaloid (vincristine; VCR or vinblastine; VBL) for 48 h. Changes in a neurite length or b number of neurite branch points were analyzed using the IncuCyte ZOOM, and measured using NeuroTrack analysis software. Mean ± SD, n = 3. NS = not significant, *P < 0.001,#P < 0.01
Fig. 6.
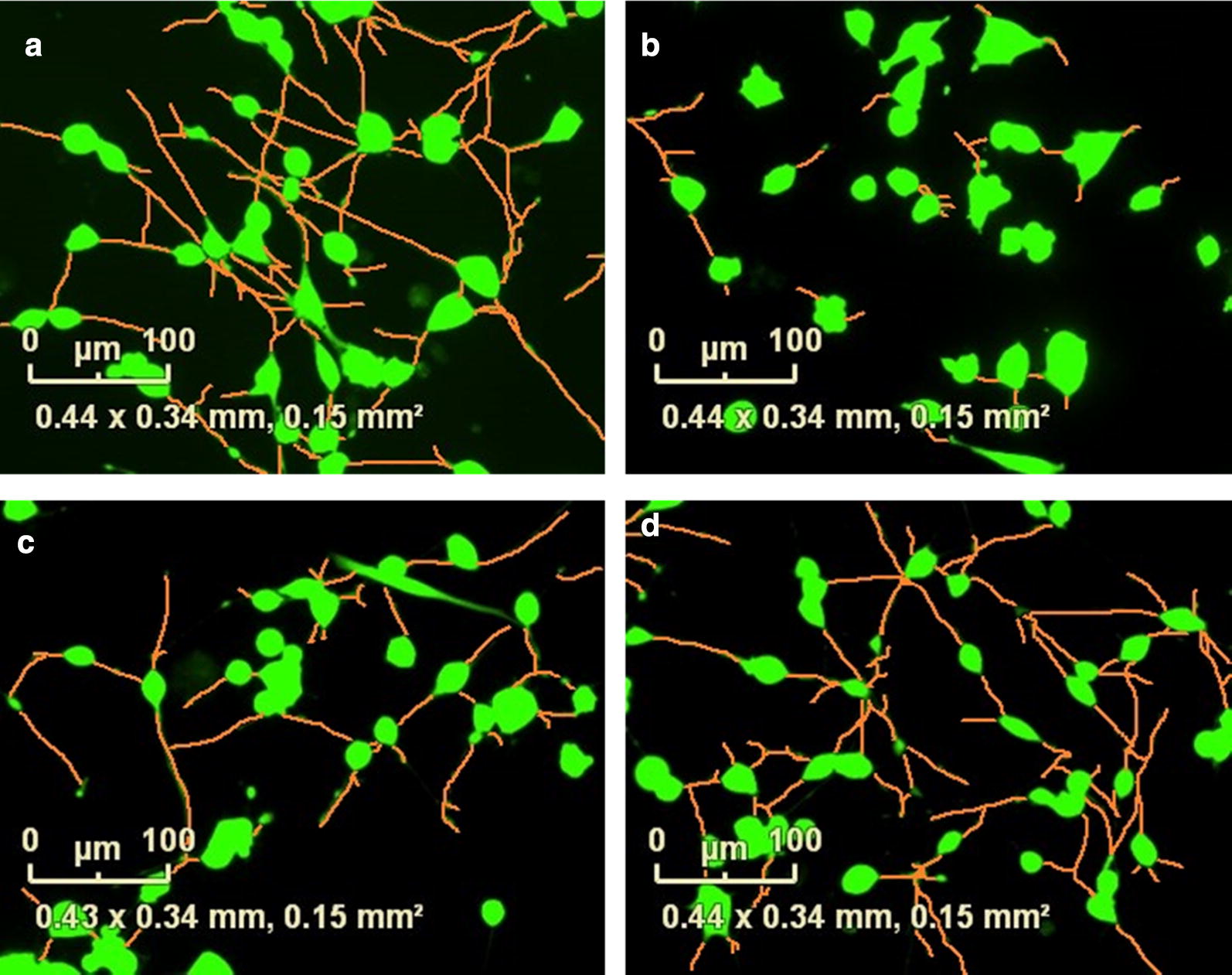
Vinca alkaloids significantly alter the neurite morphology of retinoic acid differentiated SH-SY5Y cells. Automated segmentation of neurites (orange) and calcein AM stained cell body clusters (green) of retinoic acid-differentiated SH-SY5Y cells after overnight incubation with a 1% DMSO vehicle control, or 10 nM b vincristine, c vinblastine or d HMB-isatin. Images we obtained using Neurotrack and the IncuCyte® live-cell analysis system
To further interrogate neurotoxicity, numbers of neurite branch points were examined following drug treatments (Fig. 5b). At 1 nM, all drugs exhibited minimal effect on the number of neurite branch points and were not shown to be significantly different compared to no drug controls (p > 0.05). VCR and VBL treatment at 10 nM caused a significant (p < 0.05) reduction in number of neurite branch points (46% and 33%, respectively) compared to vehicle treated control. At the same concentration HMB-isatin, I4M-isatin and N2M-isatin reduced the number of neurite branch points by only 8%–17% and were significantly different to VCR and VBL (p < 0.05). Treatment with VCR and VBL at 100 nM revealed a significant (p < 0.05) reduction in neurite branch points (55% and 45%, respectively) compared to no drug treated control. At the same concentration, HMB-isatin, I4M-isatin and N2M-isatin reduced branch points by only 25%–32% and, significantly less than VCR and VBL (p < 0.05). Upon completion of the assay, cell viability was immediately assessed by MTS assay. No significant differences in cell viability were identified across all drugs and concentrations tested (Additional file 1: Figure S2). Overall, RA differentiated SH-SY5Y cells were less affected after treatment with the N-alkylisatins compared to VCR and VBL at equivalent concentrations (p < 0.05), indicating a potential for lowered neurotoxicity.
Functional assessment by automated patch-clamp electrophysiology
Neuronal morphology with neuritic-like processes in RA differentiated SH-SY5Y cells is associated with functional features that include transmembrane currents underlying a neuronal excitability phenotype [41–43]. Such cells lines can therefore serve as a tool in neurofunctional assays. Here, we compared the effects of overnight incubation with HMB-isatin, VCR and VBL on the electrophysiological properties of non-treated RA differentiated SH-SY5Y cells and naïve SH-SY5Y (non-RA differentiated) cells. Cells from all treatment groups were studied by automated patch clamp in the perforated patch configuration using standard solutions. In all cell groups we observed rapidly activating and inactivating inward currents followed by modest delayed outward currents in response to step depolarization positive to –20 mV (Fig. 7a). The rapid inward current was characterized by: reversal potential > 48 mV, activation threshold of ~ −20 mV and maximum amplitude between −20 and 0 mV (Additional file 1: Table S1). This current could be completely inhibited by 300 nM tetrodotoxin (TTX, not shown) and thus these results are consistent with TTX-sensitive voltage gated sodium channels (VGSC) mediating the observed inward currents. The slow rising outward current, likely due to activation of delayed rectifier potassium channels, was inconsistent and thus was not further analyzed.
Fig. 7.

Cytotoxic drugs differentially affect the electrophysiological properties of SH-SY5Y RA differentiated cells. a Top traces depict whole cell voltage clamp recordings of endogenous currents from representative naïve SH-SY5Y cells (Ctr, purple), retinoic acid differentiated (RA, black), and RA-differentiated cells treated with 5 nM HMB-isatin (red), 5 nM vinblastine (VBL, blue) and 5 nM vincristine (VCR, green). Currents were elicited by 25 ms step depolarizations to 0 mV from a Vh: -80 mV preceded by a 100 ms –120 mV pre-pulse (inset under Ctr). Scale bar: 1 nA. The insets below RA, HMB-isatin and VBL show voltage traces in response to current injection (50–100 pA, ∆10 pA, inset under VCR) that elicited action potentials from their respective Vh. Scale bars are: 10 mV and 100 ms. b and c Voltage dependent parameters of inward sodium currents in all groups obtained from IV stimulation protocols from −60 to 60 mV (∆10 mV, Vh: −80 mV). IVs from representative cells are shown in the inset on the right. b Slope and activation half voltage. c Right: maximal conductance (Gmax) and inward current density estimated from the ratio of peak current amplitude observed at 0 mV to the cell membrane capacitance (pA/pF). Ctr: non differentiated; RA: Retinoic acid differentiated; HMB-isatin: 5,7-dibromo-N-(p-hydroxymethylbenzyl)isatin treated RA cells; VBL: vinblastine treated RA cells; VCR: vincristine treated RA cells (drugs were applied overnight at 10 nM). Mean ± SEM from n = 5–24 cells per condition. Unpaired t-test compares RA against all other groups with a 95% confidence interval. Data are presented as means ± SEMs (n represents the number of individual recordings). A two-tailed, paired t-test was applied to compare current density between retinoic acid differentiated cells to the other experimental groups. Data presented is compiled from 3 independent experiments involving cell differentiation and drug treatment
Naïve SH-SY5Y (Ctr) cells displayed relatively small inward currents when compared to RA differentiated cells (Fig. 7a). Treatment with RA resulted in robust inward sodium currents capable of supporting action potential firing consistent with neuronal-like functionality (Fig. 7a, inset). Current–voltage (IV) relationships were determined from peak currents elicited by 25 ms step depolarizations from −60 to 60 mV, in 10 mV steps, from a holding potential of −80 mV. A 100 ms pre-pulse to −120 mV was implemented to aid VGSC recovery from inactivation. IV relationships from representative cells from each experimental group are shown in the inset in Fig. 7c.
We compared the effects of the three cytotoxins in the electrophysiological properties of RA differentiated SH-SY5Y cells. Overnight incubation with HMB-isatin, VBL and VCR caused graded effects in the electrophysiological properties of RA differentiated SH-SY5Y cells. The voltage dependent parameters of the observed sodium current were not significantly different between RA and the HMB-Isatin and VCR groups (p = 0.7130 and 0.4758, respectively). When compared to the RA group (V0.5: −18.6 ± 0.8 mV), only small depolarizing shifts on the activation midpoint (V0.5) were observed in VBL (V0.5: −13.3 ± 3.2 mV) treated, and naïve (V0.5: −13.8 ± 1.4 mV) cells. The latter also displaying an apparent increase in the slope of the activation curve (Fig. 7 and Additional file 1: Table S1).
The magnitude of the VGSC differed between the experimental groups. This was evidenced by comparing the macroscopic conductance (from peak IV plots) and the current density (measured from the ratio of peak current amplitude observed at 0 mV to the cell membrane capacitance in pA/pF) from each group. RA-differentiated SH-SY5Y exposed to HMB-isatin displayed modestly reduced conductance (Gmax: 55.26 ± 9.3 nS, p = 0.5562) and current density (92.2 ± 6.7 pA/pF, p = 0.0885) in comparison to vehicle treated control cells (61.30 ± 5.4 nS, 111.7 ± 8.0 pA/pF, respectively), however these changes did not reach statistical significance (Fig. 6c, Additional file 1: Table S2). In contrast, the macroscopic conductance of VBL (30.81 ± 8.8 nS) and VCR (25.75 ± 5.1 nS) treated groups, as well as their current density (VBL: 65.1 ± 7.3 pA/pF, and VCR: 58.3 ± 8.2 pA/pF) were significantly reduced and comparable to those of naïve non-RA differentiated SH-SY5Y cells (26.99 ± 1.3nS and 61.8 ± 7.9 pA/pF) (Fig. 7c, Additional file 1: Table S2). A decrease in sodium channel function is invariably correlated with decreased excitability therefore; the results presented here may be related to the level of neurotoxicity of the antimitotic drugs studied.
Discussion
ALL is the most common childhood malignancy and although high remission rates have been achieved, relapse occurs in ~ 20% of patients. This is predominantly due to the development of MDR to chemotherapy [44]. Despite substantial advancements in ALL treatment, the use of Vinca alkaloids, particularly VCR, remains the gold standard of care. However, resistance to these drugs as well as their high potential to cause chemotherapy-induced peripheral neuropathy (CIPN) highlights the need to further develop strategies to treat or prevent ALL relapse. The 5,7-dibromo-N-alkylisatins are a diverse and growing class of antimitotic agents that have demonstrated the ability to retain potency against clinically resistant cancer cell lines in vitro and reduce primary tumour growth in vivo [28–30]. The current study expands on our understanding of this ability and shows three N-alkylisatins, namely HMB-isatin, I4M-isatin and N2M-isatin, to retain potency against drug resistant ALL cell lines. These N-alkylisatins also demonstrate a potential for reduced neurotoxicity compared to current clinically used Vinca alkaloids, VCR and VBL, suggesting that with further development, the N-alkylisatins could be used in the treatment of ALL with reduced potential for the onset of CIPN.
Our in vitro cytotoxicity data demonstrate that while the CEM-VCR R cell line displayed cross-resistance to an array of drugs, the CEM/2ME2-28.8R line was resistant to 2ME2 only. This confirms previous reports that the mechanisms that drive resistance in these two lines differ. 2ME2 is not considered a substrate for P-gp [17, 45] and consistent with this, the 2ME2-selected cell line did not exhibit cross-resistance to agents that are known P-gp substrates including colchicine, paclitaxel, VBL and VCR [46]. Interestingly, the CEM/2ME2-28.8R cell line contains four class I β-tubulin point mutations, S25N, D197N, A248T and K350N [17]. Of these, the S25N mutation lies within the paclitaxel-binding site, the D197N mutation is not within any known drug-binding site on β-tubulin, while A248T and K350N are located within the colchicine-binding site on β-tubulin. We proposed that this line could serve as a tool to further elucidate N-alkylisatin binding to β-tubulin, as any change in the tertiary binding site and/or its immediate environment may subsequently affect drug activity. In this study, cytotoxicity assays revealed that all three N-alkylisatins retained potency against the CEM/2ME2-28.8R cell line with IC50 values not significantly different to that of the CEM parental control cell line. Our competition binding experiments clearly indicate that N-alkylisatins compete for binding with a colchicine analog, strongly supporting their binding to the colchicine site. Therefore, the fact that the compounds retain potency against the 2ME2 (known colchicine site ligand [47]) resistant cell line indicates that residues β248 and β350 are not involved in N-alkylisatin binding to the colchicine site. Altogether these data are congruent with the observation of Liaw et al. [17] and indicate that, despite the presence of two point-mutations in the colchicine binding site, CEM/2ME2 R cells are not cross-resistant to colchicine or colchicine binding-site agents.
Multiple factors are thought to contribute to the drug resistant phenotype of the CEM-VCR R line [18, 48]. Point mutations in β-tubulin, altered tubulin polymer levels and overexpression of drug efflux-transporters have all been reported to play a role. In order to confirm P-gp-mediated resistance in this line, CEM-VCR R cells were incubated with the fluorescent P-gp substrate R123 in the absence and presence of CSA. CSA is commonly used for in vitro P-gp inhibition studies due to its specificity for P-gp [26]. In this study, treatment of VCR-resistant cells with CSA inhibited R123 extrusion by 49%. We concluded that P-gp mediated efflux is a major contributor to R123 extrusion from these cells, however additional resistance mechanisms such as the overexpression of multidrug resistance-associated proteins (MRPs) and/or lung resistance protein (LRP) may also be employed. MRPs are reported to perform similar functions to P-gp, extruding a range of toxins from cells, including Vinca alkaloids [49], however, unlike P-gp, MRP1 does not confer high levels of resistance to paclitaxel [50, 51]. LRP is a component of a protein complex located in the cytoplasm and at the nuclear pore. Given that nuclear pore complexes are able to mediate bidirectional transport of substances between the cytoplasm and nucleus, it is hypothesized that LRP may alter drug transport between these two cellular compartments and sequester drugs in the cytoplasm [52]. Clinical studies investigating the expression of these proteins in pediatric ALL patients show that MRP1 and LRP mRNA is observed in 35.3% and 41.2% of patients, respectively [53]. Moreover, high protein expression levels of MRPs have been linked to unfavorable clinical outcomes in pediatric patients [54], as well as predictive markers of relapse in adult patients [55]. When the MDR CEM-VCR R cell line was exposed to HMB-isatin, I4M-isatin or N2M-isatin, IC50 values were not statistically different to those observed for the parental control CEM cell line. This was in contrast to the dose response profiles observed for the commercial anti-cancer agents and further confirms our findings [30] that this class of molecule is not susceptible to P-gp, and potentially MRP and LRP, mediated transport. The exact mechanism by which the N-alkylisatins overcome MDR is yet to be uncovered, but we posit that polypharmacology could play a role [56].
Antimitotic agents work by causing dynamic instability of microtubules and ultimately induce tumour cell death. They also interfere with the function of microtubules in axons which mediate neuronal vesicle transport [57–59]. For this reason microtubule inhibitors often exhibit unwanted side effects including peripheral neuropathies. Measuring changes in neurite outgrowth during differentiation is one of the most widely performed phenotypic assays to assess CIPN since the ability of neuronal cells to project membrane extensions from their cell bodies is closely linked to their network function and cell health [60, 61]. We investigated the potential of our N-alkylisatins to induce neurotoxicity by comparing their effect on SH-SY5Y neurite length and branch points to VCR and VBL, cytotoxins which are known to cause significant neuronal damage [62]. Differentiated human neuroblastoma cells were treated with drug concentrations of 1, 10 and 100 nM. Previous in vitro tests have shown that treatment with VCR at these concentrations cause minor, moderate, and major neuropathic effects. In this study, the Vinca alkaloids and N-alkylisatins displayed a dose-dependent effect towards the SH-SY5Y cells, however neurotoxicity associated with VCR and VBL treatment was significantly more pronounced. N-alkylisatins induced less damage to neurites than VCR and VBL, even at 10 × higher concentrations than the Vinca alkaloids. It is important to note that while the potency of VCR and VBL is between 2.7–13-fold greater than the N-alkylisatins against the chemosensitive CEM cell line, the N-alkylisatins were up to 17 × more potent than VCR and 2.3 × more potent than VBL in the CEM-VCR R resistant line, making the concentrations used in the neurotoxicity experiments highly relevant. Of the three N-alkylisatins tested, HMB-isatin was the least neurotoxic, and was therefore chosen for further assessment in functional patch-clamp assays.
For neurofunctional experiments, we used differentiated SH-SY5Y cells as our model of neurons as they exhibit voltage-gated sodium channels essential to action potential generation and are therefore crucial for neuronal activity and cell survival. The cytotoxic drugs studied here affected sodium current magnitude with varied potency. Macroscopic conductance and current density are influenced by channel expression at the plasma membrane. Our patch-clamp results revealed that the average conductance and current density of HMB-isatin treated cells were not significantly different to those of RA differentiated control cells, which suggests that aspects of channel trafficking and expression in these two groups are akin. In contrast, the sodium currents of VBL and VCR treated cells were significantly smaller and similar in magnitude to the naïve group.
In our electrophysiological experiments, we observed complete inhibition of SH-SY5Y VGSC currents by 300 nM TTX confirming functional expression of only TTX-sensitive sodium channel isoforms [63]. A detailed molecular identification of the involved channels is beyond the scope of this work. Nevertheless, when applied directly (5 nM), none of the cytotoxic drugs produced acute modulatory effects on VGSC mediated activity in RA differentiated cells (not shown). Therefore, these drugs do not appear to act directly on the effector sodium channels. Changes in sodium channel expression in RA-differentiated SH-SY5Y cells can alter sodium current density, leading to downstream changes in action potential generation. Current clamp experiments to assess excitability of our SH-SY5Y neuronal model qualitatively reflected the differences in sodium channel current magnitude observed (Fig. 7a insets). There, it can be observed that the strength of the stimuli required to elicit APs is inversely proportional to the magnitude of the sodium current. Those groups with larger Nav currents, RA and HMB-isatin, fire action potentials upon current injection. VBL treated cells, with somehow lower Nav current, also fired APs but only upon stronger stimuli (i.e. higher current injection), whereas VCR and naïve cells failed to fire under our experimental conditions. Spontaneous firings or trains of spikes in response to prolonged stimulation were not observed in any condition precluding rigorous quantification of the resulting excitability profiles. Perhaps the absence of (repetitive) firing is indicative of incomplete neuronal maturation and/or compromised cell fitness to support firing. Thus, our functional assessment points towards an inherent detrimental role of these drugs in differentiation and/or function of neuron-like SH-SY5Y cells.
Altogether, morphological and functional indicators from this study suggest that cytotoxic drug treatment affects differentiation towards the neuronal phenotype, as tubulin disrupting drugs, HMB-isatin, VBL and VCR possibly alter channel trafficking and/or expression. Other cytotoxic drugs like platinum compounds, contribute to the development of CIPN by impairing the electrophysiological function of dorsal root ganglion neurons demonstrated by a reduction and/or loss of the sensory action potential in nerve conduction studies [64]. The reduction in sodium conductance observed in our in vitro experiments is consistent with reports of reduced excitability in peripheral motor and sensory neurons in VCR treated patients [65]. Increasing evidence supports the association between pain and ion channel modulation of neuronal activity in the peripheral nervous system. Voltage-gated sodium channels are plasma membrane proteins responsible for the upstroke phase of the action potential in electrically excitable cells. VGSC function in macromolecular signalling complexes of a pore-forming α-subunit and associated β-subunits. The β-subunits are members of the immunoglobulin (Ig) domain family of cell-adhesion molecules. They modulate multiple aspects of Nav channel behaviour and play critical roles in controlling neuronal excitability [66]. Defective association between α- and β-subunits has been shown to hinder neurite outgrowth in vitro and in vivo [67]. Furthermore, various studies suggest that clustering of VGSC is regulated by electrical activity and requires an intact actin cytoskeleton [68, 69]. Our findings provide evidence of modulation of sodium conductance whereby its dysfunction may likely contribute to neuropathy and/or neuronal death.
RA treatment significantly increased the density of the voltage-activated Na+ channels. The appearance of large inward TTX-sensitive Nav currents is an electrophysiological marker of neuronal-functionality. As a consequence, RA cells became excitable and thus fired action potentials in response to current injection (Fig. 7a, insets). We treated differentiated SH-SY5Y cells with cytotoxic drugs and upon exploration of the electrophysiological properties of their endogenous currents observed evidence supporting differential cytotoxic effect from the drugs studied. Altogether, these data highlight the usefulness of this model to evaluate the functional impact of anticancer drugs on neuronal morphology and cellular excitability using an automated patch clamp platform. In vitro cell based models continue to be indispensable tools in assessing CIPN, especially when screening large numbers of chemical compounds. Although beyond the scope of this manuscript, we acknowledge that further exploration of the mechanisms behind the phenomena are necessary. For example, studies could be conducted in rodent models where multiple behavioural, electrophysiological and pathologic measures can be utilised to evaluate neuropathy and gain further insight into the potential neurotoxicity of N-alkylisatins within a whole living organism [27].
Conclusions
The development of N-alkylisatin-based compounds with increased anti-cancer activity is ongoing and advancements in the field could lead to novel molecules that possess superior efficacy towards MDR cancers with lessened neurotoxicity profiles. A current opinion on how to best treat ALL relapse is to prevent it, which makes incorporation of these novel drugs into frontline therapy the most practical approach. Combining newer agents with conventional treatment approaches will likely overcome the challenge of balancing efficacy with toxicity and prevent the development of resistant ALL subclones. Altogether, the results obtained from this study reinforce the potential of N-alkylisatins to be considered as candidate drugs for the treatment of patients with relapsed and refractory ALL.
Supplementary information
Additional file 1: Figure S1. Growth properties of CEM, CEM-VCR R and CEM/2ME2-28.8R cell lines. Figure S2. SH-SY5Y cell viability after treatment with HMB-isatin, VCR and VBL. Figure S3. Dose dependent effect of HMB-isatin and VCR on RA-differentiated SH-SY5Y neurite morphology. Table S1. Electrophysiological parameters of endogenous sodium currents. Table S2. Current density estimates from 3 independent experiments.
Acknowledgements
This research has been conducted with additional financial support from Molecular Horizons, University of Wollongong and the Illawarra Health and Medical Research Institute, Wollongong, Australia to K.L. Vine and R.K. Finol-Urdaneta.We thank Ganadería Fernando Díaz for supplying calf brains.
Abbreviations
- 2ME2
2-Methoxyestradiol
- ABC
ATP-binding cassette
- ALL
Acute lymphoblastic leukemia
- BCRP
Breast cancer resistance protein
- CEM/2ME2-28.8R
2-Methoxyestradiol-resistant CEM cell line
- CEM
T-cell acute lymphoblastic leukemia cell line
- CEM-VCR R
Vincristine-resistant CEM cell line
- CIPN
Chemotherapy-induced peripheral neuropathy
- CSA
Cyclosporine A
- DMSO
Dimethyl sulfoxide
- HMB-isatin
5,7-dibromo-N-(p-hydroxymethylbenzyl)isatin
- I4M-isatin
5,7-dibromo-N-(1H-indol-4-ylmethyl)isatin
- LRP
Lung-resistance protein
- MDR
Multi-drug resistance
- MES-SA
Uterine sarcoma cell line
- MRP
Multidrug resistance-associated protein
- MRP1
Multi-drug resistance-associated protein 1
- MTC
2-Methoxy-5-(2,3,4-trimethoxyphenyl)-2,4,5-cycloheptatrien-1-one
- N2M-isatin
5,7-dibromo-N-(naphthalene-2-ylmethyl)isatin
- OD
Optical density
- P-gp
P-glycoprotein
- R123
Rhodamine 123
- RA
Retinoic acid
- RI
Resistance index
- SH-SY5Y
Human neuroblastoma cell line
- T-ALL
T-cell acute lymphoblastic leukemia
- TTX
Tetrodotoxin
- U937
Monocyte-like histiocytic lymphoma cell line
- VBL
Vinblastine
- VCR
vincristine
- VGSC
Voltage gated sodium channels
- Vh
Holding potential
- VIPN
Vincristine-induced peripheral neuropathy
Authors’ contributions
BK conducted cytotoxicity, R123 efflux and neurotoxicity assays, analyzed data and wrote the manuscript. RKFU conducted electrophysiology experiments, analyzed data and wrote the manuscript. AH conducted cytotoxicity assays, analyzed data and wrote the manuscript. JBB assisted with the interpretation of data and edited the manuscript. DLA, MAO and JFD designed, optimised and conducted the competition binding experiments, analyzed data and wrote the manuscript. MK provided the T-cell acute lymphoblastic leukemia cell lines and edited the manuscript. KLV conceived and designed experiments, analyzed and interpreted data, wrote and edited the manuscript. All authors read and approved the final manuscript.
Funding
European Union H2020-MSCA-ITN-ETN/0582 ITN TubInTrain to J.F. Diaz is gratefully acknowledged.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Competing interests
The authors declare there are no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12935-020-01251-6.
References
- 1.Katz AJ, et al. Acute lymphoblastic leukemia: an assessment of international incidence, survival, and disease burden. Cancer Causes Control. 2015;26(11):1627–1642. doi: 10.1007/s10552-015-0657-6. [DOI] [PubMed] [Google Scholar]
- 2.Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373(16):1541–1552. doi: 10.1056/NEJMra1400972. [DOI] [PubMed] [Google Scholar]
- 3.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 4.Stanulla M, Schrappe M. Treatment of childhood acute lymphoblastic leukemia. Semin Hematol. 2009;46(1):52–63. doi: 10.1053/j.seminhematol.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Kantarjian HM, et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18(3):547–561. doi: 10.1200/JCO.2000.18.3.547. [DOI] [PubMed] [Google Scholar]
- 6.Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood cancer J. 2017;7(6):e577–e577. doi: 10.1038/bcj.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper SL, Brown PA. Treatment of pediatric acute lymphoblastic leukemia. Pediatr Clin North Am. 2015;62(1):61–73. doi: 10.1016/j.pcl.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Den Boer ML, et al. Patient stratification based on prednisolone-vincristine-asparaginase resistance profiles in children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21(17):3262–3268. doi: 10.1200/JCO.2003.11.031. [DOI] [PubMed] [Google Scholar]
- 9.Ong V, et al. A role for altered microtubule polymer levels in vincristine resistance of childhood acute lymphoblastic leukemia xenografts. J Pharmacol Exp Ther. 2008;324(2):434. doi: 10.1124/jpet.107.128926. [DOI] [PubMed] [Google Scholar]
- 10.Vainionpaa L, et al. Vincristine therapy for children with acute lymphoblastic leukemia impairs conduction in the entire peripheral nerve. Pediatr Neurol. 1995;13(4):314–318. doi: 10.1016/0887-8994(95)00191-3. [DOI] [PubMed] [Google Scholar]
- 11.Ramchandren S, et al. Peripheral neuropathy in survivors of childhood acute lymphoblastic leukemia. J Peripher Nerv Syst. 2009;14(3):184–189. doi: 10.1111/j.1529-8027.2009.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenthal S, Kaufman S. Vincristine neurotoxicity. Ann Intern Med. 1974;80(6):733–737. doi: 10.7326/0003-4819-80-6-733. [DOI] [PubMed] [Google Scholar]
- 13.Haber M, et al. Atypical multidrug resistance in a therapy-induced drug-resistant human leukemia cell line (LALW-2): resistance to vinca alkaloids independent of P-glycoprotein. Cancer Res. 1989;49(19):5281–5287. [PubMed] [Google Scholar]
- 14.Tamai I, Safa AR. Competitive interaction of cyclosporins with the Vinca alkaloid-binding site of P-glycoprotein in multidrug-resistant cells. J Biol Chem. 1990;265(27):16509–16513. [PubMed] [Google Scholar]
- 15.Krause W. Resistance to anti-tubulin agents: from vinca alkaloids to epothilones. Cancer Drug Resist. 2019;2:82–106. doi: 10.20517/cdr.2019.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verrills NM, Kavallaris M. Improving the targeting of tubulin-binding agents: lessons from drug resistance studies. Curr Pharm Des. 2005;11(13):1719–1733. doi: 10.2174/1381612053764706. [DOI] [PubMed] [Google Scholar]
- 17.Liaw TY, et al. Class I beta-tubulin mutations in 2-methoxyestradiol-resistant acute lymphoblastic leukemia cells: implications for drug-target interactions. Mol Cancer Ther. 2008;7(10):3150–3159. doi: 10.1158/1535-7163.MCT-08-0442. [DOI] [PubMed] [Google Scholar]
- 18.Kavallaris M, et al. Multiple microtubule alterations are associated with vinca alkaloid resistance in human leukemia cells. Cancer Res. 2001;61(15):5803–5809. [PubMed] [Google Scholar]
- 19.Hwang DJ, et al. Structural optimization of indole derivatives acting at colchicine binding site as potential anticancer agents. ACS Med Chem Lett. 2015;6(9):993–997. doi: 10.1021/acsmedchemlett.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mangiatordi GF, et al. Novel chemotypes targeting tubulin at the colchicine binding site and unbiasing P-glycoprotein. Eur J Med Chem. 2017;139:792–803. doi: 10.1016/j.ejmech.2017.07.037. [DOI] [PubMed] [Google Scholar]
- 21.Stengel C, et al. Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br J Cancer. 2010;102(2):316–324. doi: 10.1038/sj.bjc.6605489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorléans A, et al. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci. 2009;106(33):13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Y, et al. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29(11):2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnst KE, et al. A potent, metabolically stable tubulin inhibitor targets the colchicine binding site and overcomes taxane resistance. Cancer Res. 2018;78(1):265–277. doi: 10.1158/0008-5472.CAN-17-0577. [DOI] [PubMed] [Google Scholar]
- 25.Singh H, et al. Triazole tethered isatin-coumarin based molecular hybrids as novel antitubulin agents: design, synthesis, biological investigation and docking studies. Bioorg Med Chem Lett. 2017;27(17):3974–3979. doi: 10.1016/j.bmcl.2017.07.069. [DOI] [PubMed] [Google Scholar]
- 26.Gupta AK, et al. Systematic review on cytotoxic and anticancer potential of N-substituted Isatins as novel class of compounds useful in multidrug-resistant cancer therapy: in silico and in vitro analysis. Topics Curr Chem. 2019;377(3):15. doi: 10.1007/s41061-019-0240-9. [DOI] [PubMed] [Google Scholar]
- 27.Varun, Sonam, and R. Kakkar, Isatin and its derivatives: a survey of recent syntheses, reactions, and applications. Medchemcomm, 2019. 10(3): p. 351-368. [DOI] [PMC free article] [PubMed]
- 28.Matesic L, et al. N-phenethyl and N-naphthylmethyl isatins and analogues as in vitro cytotoxic agents. Bioorg Med Chem. 2008;16(6):3118–3124. doi: 10.1016/j.bmc.2007.12.026. [DOI] [PubMed] [Google Scholar]
- 29.Vine KL, et al. An investigation into the cytotoxicity and mode of action of some novel N-alkyl-substituted isatins. J Med Chem. 2007;50(21):5109–5117. doi: 10.1021/jm0704189. [DOI] [PubMed] [Google Scholar]
- 30.Vine KL, et al. N-alkylated isatins evade P-gp mediated efflux and retain potency in MDR cancer cell lines. Heliyon. 2016;2(1):e00060. doi: 10.1016/j.heliyon.2015.e00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foley GE, et al. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer. 1965;18(4):522–529. doi: 10.1002/1097-0142(196504)18:4<522::aid-cncr2820180418>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 32.La Regina G, et al. Arylthioindole inhibitors of tubulin polymerization. 3. Biological evaluation, structure-activity relationships and molecular modeling studies. J Med Chem. 2007;50(12):2865–2874. doi: 10.1021/jm061479u. [DOI] [PubMed] [Google Scholar]
- 33.JF Díaz, Buey RM . Characterizing ligand-microtubule binding by competition methods. In Microtubule protocols. 2007, Springer. p. 245–260. [DOI] [PubMed]
- 34.Shipley MM, Mangold CA, Szpara ML. Differentiation of the SH-SY5Y human neuroblastoma cell line. J Visualized Exp. 2016;108:53193. doi: 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown J et al., Measuring Neurite Dynamics in Co-culture Using IncuCyte ZOOM® Live-content Imaging Platform and NeuroLight Red™ Fluorescent Label.
- 36.Priest, B.T., et al., Automated electrophysiology assays, in Assay Guidance Manual [Internet]. 2017, Eli Lilly & Company and the National Center for Advancing Translational Sciences. [PubMed]
- 37.Nuessler V, et al. Bcl-2, bax and bcl-xL expression in human sensitive and resistant leukemia cell lines. Leukemia. 1999;13(11):1864–1872. doi: 10.1038/sj.leu.2401571. [DOI] [PubMed] [Google Scholar]
- 38.Spolitu S, et al. Multidrug resistance P-glycoprotein dampens SR-BI cholesteryl ester uptake from high density lipoproteins in human leukemia cells. Am J Cancer Res. 2016;6(3):615–627. [PMC free article] [PubMed] [Google Scholar]
- 39.Courtemanche H, et al. Vincristine-induced neuropathy: atypical electrophysiological patterns in children. Muscle Nerve. 2015;52(6):981–985. doi: 10.1002/mus.24647. [DOI] [PubMed] [Google Scholar]
- 40.Radio NM, Mundy WR. Developmental neurotoxicity testing in vitro: models for assessing chemical effects on neurite outgrowth. Neurotoxicology. 2008;29(3):361–376. doi: 10.1016/j.neuro.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 41.Toselli M, Tosetti P, Taglietti V. Functional changes in sodium conductances in the human neuroblastoma cell line SH-SY5Y during in vitro differentiation. J Neurophysiol. 1996;76(6):3920–3927. doi: 10.1152/jn.1996.76.6.3920. [DOI] [PubMed] [Google Scholar]
- 42.Brown N, Kemp J, Seabrook G. Block of human voltage-sensitive Na + currents in differentiated SH-SY5Y cells by lifarizine. Br J Pharmacol. 1994;113(2):600–606. doi: 10.1111/j.1476-5381.1994.tb17032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansson S. Graded action potentials generated by differentiated human neuroblastoma cells. Acta Physiol Scand. 1994;151(3):331–341. doi: 10.1111/j.1748-1716.1994.tb09752.x. [DOI] [PubMed] [Google Scholar]
- 44.Oshima K, et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2016;113(40):11306–11311. doi: 10.1073/pnas.1608420113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Attalla H, et al. 2-Methoxyestradiol arrests cells in mitosis without depolymerizing tubulin. Biochem Biophys Res Commun. 1996;228(2):467–473. doi: 10.1006/bbrc.1996.1683. [DOI] [PubMed] [Google Scholar]
- 46.Tiberghien F, Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs. 1996;7(5):568–578. doi: 10.1097/00001813-199607000-00012. [DOI] [PubMed] [Google Scholar]
- 47.D’Amato RJ, et al. 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerization by interacting at the colchicine site. Proc Natl Acad Sci. 1994;91(9):3964–3968. doi: 10.1073/pnas.91.9.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verrills NM, et al. Proteome analysis of vinca alkaloid response and resistance in acute lymphoblastic leukemia reveals novel cytoskeletal alterations. J Biol Chem. 2003;278(46):45082–45093. doi: 10.1074/jbc.M303378200. [DOI] [PubMed] [Google Scholar]
- 49.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2012;64:138–153. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 50.Sodani K, et al. Multidrug resistance associated proteins in multidrug resistance. Chinese J Cancer. 2012;31(2):58–72. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiwari AK, et al. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol. 2011;12(4):570–594. doi: 10.2174/138920111795164048. [DOI] [PubMed] [Google Scholar]
- 52.Swerts K, et al. Prognostic significance of multidrug resistance-related proteins in childhood acute lymphoblastic leukaemia. Eur J Cancer. 2006;42(3):295–309. doi: 10.1016/j.ejca.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 53.El-Sharnouby JA, et al. Expression of lung resistance protein and multidrug resistance-related protein (MRP1) in pediatric acute lymphoblastic leukemia. J Oncol Pharm Pract. 2010;16(3):179–188. doi: 10.1177/1078155209351329. [DOI] [PubMed] [Google Scholar]
- 54.Kourti M, et al. Expression of multidrug resistance 1 (MDR1), multidrug resistance-related protein 1 (MRP1), lung resistance protein (LRP), and breast cancer resistance protein (BCRP) genes and clinical outcome in childhood acute lymphoblastic leukemia. Int J Hematol. 2007;86(2):166–173. doi: 10.1532/IJH97.E0624. [DOI] [PubMed] [Google Scholar]
- 55.Plasschaert SL, et al. Expression of multidrug resistance-associated proteins predicts prognosis in childhood and adult acute lymphoblastic leukemia. Clin Cancer Res. 2005;11(24 Pt 1):8661–8668. doi: 10.1158/1078-0432.CCR-05-1096. [DOI] [PubMed] [Google Scholar]
- 56.Krishnegowda G, et al. Synthesis and biological evaluation of a novel class of isatin analogs as dual inhibitors of tubulin polymerization and Akt pathway. Bioorg Med Chem. 2011;19(20):6006–6014. doi: 10.1016/j.bmc.2011.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartz JH. Axonal transport: components, mechanisms, and specificity. Annu Rev Neurosci. 1979;2(1):467–504. doi: 10.1146/annurev.ne.02.030179.002343. [DOI] [PubMed] [Google Scholar]
- 58.LaPointe NE, et al. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology. 2013;37:231–239. doi: 10.1016/j.neuro.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Banks P, Till R. A correlation between the effects of anti-mitotic drugs on microtubule assembly in vitro and the inhibition of axonal transport in noradrenergic neurones. J Physiol. 1975;252(1):283–294. doi: 10.1113/jphysiol.1975.sp011144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.K Hancock, M., et al., A facile method for simultaneously measuring neuronal cell viability and neurite outgrowth. Current chemical genomics and translational medicine, 2015. 9: p. 6-16. [DOI] [PMC free article] [PubMed]
- 61.Rana P, et al. Utilization of iPSC-derived human neurons for high-throughput drug-induced peripheral neuropathy screening. Toxicol In Vitro. 2017;45:111–118. doi: 10.1016/j.tiv.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 62.Geldof A, Minneboo A, Heimans J. Vinca-alkaloid neurotoxicity measured using an in vitro model. J Neurooncol. 1998;37(2):109–113. doi: 10.1023/a:1005848623771. [DOI] [PubMed] [Google Scholar]
- 63.Vetter I, et al. Characterisation of Na(v) types endogenously expressed in human SH-SY5Y neuroblastoma cells. Biochem Pharmacol. 2012;83(11):1562–1571. doi: 10.1016/j.bcp.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 64.Fukuda Y, Li Y, Segal RA. A mechanistic understanding of axon degeneration in chemotherapy-induced peripheral neuropathy. Front Neurosci. 2017;11:481. doi: 10.3389/fnins.2017.00481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grisold W, Cavaletti G, Windebank AJ. Peripheral neuropathies from chemotherapeutics and targeted agents: diagnosis, treatment, and prevention. Neuro Oncol. 2012;14(4):45–54. doi: 10.1093/neuonc/nos203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Namadurai S, et al. A new look at sodium channel beta subunits. Open Biol. 2015;5(1):140192. doi: 10.1098/rsob.140192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brackenbury WJ, et al. Functional reciprocity between Na + channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A. 2010;107(5):2283–2288. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaplan MR, et al. Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30(1):105–119. doi: 10.1016/s0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- 69.Kaplan MR, et al. Induction of sodium channel clustering by oligodendrocytes. Nature. 1997;386(6626):724–728. doi: 10.1038/386724a0. [DOI] [PubMed] [Google Scholar]
- 70.Diaz JF, Andreu JM. Kinetics of dissociation of the tubulin-colchicine complex. Complete reaction scheme and comparison to thermodynamic measurements. J Biol Chem. 1991;266(5):2890–2896. [PubMed] [Google Scholar]
- 71.Antunez-Mojica M, et al. Structural and biochemical characterization of the interaction of tubulin with potent natural analogues of podophyllotoxin. J Nat Prod. 2016;79(8):2113–2121. doi: 10.1021/acs.jnatprod.6b00428. [DOI] [PubMed] [Google Scholar]
- 72.Andreu JM, et al. Interaction of tubulin with bifunctional colchicine analogs: an equilibrium study. Biochemistry. 1984;23(8):1742–1752. doi: 10.1021/bi00303a025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Growth properties of CEM, CEM-VCR R and CEM/2ME2-28.8R cell lines. Figure S2. SH-SY5Y cell viability after treatment with HMB-isatin, VCR and VBL. Figure S3. Dose dependent effect of HMB-isatin and VCR on RA-differentiated SH-SY5Y neurite morphology. Table S1. Electrophysiological parameters of endogenous sodium currents. Table S2. Current density estimates from 3 independent experiments.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.