

Abstract
As an emerging approach to protein perturbation, small molecule-induced protein degradation has gained significant attention as both a chemical tool and a potential therapeutic. To enable discreet control over its function, we have developed a broadly applicable approach for the optical activation of small molecule-induced protein degradation. By installing two different photolabile protecting groups, so called caging groups, onto two different ligands recruiting Von Hippel-Lindau (VHL) and cereblon (CRBN) E3 ubiquitin ligases, our strategy enables light-triggered protein degradation for any small molecule warhead.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
While small molecule inhibitors have traditionally been the most accessible approach to conditionally modulating protein function, their scope in regards to ‘druggable’ protein targets has been limited to less than 20% of the proteome.1 The development of proteolysis targeting chimera (PROTAC) technology has the potential to overcome this limitation, enabling rapid and selective degradation of protein targets using small molecule-based reagents to hijack the endogenous ubiquitin proteasome system. PROTACs are heterobifunctional constructs containing a ‘warhead’, capable of binding the protein of interest, connected via a linker to an E3 ligase ligand. Recruitment of an E3 ubiquitin ligase into close proximity to the protein of interest results in ubiquitination of surface accessible lysines and subsequent degradation of the protein of interest via the ubiquitin proteasome system. This approach has been successfully applied to degrade numerous protein targets, including bromodomain-containing protein 4,2–4 estrogen-related receptor α,5 CDK9,6–7 and TANK-binding kinase 1.8
While PROTACs have demonstrated robust protein degradation both in vitro and in vivo, a lack of external control over their function remains a limitation to their applications as both chemical tools and therapeutics. Potential toxicity from systemic degradation of proteins in healthy cells, as well as diseased cells, is a critical concern.9 For example, one study of mice treated with a BRD4 degrader, ARV-771, exhibited noticeable deterioration of skin health, hunching of the spine, lethargy, and decreased mobility as a result of treatment.10 In addition, undesirable ligase-mediated off-target effects pose a significant risk and are not yet well understood.11 Achieving conditional control of PROTAC function may provide the ability to tightly regulate PROTAC activity, leading to an improved therapeutic index, reduced off-target effects, and a highly targetable therapy. To achieve this goal, we sought out to develop a broadly applicable approach to controlling PROTAC activity using light. Light is an excellent external control element for biological processes, since it acts non-invasively, rapidly, and with spatiotemporal precision.12 Recent reports have demonstrated successful regulation of PROTAC-mediated degradation of BET proteins and FKBP12 through the incorporation of photoswitchable azobenzene linkers.13–14 Herein, we report the development of photocaged E3 ligase ligands for both Von Hippel-Lindau (VHL) and cereblon (CRBN) E3 ubiquitin ligases. By utilizing a general photocaging strategy (Figure 1A), this approach is applicable to all PROTACs utilizing the VHL or CRBN E3 ligands.
Figure 1.
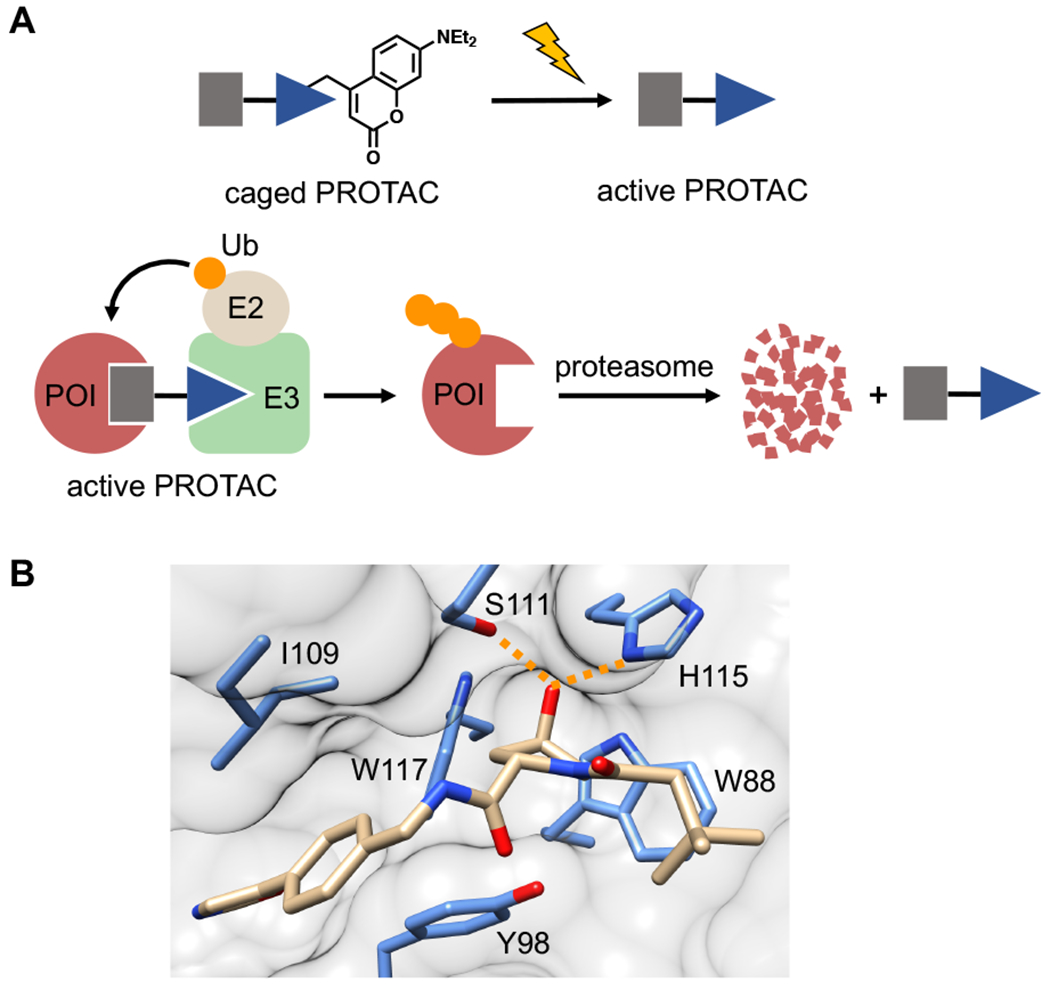
Design of light-activatable PROTACs. A) Placement of a caging group on the E3 ligase ligand (blue triangle) prevents ternary complex formation between the E3 ubiquitin ligase (E3) and the protein of interest (POI). Treatment with light removes the caging group, enabling ternary complex formation, resulting in ubiquitin (Ub, orange circle) transfer and subsequent proteasomal degradation. B) Crystal structure analysis of the VHL ligand bound to VHL protein (PDB: 4W9C) identifies the hydroxyl group on hydroxyproline as critical for the ligand-protein interaction, evidenced by key hydrogen bond interactions (orange dashed line).
To develop a caged VHL ligand, we first determined the optimal location for placement of the light-cleavable caging group onto the VHL ligand by analyzing a crystal structure of the protein-ligand complex (PDB: 4W9C). We observed that the hydroxyproline moiety is buried into the binding cleft and is also involved in two hydrogen bond interactions with Ser111 and His115 (Figure 1B). This hydrogen bond network has been established as critical for VHL recognition of its endogenous protein target, the oxygen-sensitive hypoxia-inducible factor protein, HIF1-α.15–17 Furthermore, it has been shown that inversion of the stereochemistry at the hydroxyl group abolishes all PROTAC activity.4–5 Taken together, we hypothesized that placement of a bulky caging group on the hydroxyl moiety would render the caged ligand unable to bind VHL until removal of the caging group via light irradiation. To test this hypothesis, we synthesized the VHL-based ERRα PROTAC 1 that targets estrogen related receptor α (ERRα),5 an orphan nuclear hormone receptor involved in cellular metabolism and with implications in cancer initiation and progression.18 As a photolabile caging group, we installed a diethylamino coumarin (DEACM) at the hydroxyl group via a carbonate linkage to yield the caged ERRα PROTAC 2 (Figure 2A). The DEACM caging group enables activation by photolysis with ≤405 nm light and release of acidic functional groups with a pKa below 5.19 For the DEACM-caged ERRα PROTAC 2, we performed a UV exposure time course and monitored decaging by HPLC (Supporting Figure S1). Starting material and products were confirmed by mass spectrometry. Upon UV treatment, we observe conversion of 2 to the native ERRα PROTAC 1, as well as the release of the coumarin caging group fragment after a 3 min irradiation. This confirms rapid and efficient decaging of the VHL ligand.
Figure 2.
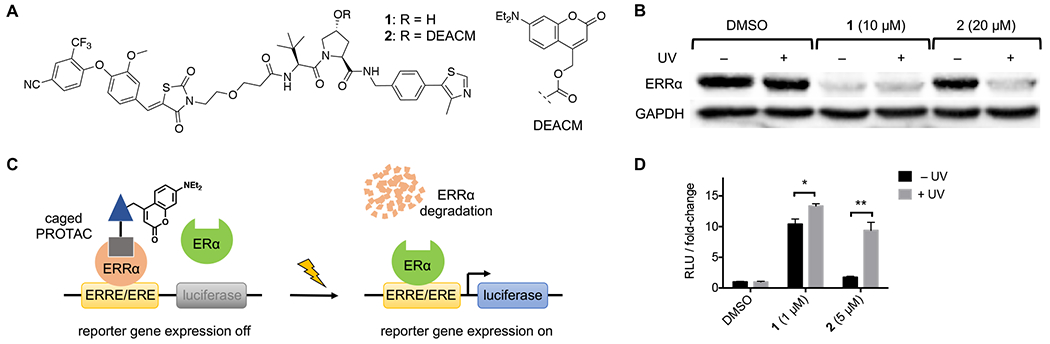
A) Structures of ERRα PROTAC 1 and DEACM-caged ERRα PROTAC 2. B) Western blot analysis of ERRα protein levels from MCF-7 cells treated with DMSO, 1, or 2, and either kept in the dark (− UV) or irradiated with UV light (+ UV). C) ERRα represses luciferase expression by occupying the shared ERRE/ERE consensus sequence preventing ERα transactivation. Upon treatment with UV light, the caging group is removed resulting in degradation of ERRα and activation of luciferase expression by ERα. D) MCF-7 cells transfected with 3xERRE/ERE-luc reporter were treated with DMSO, 1, or 2, and either kept in the dark or irradiated with UV light. Relative luciferase units (RLU) represent firefly luciferase signal normalized to Renilla luciferase control signal. Data represents the means ± standard deviations from at least three independent experiments. Statistical significance was determined using an unpaired t test; *P< 0.05, **P < 0.001.
To test the ability of the DEACM group to block ERRα degradation, MCF-7 cells were treated with DMSO, 1, or 2 followed by presence or absence of UV irradiation (365 nm, 180 s). After an 8 h incubation, ERRα levels were determined via western blot. As expected, treatment with PROTAC 1 resulted in a significant reduction in ERRα protein relative to DMSO control, matching literature reported results.5 Gratifyingly, treatment with the DEACM-caged ERRα PROTAC 2 at twice the concentration of 1 led to no reductions in ERRα protein levels in the absence of UV light. This confirms that the caged compound is completely inactive with regard to E3 ligase recruitment and that the incorporation of the caging group allows for increased dosing concentrations without any observed activity. In contrast, cells treated with 2 and subjected to UV irradiation exhibited comparable reductions in ERRα protein to that of cells treated with 1, demonstrating triggering of degradation activity following photolysis of the DEACM caging group (Figure 2B). The degradation of ERRα was blocked in the presence of either the proteasome inhibitor MG132 or the neddylation inhibitor MLN4924 (cullin-RING ubiquitin ligases need to be neddylated in order to be active), confirming that the degradation observed by these PROTACs is both proteasome- and E3 ligase-mediated (see Supporting Figure S3). MCF-7 cells treated with the coumarin caging group fragment released upon photolysis exhibited no effects on ERRα levels, demonstrating that the observed degradation is entirely mediated by the active, non-caged PROTAC generated via decaging (Supporting Figure S5).
In MCF-7 cells, ERRα acts as an antagonist to estrogen receptor α (ERα) by directly competing for binding to their shared consensus palindromic DNA response element (ERRE/ERE).20 Structural studies have identified that ERRα is constitutively active and is not regulated by traditional estrogen-receptor ligands,21 but rather is regulated by protein co-regulators.22 While the caged ERRα PROTAC 2 is capable of binding ERRα, binding alone is not capable of abolishing ERRα-mediated repression. Transcriptional repression by ERRα has been shown to be alleviated through siRNA knockdown.23 Taking advantage of this mechanism, degradation of ERRα with PROTAC technology would be capable of activating ERα-mediated transcription in MCF-7 cells (Figure 2C). To this end, a luciferase reporter containing the ERRE/ERE response element placed upstream of a firefly luciferase gene was transfected into MCF-7 cells, followed by treatment with DMSO, 1, or 2 in the presence or absence of UV light treatment. Cells treated with 1 demonstrated a 10-fold increase in luciferase signal, indicative of a loss in ERRα-mediated repression following degradation of ERRα (Figure 2D). Treatment with the caged ERRα PROTAC 2 at concentrations 5-fold higher than 1 showed no significant increase in luciferase signal in the absence of light, supporting that ERRα ligand binding alone is incapable of inhibiting ERRα repression. However, following treatment with light (405 nm, 30 s), we observed rescue in luciferase expression to levels similar to that seen following treatment with 1. Taken together, these results demonstrate the ability to control VHL-based PROTAC-mediated protein degradation in cells using both 365 nm and 405 nm light.
With the goal of expanding this technology to encompass CRBN-based PROTACs as well, we next turned our attention to developing a photocaged CRBN ligand. Crystal structure analysis of the thalidomide-CRBN protein complex depicts the imide moiety on the glutarimide ring to be buried into the hydrophobic binding pocket and involved in a hydrogen bond interaction with the peptide backbone of His380 (Figure 3A). We again speculated that installation of a caging group would disrupt a crucial hydrogen-bonding network in addition to introducing a significant amount of steric bulk which could not be accommodated by the binding pocket. This speculation was supported by a previous report which showed that methylation of the imide nitrogen abolished degradation activity of a CRBN-based PROTAC.24 Thus, we synthesized the known CRBN-based BRD4 PROTAC 3, which is capable of targeting and degrading bromodomain-containing protein 4 (BRD4).25 Using chemistry previously established by our lab,26–27 we successfully installed a 6-nitropiperonyloxymethyl (NPOM) group onto the glutarimide nitrogen to generate the caged BRD4 PROTAC 4 (Figure 3B). The NPOM group was chosen for its stability under aqueous conditions, as the relatively high acidity of imide nitrogens (pKa = 14) compared to aliphatic amines (pKa = 30 - 40) has previously proven problematic.26, 28 The NPOM caging group undergoes efficient photolysis by treatment with 365 nm light and has been employed as a photocaging group in numerous biological applications.29 We tested light-activation of the NPOM-caged BRD4 PROTAC 4 through a UV exposure time course and monitored product formation by HPLC, followed by confirmation through mass spectrometry (Supporting Figure S2). We observed clean formation of the non-caged BRD4 PROTAC 3 and the corresponding nitrosoketone caging group product after a 2 min irradiation, suggesting efficient release of the native CRBN ligand upon photolysis.
Figure 3.
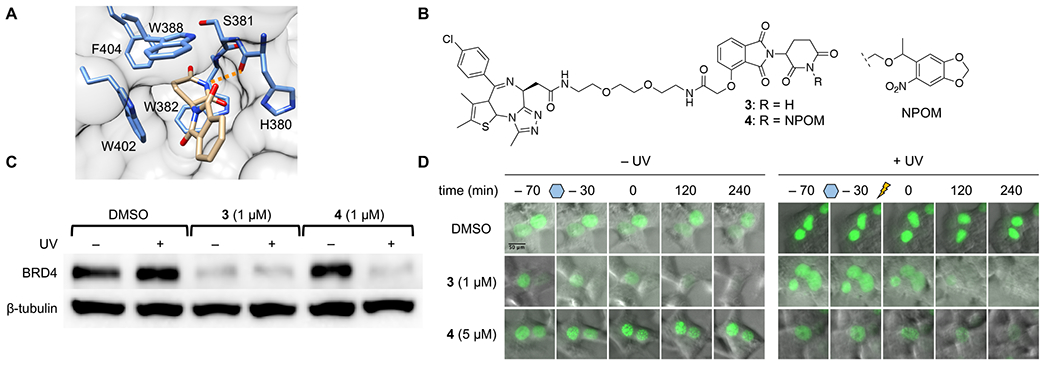
A) Crystal structure analysis of the CRBN ligand bound to CRBN (PDB: 4CI1) identifies the imide moiety buried into the hydrophobic binding pocket, while also forming a hydrogen bond interaction with the backbone of His380 (orange dashed line). B) Structures of BRD4 PROTAC 3 and NPOM-caged BRD4 PROTAC 4. C) Western blot analysis of BRD4 protein levels in HEK293T cells treated with DMSO, 3, or 4, and then either kept in the dark (− UV) or irradiated with UV light (+ UV). D) HEK293T cells transfected with GFP-BRD4 were incubated with either DMSO, 3, or 4 for 1 h. Following incubation, cells were either kept in the dark or irradiated with UV light. GFP levels were monitored via fluorescence imaging over 4 h post-irradiation.
With the caged PROTAC 4 in hand, we tested the ability to optically control CRBN-mediated degradation of BRD4 in cells. Treatment of HEK293T cells with the BRD4 PROTAC 3 resulted in significant degradation of BRD4 within 5 h, as evidenced by western blot. Treatment with caged 4 in the absence of light resulted in no degradation of BRD4, confirming that installation of the NPOM caging is capable of blocking E3 ligase recruitment. However, following irradiation with UV light (365 nm, 180 s), we observed activation of the PROTAC and subsequent degradation of BRD4 by western blot (Figure 3C). No reduction in BRD4 protein levels was observed upon treatment with either MG132 or MLN4924, validating again that the observed degradation is both proteasome- and E3 ligase-mediated (see Supporting Figure S4). Treatment with the nitrosoketone caging group fragment exhibited no effect on BRD4 levels in HEK293T cells, confirming that the protein degradation is entirely mediated by the active BRD4 PROTAC 3, generated upon photolysis (Supporting Figure S5). Furthermore, we were able to optically trigger rapid degradation of a GFP-BRD4 fusion expressed in HEK293T cells (Figure 3D). Upon treatment with 3, nearly full degradation of GFP is observed after 4 h. In cells treated with NPOM-caged BRD4 PROTAC 4, degradation of GFP is only observed after UV irradiation (365 nm, 180 s), demonstrating that with these tools we can temporally control induced degradation of a target protein. Together, these results successfully demonstrate the ability to conditionally control protein degradation in cells using a CRBN ligand-based PROTAC reagent.
In order to demonstrate optical control of the functional consequences of BRD4 degradation, we next investigated the therapeutic response elicited following photoactivation of PROTACs in a cellular model. It has previously been reported that PROTAC-induced degradation of BRD4 is on the order of 10- to 500-fold more potent at inhibiting cell proliferation than treatment with a traditional BRD4 inhibitor, JQ1, in castration-resistant prostate cancer cell lines.10 Taking advantage of the marked increase in efficacy observed for BRD4 degradation agents, we treated 22Rv1 cells, a castration-resistant prostate cancer cell line, with 3 and 4 in the presence and absence of light. Treatment with 3 for 72 h resulted in a 51% reduction in cell viability, while cells treated with caged PROTAC 4 in the absence of light showed no significant reductions in cell viability, demonstrating that BRD4 degradation likely governs the therapeutic response observed with BRD4 PROTAC 3 – in contrast to mere protein inhibition. As expected, removal of the caging group through light exposure (365 nm, 180 s) rescued therapeutic efficacy leading to a 39% decrease in cell viability (Figure 4A). To confirm the mechanism of cell death, we also monitored caspase-3/7 activation as an indicator of apoptosis. Treatment of 22Rv1 cells with 3 resulted in a nearly 3-fold increase in caspase-3/7 activity, while treatment with the caged PROTAC 4 in the absence of light had no significant effect. Following irradiation with UV light, caspase-3/7 activity was rescued to levels similar to that observed with PROTAC 3 (Figure 4B). Taken together, these results demonstrate that optical control of PROTAC activity is a feasible approach to controlling the therapeutic effects of PROTACs.
Figure 4.
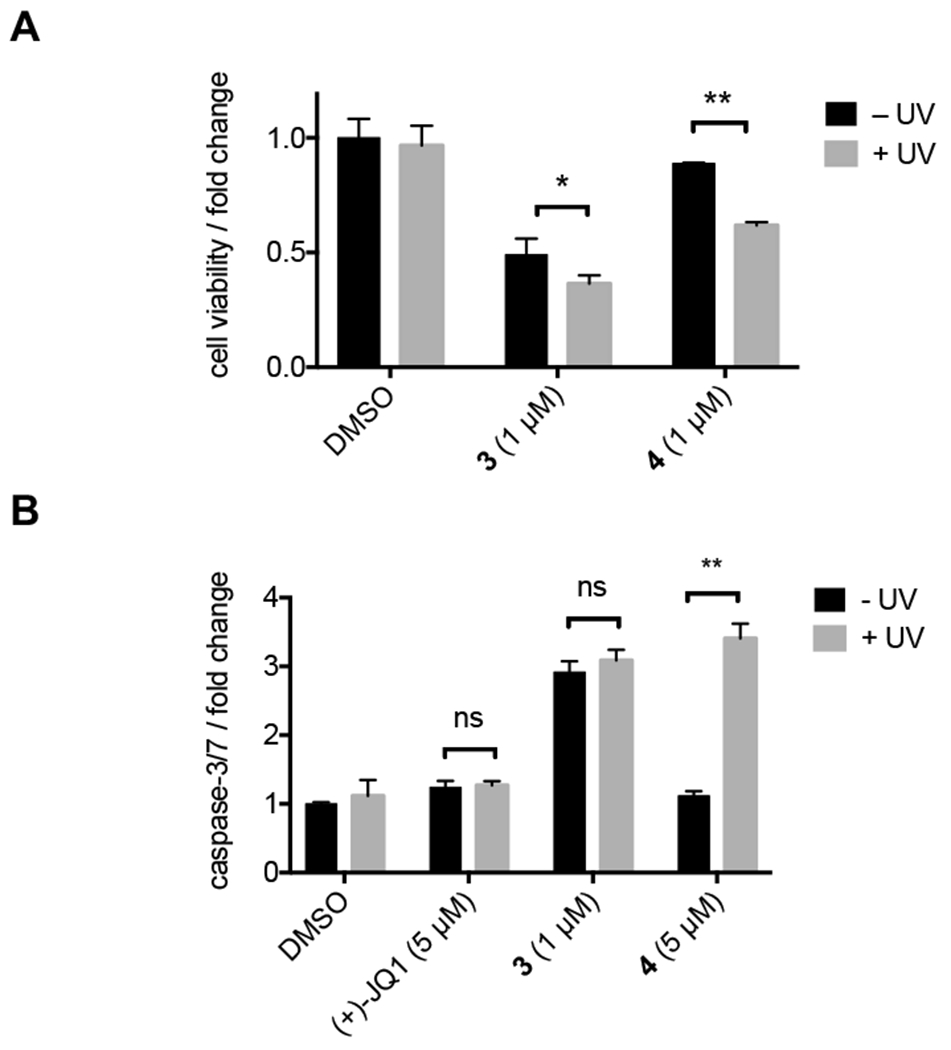
A) 22Rv1 cells were treated with DMSO, 3, or 4 in the presence (+ UV) or absence (− UV) of UV light. Cells were incubated 72 h before determining cell viability using a CellTiterGlo assay. Treatment normalized to DMSO control. B) 22Rv1 cells were treated with DMSO, JQ1, 3, or 4 in the presence or absence of UV light. Cells were incubated for 24 h before determining caspase-3/7 activity using a CaspaseGlo-3/7 assay. Statistical significance was determined using an unpaired t test; ns P>0.05, *P<0.05, **P<0.001.
In summary, we have successfully developed two approaches to optically controlling PROTAC function through the strategic installation of photocaging groups onto E3 ligase ligands recruiting VHL and CRBN. Introduction of the caging groups completely abolishes degradation activity of PROTACs until treatment with light unmasks the E3 ligands returning them to their native states. We demonstrated robust photoactivation of protein degradation in cells and showed that photocontrol of degradation can also function as an effective approach to conditionally control therapeutic response to treatment. These approaches afford enhanced control over biological processes and has far reaching implications for the development of highly targeted therapies based on precision drug activity profiles. Continued efforts are being made to improve the spectral properties of these reagents with the hopes of shifting absorption into the near IR region for enhanced tissue penetration.
Supplementary Material
ACKNOWLEDGMENT
The authors wish to thank the lab of Shivendra Singh (University of Pittsburgh, Hillman Cancer Center) for providing the 22Rv1 prostate cancer cell line. The plasmids 3xERRE/ERE-luciferase (Addgene #37852) and GFP-BRD4 (Addgene #65378) were gifts from Drs. Rebecca Riggins and Kyle Miller, respectively. The corresponding plasmid maps can be found in Supporting Figure S6.
Funding Sources
This work was supported by the University of Pittsburgh and the National Institutes of Health (R21AI130815 and R01GM127153).
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Chemical synthesis procedures and full characterization, biological protocols, and supporting figures.
The authors declare no competing financial interest.
REFERENCES
- (1).Ottis P; Crews CM, Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol 2017, 12 (4), 892–898. [DOI] [PubMed] [Google Scholar]
- (2).Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM, Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol 2015, 22 (6), 755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE, Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zengerle M; Chan KH; Ciulli A, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol 2015, 10 (8), 1770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM, Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 2015, 11 (8), 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Robb CM; Contreras JI; Kour S; Taylor MA; Abid M; Sonawane YA; Zahid M; Murry DJ; Natarajan A; Rana S, Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb) 2017, 53 (54), 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Olson CM; Jiang B; Erb MA; Liang Y; Doctor ZM; Zhang Z; Zhang T; Kwiatkowski N; Boukhali M; Green JL; Haas W; Nomanbhoy T; Fischer ES; Young RA; Bradner JE; Winter GE; Gray NS, Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nature Chemical Biology 2017, 14, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Crew AP; Raina K; Dong H; Qian Y; Wang J; Vigil D; Serebrenik YV; Hamman BD; Morgan A; Ferraro C; Siu K; Neklesa TK; Winkler JD; Coleman KG; Crews CM, Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem 2018, 61 (2), 583–598. [DOI] [PubMed] [Google Scholar]
- (9).Gu S; Cui D; Chen X; Xiong X; Zhao Y, PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. Bioessays 2018, 40 (4), e1700247. [DOI] [PubMed] [Google Scholar]
- (10).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AM; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG, PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2016, 113 (26), 7124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Churcher I, Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J Med Chem 2018, 61 (2), 444–452. [DOI] [PubMed] [Google Scholar]
- (12).Mayer G; Heckel A, Biologically active molecules with a “light switch”. Angewandte Chemie International Edition 2006, 45 (30), 4900–4921. [DOI] [PubMed] [Google Scholar]
- (13).Pfaff P; Samarasinghe KTG; Crews CM; Carreira EM, Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable PhotoPROTACs. ACS Central Science 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Trauner D; Pagano M; Marzio A; Simoneschi D; Bérouti M; Matsuura B; Reynders M, PHOTACs Enable Optical Control of Protein Degradation. ChemRxiv 2019, Preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yu F; White SB; Zhao Q; Lee FS, HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proceedings of the National Academy of Sciences 2001, 98 (17), 9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Min JH; Yang H; Ivan M; Gertler F; Kaelin WG Jr.; Pavletich NP, Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 2002, 296 (5574), 1886–9. [DOI] [PubMed] [Google Scholar]
- (17).Hon W-C; Wilson MI; Harlos K; Claridge TDW; Schofield CJ; Pugh CW; Maxwell PH; Ratcliffe PJ; Stuart DI; Jones EY, Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 2002, 417, 975. [DOI] [PubMed] [Google Scholar]
- (18).Ranhotra HS, The estrogen-related receptor alpha: the oldest, yet an energetic orphan with robust biological functions. J Recept Signal Transduct Res 2010, 30 (4), 193–205. [DOI] [PubMed] [Google Scholar]
- (19).Schmidt R; Geissler D; Hagen V; Bendig J, Mechanism of photocleavage of (coumarin-4-yl)methyl esters. J Phys Chem A 2007, 111 (26), 5768–74. [DOI] [PubMed] [Google Scholar]
- (20).Kraus RJ; Ariazi EA; Farrell ML; Mertz JE, Estrogen-related receptor alpha 1 actively antagonizes estrogen receptor-regulated transcription in MCF-7 mammary cells. J Biol Chem 2002, 277 (27), 24826–34. [DOI] [PubMed] [Google Scholar]
- (21).Kallen J; Schlaeppi JM; Bitsch F; Filipuzzi I; Schilb A; Riou V; Graham A; Strauss A; Geiser M; Fournier B, Evidence for ligand-independent transcriptional activation of the human estrogen-related receptor alpha (ERRalpha): crystal structure of ERRalpha ligand binding domain in complex with peroxisome proliferator-activated receptor coactivator-1alpha. J Biol Chem 2004, 279 (47), 49330–7. [DOI] [PubMed] [Google Scholar]
- (22).Schreiber SN; Knutti D; Brogli K; Uhlmann T; Kralli A, The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor alpha (ERRalpha). J Biol Chem 2003, 278 (11), 9013–8. [DOI] [PubMed] [Google Scholar]
- (23).Watanabe A; Kinoshita Y; Hosokawa K; Mori T; Yamaguchi T; Honjo H, Function of estrogen-related receptor alpha in human endometrial cancer. J Clin Endocrinol Metab 2006, 91 (4), 1573–7. [DOI] [PubMed] [Google Scholar]
- (24).Hatcher JM; Wang ES; Johannessen L; Kwiatkowski N; Sim T; Gray NS, Development of Highly Potent and Selective Steroidal Inhibitors and Degraders of CDK8. ACS Med Chem Lett 2018, 9 (6), 540–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Winter GE; Mayer A; Buckley DL; Erb MA; Roderick JE; Vittori S; Reyes JM; di Iulio J; Souza A; Ott CJ; Roberts JM; Zeid R; Scott TG; Paulk J; Lachance K; Olson CM; Dastjerdi S; Bauer S; Lin CY; Gray NS; Kelliher MA; Churchman LS; Bradner JE, BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell 2017, 67 (1), 5–18 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lusic H; Deiters A, A new photocaging group for aromatic N-heterocycles. Synthesis-Stuttgart 2006, 2006 (13), 2147–2150. [Google Scholar]
- (27).Young DD; Lively MO; Deiters A, Activation and deactivation of DNAzyme and antisense function with light for the photochemical regulation of gene expression in mammalian cells. J Am Chem Soc 2010, 132 (17), 6183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bordwell FG, Equilibrium Acidities in Dimethyl-Sulfoxide Solution. Accounts of Chemical Research 1988, 21 (12), 456–463. [Google Scholar]
- (29).Ankenbruck N; Courtney T; Naro Y; Deiters A, Optochemical Control of Biological Processes in Cells and Animals. Angew Chem Int Ed Engl 2018, 57 (11), 2768–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.