


Abstract
SIRT6 deacetylase activity improves stress resistance via gene silencing and genome maintenance. Here, we reveal a deacetylase-independent function of SIRT6, which promotes anti-apoptotic gene expression via the transcription factor GATA4. SIRT6 recruits TIP60 acetyltransferase to acetylate GATA4 at K328/330, thus enhancing its chromatin binding capacity. In turn, GATA4 inhibits the deacetylase activity of SIRT6, thus ensuring the local chromatin accessibility via TIP60-promoted H3K9 acetylation. Significantly, the treatment of doxorubicin (DOX), an anti-cancer chemotherapeutic, impairs the SIRT6–TIP60–GATA4 trimeric complex, blocking GATA4 acetylation and causing cardiomyocyte apoptosis. While GATA4 hyperacetylation-mimic retains the protective effect against DOX, the hypoacetylation-mimic loses such ability. Thus, the data reveal a novel SIRT6–TIP60–GATA4 axis, which promotes the anti-apoptotic pathway to prevent DOX toxicity. Targeting the trimeric complex constitutes a new strategy to improve the safety of DOX chemotherapy in clinical application.
INTRODUCTION
Anthracyclines, including doxorubicin (DOX), are widely used as efficacious chemotherapeutics, but cause severe dose-dependent cardiotoxicity (1,2). Anthracycline-induced heart failure is largely due to myocyte apoptosis (3). GATA4, a member of the GATA transcription factor family with the ability to bind the consensus DNA motif ‘GATA’, plays critical roles in myocardial differentiation and function (4–6). GATA4 activates the transcription of anti-apoptotic gene Bcl-2 and Bcl-xL etc., which protect against myocyte death induced by DOX (3,7–9). Upon the DOX treatment, GATA4 is rapidly downregulated at both both transcript and protein levels (3,10–12). Intriguingly, GATA4 overexpression causes cardiac hypertrophy (13). These findings suggest that GATA4 might undergo additional layers of fine-tuned regulation, which merits further examination before applying GATA4 restoration as a clinical strategy to prevent DOX-induced cardiotoxicity (3,7,14).
SIRT6 belongs to the highly conserved family of NAD+-dependent sirtuins, which deacetylate histones and non-histone substrates to modulate chromatin stability and restrict transcription (15–17). Through these functions, SIRT6 maintains organismal health and protects against aging and various diseases, including cancers and metabolic disorders (18–21). SIRT6 is implicated in protecting against cardiac hypertrophy and heart failure by deacetylating H3K9 to repress IGF-Akt (22,23) and NF-κβ signaling (24,25). Cardiac Sirt6 is sensitive to stress stimuli, i.e. angiotensin II, isoproterenol and ischemia/reperfusion-induced reactive oxygen species (ROS) and DOX (23,26–28). Exercise during pregnancy protects neonatal cardiomyocytes against DOX toxicity, accompanied by the increased expression of SIRT6 (29). Despite these advances, how SIRT6 protects cardiomyocytes against DOX are unclear.
Here, we demonstrated a novel, deacetylase-independent mechanism by which SIRT6 protects against DOX-induced cardiomyocyte death. Our data suggest that targeting the non-catalytic function of SIRT6 may enhance the safety of DOX chemotherapy.
MATERIALS AND METHODS
Cell culture and treatments
HEK293 (CRL-1573) and H9C2 (GNR-5) cells were purchased from ATCC. Wild-type (WT) and Sirt6−/− mouse embryonic fibroblasts (MEFs) were obtained as previously described (30). SIRT6 knockout (KO) HEK293 cell lines were generated using the CRISPR/Cas9 system, as described previously (21). Primary neonatal mouse cardiomyocytes were prepared with a standard procedure (31). Briefly, hearts from 1- to 3-day-old C57BL/6 mice were isolated and incubated with digestion medium. After centrifuging and plating, the viable cardiomyocytes formed a monolayer with synchronized beating within two days of culture. All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM, Life Technologies, USA) supplemented with 15% fetal bovine serum, 100 U/ml penicillin and streptomycin at 37°C in 5% CO2 and atmospheric oxygen. The cells were treated with DOX at the indicated doses for specific analyses.
Mice and DOX administration
Sirt6f/f mice were crossed with Myh6-cre/Esr1 mice to generate Sirt6f/f;Myh6-cre mice. To generate cardiac-specific Sirt6 KO mice, 4-hydroxytamoxifen was injected intraperitoneally (i.p.) daily in Sirt6f/f;Myh6-cre mice for three consecutive days. After 1 month, the heart tissues were harvested and analyzed by PCR-based genotyping and quantitative PCR (qPCR). All mice were housed and handled in the Laboratory Animal Research Center of Shenzhen University. All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC).
Plasmids and RNA interference
Human FLAG-GATA4 and FLAG-TIP60 constructs were purchased from Vigene Biosciences (China). Human FLAG-SIRT1, 2, 6 and 7 constructs were purchased from Addgene (USA). FLAG-GATA4 and FLAG-SIRT6 with amino acid substitutions were generated by PCR-based mutagenesis using a KOD-PLUS kit (Toboyo, Japan). The truncated SIRT6 and GATA4 plasmids were constructed by PCR-based deletion based on the HA-SIRT6 and HA-GATA4 templates. The primers used for mutagenesis are listed in Supplemental Table S1.
For RNA interference, the cells were transfected with small interfering RNAs (siRNAs) for 48 h using Lipofectamine® 3000 (Invitrogen, USA), according to the manufacturer's instructions. Scrambled siRNAs were used as a negative control. All siRNAs were purchased from Gene Pharma (China), and their specific sequences are included in Supplemental Table S1.
Cell transfection and colony formation assay
To establish stably transfected H9C2 cell lines, the pcDNA3.1, GATA4, GATA4 2KR (K328/330R) and GATA4 2KQ (K328/330Q) plasmids were digested with PvuI nuclease for 30 min to linearize, and then individually transfected into H9C2 cells with Lipofectamine®3000. The medium was replaced after 24 h and supplemented with 2 mg/ml G418 for selection. After 10 days, stably transfected cells were obtained, and their expression was confirmed by western blotting.
For the colony-formation assay, the cells were seeded in six-well plates in triplicate and cultured under normal growth conditions in the presence or absence of DOX at the indicated doses. After culturing for a further 10–14 days, the cell colonies were stained with 0.5% crystal violet solution. The number of colonies in each well was quantitated and the surviving fraction was calculated.
Chromatin-bound fraction assay
The cells were carefully detached from the culture vessel in 1 ml cold PBS buffer and then pelleted by centrifugation at 3000 × g for 1 min. The cell pellets were resuspended with 500 μl Buffer A (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X-100, 10 mM Na3VO4 and protease inhibitor cocktail) and incubated on ice for 10 min. The cell lysates were then centrifuged at 1300 × g for 5 min and supernatant containing the cytosolic proteins was collected. The pellet was washed once with Buffer A, and then lysed in 250 μl Buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 10 mM Na3VO4 and protease inhibitor cocktail). Following incubation and centrifugation (1700 × g, 5 min), the pellet (chromatin-bound fraction) was obtained and denatured with 1× SDS loading buffer.
Protein extraction, western blotting and antibodies
The mouse tissues were homogenized in 1 ml ice-cold tissue lysis buffer (25 mM Tris–HCl, pH 7.5, 10 mM Na3VO4, 100 mM NaF, 50 mM Na4P2O7, 5 mM EGTA, 5 mM EDTA, 0.5% SDS, 1% NP-40 and protease inhibitor cocktail). Following sonication, the lysates were centrifuged at 16 000 × g for 10 min and then carefully collected. The protein concentrations were determined using a Bicinchoninic Acid kit (BCA; Thermo). Western blotting was performed following a standard protocol. Anti-acetyl lysine (CST, Ac-K-100, #6952), anti-poly/mono ADP ribose (CST, #83732) and anti-Tip60 (CST, #12058) antibodies were purchased from Cell Signaling Technology (USA). Anti-GATA4 (ab84593), anti-SIRT6 (ab62739) and anti-H3 (ab1791) antibodies were purchased from Abcam (UK). Anti-P300 (sc48343) and anti-GST (sc138) antibodies were purchased from Santa Cruz Biotechnology (USA). Anti-H3K9ac (ABE18) was obtained from EMD Millipore (USA) and anti-FLAG M2 was purchased from Sigma-Aldrich (USA).
Co-immunoprecipitation (Co-IP) and pull-down assays
The co-immunoprecipitation assay was performed as previously described (32). Briefly, the cells were washed with cold PBS and harvested. The cell pellets were then resuspended with 500 μl IP lysis buffer (20 mM Tris–HCl PH 8.0, 250 mM NaCl, 5 M sodium pyrophosphate, 1 mM sodium fluoride, 10% glycerol, 2 mM EDTA, 10 mM Na3VO4, protease inhibitor cocktail and 1 mM nicotinamide). Following sonication and centrifugation, the supernatant was collected and incubated with anti-HA or anti-FLAG M2 agarose (Sigma, USA) overnight at 4°C on a rotating platform. The beads were then washed three times with 1 ml cold lysis buffer. To denature, 1× SDS loading buffer was added to the samples and boiled at 98°C for 6 min. The samples, including the input (5%), were then analyzed by western blotting.
For the in vitro pull-down assay, GST or GST-SIRT6 (1 μg)-conjugated GSH beads were suspended in NETN buffer (20 mM Tris–HCl pH 8, 100 mM NaCl, 1 mM EDTA, 0.5% NP40 and phosphatase and protease inhibitor cocktail). The purified His-GATA4 proteins (0.2 μg) were then added and incubated overnight at 4°C, followed by three washes with NETN buffer. The boiled eluates were then separated and detected by Coomassie staining and western blotting.
Luciferase reporter assay
HEK293 cells were transfected with a pGL3-Bcl-2-promoter reporter, together with the expression plasmids and a control pCMXβgal plasmid. The luciferase activity was measured using a luciferase reporter assay kit (Promega, #E1501), according to the manufacturer's instructions. The luciferase values were normalized according to β-gal activity.
RNA preparation and real-time qPCR analysis
Total RNAs were extracted from cells or mouse tissues using Trizol® reagent RNAiso Plus (Takara, Japan) following the phenol-chloroform extraction method. A cDNA library was constructed from the purified RNAs using Prime Script RT Master Mix (Takara, Japan) and the following thermal cycling conditions: 37°C for 40 min; 85°C for 5 s. Gene expression was analyzed using a CFX Connected™ Real-time PCR Detection System (Bio-Rad, USA) with SYBR Ex Taq Premixes (Takara, Japan). Each experiment was performed at least three times. The values were standardized to 18s rRNA. The primers for qPCR are listed in Supplemental Table S1.
Chromatin immunoprecipitation (ChIP) assay
The ChIP assay was performed as previously described (21). Briefly, the cells were cross-linked and lysed with lysis buffer (50 mM Tris–HCl pH 8.0, 2 mM EDTA, 15 mM NaCl, 1% SDS, 0.5% deoxycholate, protease inhibitor cocktail and 1 mM PMSF). Following sonication and centrifugation, the supernatant was collected and precleared with protein A/G Sepharose. The precleared samples were incubated overnight with the indicated antibodies: anti-H3K9ac, anti-SIRT6, anti-GATA4 antibody (2 μg/sample) and appropriate control IgGs. The bound DNA fragments were precipitated by supplementing with 50% suspension of protein A/G Sepharose. Following washing sequentially with a series of buffers, the beads were heated to 65°C to reverse the cross-links. The DNA fragments were purified and analyzed by qPCR. The primers used in this study are listed in Supplemental Table S1.
TUNEL staining assay
DOX-induced cardiomyocyte apoptosis was evaluated using a Dead End™ Fluorometric TUNEL System (Promega, G3250), according to the manufacturer's instructions. Briefly, the cells were fixed and incubated with a reaction mixture containing terminal reoxynucleotidyl transferase and Fluorescein-12-dUTP at 37°C for 1 h. DAPI was used to counterstain the nuclei. Five fields-of-view were randomly selected, and images were captured under the microscope to calculate the TUNEL-positive staining rate for each slice.
Fluorescence-activated cell sorting (FACS) assay
The H9C2 cells were treated with 1 μM DOX for 12 h and then stained with FITC-Annexin V (Beyotime, C1062L) and 7-AAD (BD Pharmingen) for live/dead cell discrimination. Flow cytometry analyses were performed using an LSR-II flow cytometer and analyzed with FlowJo x7.5.
In vitro SIRT6 deacetylation assay
SIRT6 deacetylation assay was performed with the SIRT6 Activity Assay Kit (Fluorometric) (ab 156068, Abcam, UK) following the manufacturer's instructions. Briefly, bacterially purified GST-SIRT6 or GST-SIRT6 H133Y was pre-incubated with purified His-GATA4, G4-13 peptide (RKRKPKNLNKSKT) or G4-24 peptide (ACGLYMKLHGVPRPLAMRKEGIQT) (GL Biochem LTD, Shanghai, China), in the reaction buffer at 4°C for 30 min. Once the fluorophore-conjugated synthetic Ac-peptide was added, the plate was moved to measure the fluorescence intensity.
Statistical analyses
All values are expressed as the mean ± s.e.m. Statistical differences among groups were determined using Student's t-test or two-way ANOVA using GraphPad Prism software. Survival curve data were analyzed using a log-rank (Mantel–Cox) test.
RESULTS
Sirt6 enhances anti-apoptotic gene expression against DOX via Gata4
To determine how Sirt6 protects cell survival against DOX, we assessed the RNA-seq data sets from two independent Sirt6 null cell lines: mouse embryonic stem cells (mESCs) (33) and mouse embryonic fibroblast cells (MEFs) (21). As shown, Sirt6 depletion activates apoptotic pathway, with dramatic decrease of anti-apoptotic gene expression, like Bcl-2 and Bcl-xL (Supplemental Figure S1A, B). GATA4 transcription factor is pivotal for postnatal cardiomyocyte survival against DOX by direct targeting the promoter regions of anti-apoptotic genes (3,7–9). Indeed, mRNA levels of Gata4, Bcl-2 and Bcl-xL were all rapidly downregulated in H9C2 cells and primary mouse cardiomyocytes upon DOX treatment (Figure 1A and Supplemental Figure S1C). To further understand the causal link between Sirt6 and the Gata4/Bcl-2 pathway, we generated cardiac-specific Sirt6 knockout in adult mice using the tamoxifen-inducible α-myosin heavy chain (Myh6) Cre line, i.e. Sirt6f/f;Myh6-Cre, which spontaneously develop heart failure (23). qPCR analysis showed that Sirt6 depletion in mouse hearts significantly reduced Bcl-2 and Bcl-xL levels, with little effect on GATA4 mRNA or protein expression (Figure 1B and Supplemental Figure S1D). Sirt6 knockdown by siRNAs further exacerbated the DOX-induced reduction of Bcl-2 and Bcl-xL mRNA expression in H9C2 cells (Figure 1C and Supplemental Figure S1E). By contrast, Sirt6 overexpression enhanced Bcl-2 and Bcl-xL expression levels and protected against the decline induced by DOX in mice (Figure 1D and Supplemental Figure S1F).
Figure 1.

SIRT6 antagonizes DOX-induced apoptotic pathway in cardiomyocytes via GATA4. (A) Quantitative PCR (qPCR) showing the mRNA levels of Bcl-2, Bcl-xL, Gata4 and Sirt6 in primary mouse cardiomyocytes after the treatment of DOX (1 μM) for 6 h. ***P < 0.001. (B) qPCR results showing the mRNA levels of Bcl-2, Bcl-xL, Sirt6 and Gata4 in heart tissues isolated from Sirt6f/f and Sirt6f/f;Myh6-Cre mice treated with tamoxifen. *P < 0.05, **P < 0.01, ***P < 0.001, ‘ns’ indicates no significance. (C) H9C2 cells with si-NC (negative control) or si-Sirt6 small interfering (si)RNAs were exposed to DOX (1 μM, 6 h). The mRNA levels of Bcl-2, Bcl-xL and Gata4 were determined by quantitative PCR. ***P < 0.001, ‘ns’ indicates no significance. (D) qPCR results showing the mRNA levels of Bcl-2, Bcl-xL and Sirt6 in wild-type (WT) and Sirt6 transgenic (Sirt6-TG) mice after intraperitoneal injection of DOX (n = 5/group). *P < 0.05, **P < 0.01, ***P < 0.001, ‘ns’ indicates no significance. (E) Schematic illustration of the GATA consensus motif on Bcl-2 promoter region. P1 and P2 indicate –266 GATA motif and –1025 GATA motif respectively. TSS represents the transcription start site of the Bcl-2 gene. (F) ChIP was performed using anti-SIRT6 antibodies and IgG. Enrichment of SIRT6 on the Bcl-2 promoter region was analyzed by qPCR with primers targeting the P1 and P2 motifs. **P < 0.01. (G) HEK293 cells were transfected with indicated plasmids, and the luciferase activities were measured and normalized to β-gal activities. **P < 0.01. The western blots indicate the expression levels of FLAG-GATA4 and FLAG-SIRT6. (H) ChIP was performed to test GATA4 enrichment on the Bcl-2 promoter using anti-GATA4 antibody in HEK293 cells with or without FLAG-SIRT6 overexpression. *P < 0.05. (I) ChIP was performed to test GATA4 enrichment on the Bcl-2 promoter using anti-GATA4 antibody in HEK293 cells with si-NC or si-SIRT6. *P < 0.05.
We then asked whether Sirt6 regulates anti-apoptotic pathway via Gata4. To this end, we first compared the down-regulated genes in Sirt6 null mESCs and the genes that are bound by Gata4 obtained from chromatin immunoprecipitation and sequencing (ChIP-seq) in mESCs-derived mesoderm (34). Intriguingly, almost 50% of down-regulated genes caused by Sirt6 deficiency harbor Gata4 binding sites and gene ontology analysis revealed significant enrichment of pathways regulating apoptosis (Supplemental Figure S1G, H and Supplemental Table S2). We thus speculate that Sirt6 and Gata4 might cooperate in regulating anti-apoptotic gene expression. As described, GATA4 regulates cardiac Bcl-2 transcription by binding to the proximal −266 GATA motif (P1) rather than the distal −1025 GATA motif (P2) (8). Interestingly, ChIP analysis demonstrated that SIRT6 was also highly enriched at the P1 site (Figure 1E, F). We then determined the luciferase activity under transcriptional control of Bcl-2 promoter containing the P1 motif. We found that ectopically expressed SIRT6 significantly elevated the Bcl-2 promoter (P1) -driven luciferase activity (Figure 1G). Moreover, SIRT6 overexpression enhanced the GATA4 occupancy at the P1 site (Figure 1H and Supplemental Figure S1I), but SIRT6 KD compromised it (Figure 1I and Supplemental Figure S1J). Together, these data suggest that SIRT6 enhances GATA4 chromatin binding to promote anti-apoptotic gene expression.
GATA4 physically associates with SIRT6
As shown above, SIRT6 and GATA4 are both enriched at the P1 promoter region and coordinate the transcription of Bcl-2, suggesting that these two proteins may directly interact. Indeed, we detected FLAG-GATA4 in anti-HA-SIRT6 immunoprecipitates in HEK293 cells expressing FLAG-GATA4 together with or without HA-SIRT6 (Figure 2A). Reciprocally, HA-SIRT6 was present in the purified pool of FLAG-GATA4 from HEK293 cells expressing HA-SIRT6 together with or without FLAG-GATA4 (Figure 2B). To confirm such interaction under physiological condition, we did endogenous co-immunoprecipitation (CoIP) and immunofluorescence microscopy in H9C2 cardiomyocytes. Again, SIRT6 was detected in the anti-GATA4 immunoprecipitates and co-stained with GATA in the nucleus (Figure 2C, D). Next, we performed in vitro pull-down assay using purified E. coli-expressing His-GATA4 and GST-SIRT6. As shown, the result suggested a direct interaction between GATA4 and SIRT6 (Figure 2E). GATA4 possesses two zinc finger (Zn-finger) domains; the C-terminal Zn-finger is essential for DNA and cofactor binding (35,36) (Figure 2F). Domain mapping showed that SIRT6 specifically bound GATA4 at C-terminal Zn-finger region (a.a. 251–350) (Figure 2G).
Figure 2.

SIRT6 physically interacts with GATA4. (A) Co-immunoprecipitation using anti-HA agarose was performed in HEK293 cells transfected with indicated constructs. The level of FLAG-GATA4 was detected by western blotting in purified pool of HA-SIRT6. (B) Co-immunoprecipitation using anti-FLAG agarose was performed in HEK293 cells transfected with indicated constructs. The level of HA-SIRT6 was detected by western blotting in purified pool of FLAG-GATA4. (C, D) Endogenous interaction of GATA4 and SIRT6 in H9C2 cells was evaluated by co-immunoprecipitation (Co-IP) and immunostaining with anti-GATA4 and anti-SIRT6 antibodies. Scale bar, 50 μm. (E) GST pull-down assay was performed to test the in vitro binding of purified His-GATA4 and GST-fused SIRT6 from E. coli. Coomassie blue staining indicates the loading levels of GST and GST-SIRT6. (F) Diagram of full-length (F-L) and fragmented human GATA4 with one or two zinc finger DNA-binding domains. (G) Domain-based truncations of HA-GATA4 were co-expressed with FLAG-SIRT6 in HEK293 cells. HA-GATA4 protein was precipitated and FLAG-SIRT6 was analyzed by western blotting.
SIRT6 facilitates GATA4 acetylation independently of its deacetylase activity
We next sought to figure out how SIRT6 entrains GATA4-targeted gene expression. Of note, post-translational modifications, particularly phosphorylation and acetylation, are critical to GATA4 DNA-binding capacity and activation (37–39). To examine whether SIRT6 modulates the acetylation level of GATA4, we IPed FLAG-GATA4 in HEK293 cells co-overexpressing GATA4 and SIRT6 and examined its acetylation level using pan anti-acetyl lysine antibodies. Rather surprisingly, the acetylation level of GATA4 was markedly increased in the presence of ectopic SIRT6 (Figure 3A). Low level of GATA4 acetylation was detected in the Sirt6-depleted cells (Supplemental Figure S2A). In addition to deacetylase activity, SIRT6 also possesses mono-ADP-ribosylase (MAR) activity and could auto-MAR itself (40). However, no GATA4 MAR were observed, indicating that GATA4 is less likely a MAR target of SIRT6 (Supplemental Figure S2B).
Figure 3.

SIRT6 enhances GATA4 acetylation and protects against DOX-induced myocyte apoptosis independently of its deacylase activity. (A) Western blotting showing the acetylation level of GATA4 immunoprecipitated from HEK293 cells overexpressing of FLAG-SIRT6 and HA-GATA4. Acetylation level was determined with anti-acetyl lysine (AcK) antibodies. Lower, fold change of the acetylation level of GATA4 relative to HA-GATA4 only control, determined by ImageJ. n = 6. ***P < 0.001. (B) HEK293 cells co-expressing FLAG-SIRT6 and HA-GATA4 were treated with 10 μM nicotinamide (NAM) for 4 h. The acetylation level of GATA4 was examined using anti-AcK antibodies following immunoprecipitation. Lower, fold change of the acetylation level of GATA4 relative to HA-GATA4 only control, determined by ImageJ. n = 3. ***P < 0.001. ‘ns’ indicates no significance. (C) HA-GATA4 co-expressed with different forms of FLAG-SIRT6 (WT, D63Y, D116N and H133Y mutant) separately in HEK293 cells was immunoprecipitated with anti-HA beads. The blots of FLAG-SIRT6 in IPs indicate the association of GATA4 and SIRT6. Lower, fold change of the acetylation level of GATA4 relative to HA-GATA4 only control, determined by ImageJ. n = 3. **P < 0.01. *P < 0.05. ‘ns’ indicates no significance. (D) Real-time PCR analysis of mRNA levels of Bcl-2 in WT and Sirt6−/− MEFs re-expressing FLAG-SIRT6, or enzyme-dead mutations. **P < 0.01; ***P < 0.001. 18srRNA was used as a reference gene. (E, F) Representative flow plots and quantification showing the percentages of apoptotic H9C2 cells overexpressing the indicated genes with the treatment of DOX (1 μM, 12 h) following fluorescence-activated cell sorting assay. *P < 0.05; **P < 0.01; ***P < 0.001.
We next examined whether the increase of GATA4 acetylation requires deacetylase activity of SIRT6. Nicotinamide (NAM) binds to a conserved pocket adjacent to that of NAD+ to block deacetylase and mono-ADP-ribosylase activities of SIRT6 (41–43). We observed that NAM abrogated sirtuin-mediated H3K9 deacetylation but was unable to inhibit the increased level of GATA4 acetylation induced by SIRT6 overexpression (Figure 3B). Further, the catalytically inactive SIRT6 mutants, D63Y and D116N (26), remained capable of binding to and enhancing GATA4 acetylation (Figure 3C). More importantly, the overexpression of SIRT6 WT, D63Y and D116N restored Bcl-2 and Bcl-xL expression in Sirt6-deficient MEFs (Figure 3D and Supplemental Figure S2C), and significantly inhibited apoptosis in H9C2 cells exposed to DOX (Figure 3E, F). These data thus implicate that SIRT6 promotes GATA4 acetylation and thus transcription activity independent of its deacetylase activity.
TIP60 is required for SIRT6-induced GATA4 acetylation
The data that SIRT6 interacts with GATA4 and enhances its acetylation level suggests that additional factors, most likely histone acetyltransferase (HAT), are involved. To determine which HAT is responsible for enhanced GATA4 acetylation, we examined the effects of SIRT6-regulated GATA4 acetylation upon exposure to a series of HAT inhibitors, including Cpth2 to inhibit Gcn5 (44), Mg149 to inhibit TIP60 (45) and C646 to inhibit p300 (46,47). Interestingly, while H3K56ac levels were further reduced by the treatment of all HAT inhibitors, only Mg149 treatment significantly inhibited the increased acetylation of GATA4 induced by SIRT6 (Figure 4A). Specific Tip60 deletion in mouse hearts shortens lifespan in mice, accompanied with increased myocyte density and apoptosis (48). Depletion of Tip60–p400 HAT complex severely affected Gata4-regulated primitive endoderm differentiation in ESCs (49). However, whether GATA4 is a direct target of TIP60 remains unknown. We found that TIP60 overexpression increased GATA4 acetylation level, and that this effect was eliminated upon application of Mg149 (Supplemental Figure S3A, B). TIP60 KD by siRNA also markedly decreased GATA4 acetylation levels (Supplemental Figure S3C). These results indicate that TIP60 is a novel acetyltransferase of GATA4.
Figure 4.

SIRT6 recruits TIP60 for GATA4 acetylation at Lysine 328/330. (A) Immunoblots of anti-HA immunoprecipitates showing the acetylation level of HA-GATA4 in HEK293 cells overexpressing ectopic HA-GATA4 treated with indicated histone acetyltransferase (HAT) inhibitors, i.e. Cpth2 (20 μM), Mg149 (20 μM) and C646 (10 μM), for 6 h. Lower, fold change of the acetylation level of HA-GATA4 relative to HA-GATA4 only control, determined by ImageJ. n = 3. ***P < 0.001. ‘ns’ indicates no significance. (B) Immunoblots of anti-HA immunoprecipitates showing the acetylation level of HA-GATA4 in HEK293 cells overexpressing ectopic HA-GATA4 together with FLAG-TIP60 or empty vector HA-GATA4 proteins were purified from si-NC- or si-SIRT6-treated HEK293 cells co-expressing HA-GATA4. The acetylation status of GATA4 was determined by western blotting with anti-AcK antibodies. Right, fold change of the acetylation level of HA-GATA4 relative to HA-GATA4 only control, determined by ImageJ. n = 4. ***P < 0.001. (C) Western blotting showing the levels of endogenous Tip60 and Sirt6 in anti-Gata4 IPs from primary mouse cardiomyocytes. IgG serves as an antibody control. (D) FLAG-TIP60 was co-expressed with or without HA-GATA4 in wild-type (WT) or SIRT6 knockout (KO) HEK293 cells. Co-IP analysis of HA-GATA4 was performed and the levels of FLAG-TIP60 and GATA4 acetylation were assessed. Right, fold change of FLAG-TIP60 protein bound to HA-GATA4 relative to WT HEK293 cell control, determined by ImageJ. n = 4. **P < 0.01. (E) Truncated HA-GATA4 constructs were co-transfected with or without FLAG-SIRT6. Western blotting was performed with the indicated antibodies. Lower, fold change of HA-GATA4 acetylation level relative to HA-GATA4 (F-L) only control, determined by ImageJ. n = 3. ***P < 0.001. ‘ns’ indicates no significance. (F) Protein sequence alignment of C-terminal Zn-finger domain of human GATA4 and its orthologues in mouse, rat and Xenopus. All K residues within this region targeted for acetylation were highlighted. (G) The constructs of WT and site-mutated (K328/330R) HA-GATA4 were co-expressing with FLAG-TIP60 in HEK293 cells. The GATA4 acetylation levels were determined. Right, fold change of HA-GATA4 acetylation level relative to HA-GATA4 WT only control, determined by ImageJ. n = 4. ***P < 0.001. ‘ns’ indicates no significance. (H) Western blotting showing the levels of HA-SIRT6 protein and TIP60 acetylation in HEK293 cells co-transfected with FLAG-TIP60 and HA-SIRT6 or empty vector. Right, fold change of FLAG-TIP60 acetylation level relative to FLAG-TIP60 only control, determined by ImageJ. n = 3, ‘ns’ indicates no significance.
To confirm the role of TIP60 in SIRT6-enhanced GATA4 acetylation, we examined the acetylation level of GATA4 in TIP60 KD cells with SIRT6 overexpression. We found that the SIRT6-enhanced acetylation of GATA4 was significantly abolished in TIP60 KD cells (Supplemental Figure S3D). In the other way around, overexpression of TIP60 in SIRT6-depleted cells was unable to fully acetylate GATA4 (Figure 4B). Importantly, endogenous Sirt6 and Tip60 were present in the anti-Gata4 immunoprecipitates from primary mouse cardiomyocytes (Figure 4C), implying that these proteins form a trimeric complex. Moreover, low level of GATA4 acetylation in SIRT6 KO cells was associated with an undetectable interaction between TIP60 and GATA4 (Figure 4D). These data suggest that SIRT6 acts as a scaffold to facilitate TIP60-mediated GATA4 acetylation.
TIP60 acetylates GATA4 at K328/330
The C-terminal Zn-finger of GATA4 is often acetylated (36), which is, as shown above, the binding site for SIRT6. Deletion of the C-terminal Zn-finger domain eliminated both acetylation and chromatin association of GATA4, as determined by anti-AcK and anti-histone H3 immunoblotting in the anti GATA4 immunoprecipitates and the chromatin fractionation assay (Figure 4E and Supplemental Figure S3E). There are six evolutionally conserved lysine (K) residues within this region (Figure 4F). It has been reported that p300 acetylates GATA4 on K313/320/322/324 and thus enhances the DNA-binding activity, resulting in stress-induced cardiac hypertrophy (36,50). However, by K-to-R mutation, we noticed that K313/320/322/324 were less likely acetylated by TIP60. Instead, the K328/330R mutant (2KR) almost totally abolished the increased GATA4 acetylation induced by either ectopic SIRT6 or TIP60 (Figure 4G and Supplemental Figure S3F). Together, these data suggest that TIP60 mediates GATA4 acetylation at K328/330.
Auto-acetylation activates TIP60, which is negatively regulated by SIRT1 (51). We thus examined whether SIRT6 modulates the acetylation status of TIP60. As shown, SIRT6 overexpression had marginal effect on the acetylation level of TIP60 (Figure 4H). Further Co-IP analysis showed that both WT and enzyme-dead SIRT6 bound to TIP60 (Supplemental Figure S3G). We also examined the MAR levels of TIP60 when SIRT6 was overexpressed, but hardly observed TIP60 MAR, suggesting that TIP60 is less likely MARed (Supplemental Figure S3H). The data support a deacetylase-independent and structural role of SIRT6 in GATA4 acetylation.
GATA4 acetylation at K328/330 is critical for DOX resistance
Restoring GATA4 transcriptional activity has been highly recognized as a promising approach to prevent DOX-induced heart failure (7,12). We next investigated the cardioprotective role of TIP60-mediated GATA4 acetylation. We co-expressed HA-GATA4 and FLAG-SIRT6, and then treated the cells with DOX. Significantly, the DOX treatment disrupted the interaction of GATA4 and SIRT6, and simultaneously reduced the levels of GATA4 acetylation in a dose-dependent manner (Figure 5A). In primary cardiomyocyte, DOX treatment dramatically reduced the Bcl-2 expression, which was largely rescued by overexpression of GATA4 WT and K328/330Q (2KQ), but not 2KR (Figure 5B). Consequently, overexpression of GATA4 WT or 2KQ was able to alleviate DOX-induced myocyte apoptosis as determined by TUNEL assay, but 2KR mutant failed (Figure 5C). We further generated H9C2 cell lines stably overexpressing GATA4 WT, 2KR mutant or 2KQ mutant (Supplemental Figure S4A). While the ectopic GATA4 WT and 2KQ mutant significantly attenuated the decline of Bcl-2 and Bcl-xL levels in H9C2 cells exposed to DOX, the 2KR mutant had little effect (Figure 5D and Supplemental Figure S4B). Consistently, ChIP data showed that the 2KR, but not the 2KQ mutant, compromised the DNA-binding capacity of GATA4, either in presence or absence of DOX (Figure 5E). GATA4 overexpression alleviates DOX-induced cardiomyocyte apoptosis and resultant myocardial atrophy; however, this benefit was abrogated by the 2KR mutation (Figure 5F, G and Supplemental Figure S4C, D). Further colony formation assay demonstrated that GATA4 2KQ mutant promoted myocyte survival to a similar extent as WT in response to DOX, whereas the 2KR mutant failed to do so (Figure 5H). These data support the notion that TIP60-mediated acetylation is critical for the cardioprotective role of GATA4.
Figure 5.

Hypoacetylation of GATA4 diminishes cardio-protection against DOX. (A) Western blotting analysis of the levels of FLAG-SIRT6 protein and HA-GATA4 acetylation in the precipitated pool of HA-GATA4 with DOX treatment at 0, 1, 2 μM for 6 h (left). Percent level of acetylated GATA4 relative to vehicle/no FLAG-SIRT6 control (middle) and percent FLAG-SIRT6 bound to HA-GATA4 relative to vehicle control (right). Quantification was performed by ImageJ. n = 3. *P < 0.05. **P < 0.01. (B) qPCR analysis of Bcl-2 mRNA levels in primary mouse neonatal cardiomyocytes ectopically expressing GATA4 WT, K328/330R (2KR) and K328/330Q (2KQ) upon exposure to DOX (1 μM, 6 h). ***P < 0.001, ‘ns’ represents no significance. (C) Percent TUNEL positively stained primary cardiomyocytes after treatment with DOX (1 μM) for 24 h. **P < 0.01, ***P < 0.001, ‘ns’ indicates no significance. (D) H9C2 cells stably expressing GATA4 WT, K328/330R (2KR) and K328/330Q (2KQ) were exposed to DOX (1 μM, 6 h). The mRNA transcripts of Bcl-2 were evaluated by qPCR. **P < 0.01, ‘ns’ represents no significance. 18srRNA was used as a reference gene. (E) ChIP analysis was performed in the stably transfected H9C2 cells by using anti-GATA4 antibody in the presence/absence of DOX. GATA4 enrichment on the Bcl-2 promoter region was measured by qPCR. ***P < 0.001, **P < 0.01, ‘ns’ represents no significance. (F, G) Stably transfected H9C2 cells were exposed to DOX (1 μM) for 24 h and stained with TUNEL or TRITC-phalloidin. Images of the stained cells were captured. The quantification of the number of positively TUNEL stained H9C2 cells (F). Cell area was measured and quantified (G). Scale bar, 20 μm. ***P < 0.001, ‘ns’ represents no significance. (H) Cell colony formation showing the survival fraction of H9C2 cells after the indicated doses of DOX for 1 h. The plates (n = 3/group) were stained with crystal violet and the colony numbers were counted. **P < 0.01, ***P < 0.001, ‘ns’ represents no significance.
The SIRT6–TIP60–GATA4 axis epigenetically ensures gene expression
SIRT6 and TIP60 are the main HDAC and HAT for H3K9 and/or H3K56 acetylation, which dictates open chromatin structure to facilitate transcription (52,53). We therefore asked whether/how the SIRT6–TIP60–GATA4 axis integrate chromatin remodeling to gene transcription. Again, using the promoter of Bcl2 as an example, the ChIP data showed that TIP60 was highly enriched on the P1 region, which was significantly dampened when SIRT6 was knocked down (Figure 6A). Consistently, overexpression of TIP60 significantly enhanced the level of H3K9ac at P1 (Figure 6B), which was diminished by SIRT6 depletion (Figure 6C). Interestingly, despite the global levels of H3K9ac and H3K56ac were remarkably decreased upon ectopically expressed SIRT6 (Supplemental Figure S5A), ChIP assay showed that the enrichment of H3K9ac and H3K56ac at the P1 motif was unchanged (Figure 6C). Likewise, SIRT6 knockdown (KD) unaffected the enrichment of H3K9ac at the P1 motif (Supplemental Figure S5B). By contrast, GATA4 KD significantly downregulated the level of H3K9ac at the promoter locus without affecting SIRT6 and TIP60 enrichment (Figure 6D–F).
Figure 6.

The SIRT6–TIP60–GATA4 axis facilitates chromatin accessibility. (A, B) ChIP assay was performed using anti-TIP60 or anti-H3K9ac antibody in WT and SIRT6 KO HEK293 cells with or without overexpression of FLAG-TIP60. The enrichment of TIP60 and H3K9ac on Bcl-2 promoter was analyzed by qPCR. **P < 0.01, ***P < 0.001. (C) ChIP analysis was performed using anti-H3K9ac and H3K56ac antibodies in HEK293 cells with or without FLAG-SIRT6 overexpression, showing enrichment of H3K9ac and H3K56ac on the P1 motif of the Bcl-2 promoter region. ‘ns’ indicates no significance. (D–F) ChIP analysis was performed to test the enrichment of H3K9ac, SIRT6 and TIP60 on the Bcl-2 promoter region by using anti-H3K9ac and antibodies in HEK293 cells with si-NC or si-GATA4. *P < 0.05. ‘ns’ indicates no significance. (G) Co-IP using anti-FLAG agarose was performed in HEK293 cells expressing HA-GATA4 alone, or with wild-type FLAG-SIRT6 or the FLAG-SIRT6 H133Y mutant. HA-GATA4 levels were analyzed in FLAG-SIRT6 precipitates by western blotting. (H) Deacetylation activity of recombinant SIRT6 after pre-incubation with or without purified His-GATA4 in an in vitro assay. ***P < 0.001. (I) Deacetylation activity of recombinant SIRT6 WT and SIRT6 H133Y after pre-incubation with indicated doses of G4-13 peptides (0, 0.2, 2, 20, 200 μM) in an in vitro assay. ***P < 0.001. ‘ns’ indicates no significance.
We noticed that the enzyme-dead mutant H133Y completely abolished the interaction between SIRT6 and GATA4 (Figure 6G), and the ability against DOX-induced cardiomyocyte apoptosis (Supplemental Figure S5C). We therefore reasoned that the association of GATA4 might suppress deacetylase activity of SIRT6 on the target promoter. To gain more experimental evidences, we performed in vitro deacetylation assay and found that purified His-tagged GATA4 significantly blocked deacetylase activity of recombinant SIRT6 (Figure 6H). Considering that the SIRT6/TIP60 complex mediates GATA4 acetylation on K328/330 and the C-terminal Zn-finger of GATA4 is essential for SIRT6 binding, we synthesized two peptides in this region (a.a. 295–331.) of GATA4: ACGLYMKLHGVPRPLAMRKEGIQT (a.a. 295–318) termed G4-24 and RKRKPKNLNKSKT (a.a. 319–331) termed G4-13. The in vitro deacetylation assay showed that the G4-13, but not G4-24, dramatically inhibited the deacetylase activity of SIRT6 (14 μM for 50% inhibition) (Figure 6I and Supplemental Figure S5D).
Considering that H133 site is also required for the nucleosome-association of SIRT6 (54) and together with our findings, we proposed that SIRT6 occupies the promoter region via a general nucleosome-binding property; SIRT6 recruits TIP60, globally or at specific gene promoter, to balance dynamic acetylation of histones; when GATA4 is recruited and blocks SIRT6 deacetylase activity, TIP60 acetylates GATA4 to enhance its transcription activity, and to acetylate histones, e.g. H3K9ac, thus ensuring an open chromatin structure for transcription (Figure 7).
Figure 7.
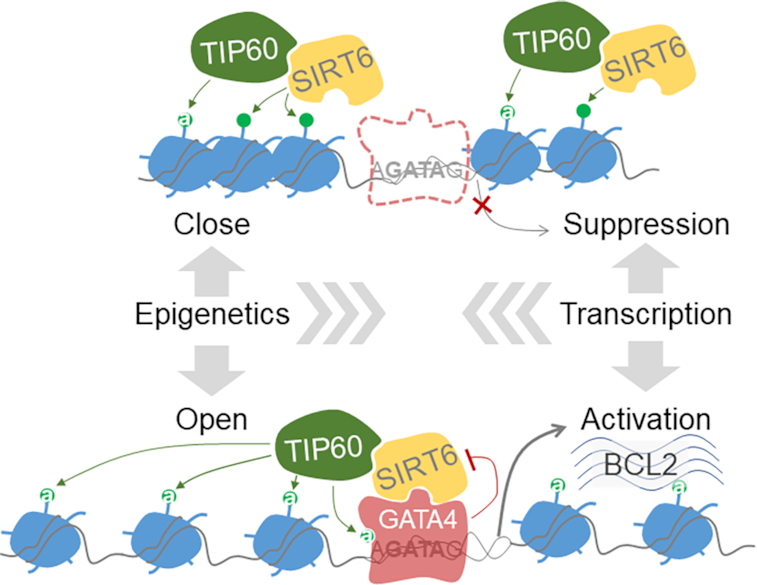
A schematic model: a SIRT6–TIP60–GATA4 axis couples gene transcription and epigenetic activation. SIRT6 initially occupies the promoter region via a nucleosome-binding property and recruits TIP60 to balance dynamic acetylation of local histones. When GATA4 comes by recognizing the GATA sequence and simultaneously interacts and blocks SIRT6 deacetylase activity, TIP60 acetylates GATA4 to enhance its transcription activity. The inhibition of SIRT6 disrupts local histone acetylation balance, thus ensuring an open chromatin structure for transcription.
DISCUSSION
The clinical use of DOX against malignancies is limited due to severe adverse effect—cardiotoxicity. Multiple mechanisms are involved in the DOX-induced heart failure, including oxidative stress, DNA damage and apoptosis (55,56). In response to DOX, GATA4 is downregulated and fails to promote cardiomyocyte survival (3,7). Here, we revealed that dynamic GATA4 acetylation, which is independent of p300 (36,50), is required for its transactivation against DOX-induced cardiotoxicity. SIRT6 acts as a scaffold to recruit TIP60, which acetylates GATA4 to enhance transcriptional activity. SIRT6/TIP60-regulated GATA4 acetylation might be considered as potential targets to prevent DOX cardiotoxicity.
The regional epigenetic regulation is a fundamental mechanism that modulates gene transcription. SIRT6 is well-recognized as a transcriptional repressor, which catalyzes histone deacetylation using NAD+ as a coenzyme or substrate (57,58). The D63Y and D116N mutations disrupt the affinity of SIRT6 for NAD+ (59), thus eliminating SIRT6 deacetylase activity. NAM binds to a conserved pocket adjacent to that of NAD+ to inhibit SIRT6 deacetylase (43). Here, we found that neither NAM, D63Y nor D116N disrupts the SIRT6–GATA4 interaction or inhibit the increase of GATA4 acetylation. Interestingly, the level of H3K9ac remains unaffected at promoter locus upon SIRT6 overexpression. These findings raise a question: how SIRT6 deacetylase activity is silenced at local chromatin? We noticed that SIRT6 H133Y loses the ability to interact with GATA4. GATA4 and synthesized peptide inhibit SIRT6 deacetylase activity in vivo and in vitro. Supporting our findings, SIRT6 regulates the mTOR signaling by repressing the transcriptional activity of Sp1, independent of its deacetylase activity (60). SIRT1 enhances glucocorticoid receptor transcription independently of its deacetylase activity (61). We propose a model of the SIRT6–TIP60–GATA4 axis, which couples the epigenetic activation and transcription (Figure 7). However, it is unclear how SIRT6 and TIP60 cooperate to maintain the balance of histone acetylation in the absence of GATA4. A future study investigating this model in the context of other HDACs, HATs and transcription factors is also warranted.
Structural studies of mouse GATA3 C-finger suggest a dimerizing property of the GATA in the DNA binding mechanisms (62). Compared to the cells expressing empty vector, cardiomyocytes expressing GATA 2KR mutant exhibit little difference in Bcl-2 expression and TUNEL-staining upon DOX treatment, suggesting that the 2KR loses chromatin binding capacity without interfering the function of endogenous GATA4. This finding suggests that either dimerization of GATA4 is not required for the transcription activity, which is different from GATA3, or the 2KR mutation loses the capacity of dimerization with another 2KR or with WT GATA4. These hypotheses require more evidences to support in future study.
TIP60 belongs to the MYST family of acetyltransferases. Heart-specific depletion of Tip60 increases myocyte density and apoptosis and shortens lifespan in mice (48); however, the underlying mechanism remains to be fully elucidated. Here, we identify TIP60 as a novel acetyltransferase for GATA4 acetylation on K328/330 and protects against stress-induced heart failure. The data provide an explanation by which TIP60 maintains heart homeostasis and a potential target for the prevention of chemotherapy side effects.
In summary, we identify a novel Sirt6–Tip60–Gata4 axis that regulates anti-apoptotic pathway and thus prevent DOX cardiotoxicity. Targeting this axis might constitute a new approach to improve the safety and efficacy of DOX chemotherapy. The data highlight a cooperative and fine-tuned action of HAT, HDAC and transcription factor in the regulation of gene transcription.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Jessica Tamanini (Shenzhen University and ETediting) for editing the manuscript prior to submission.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [91849208, 91949124, 81871114, 81571374, 81972602, 81702909]; National Key R&D Program of China [2017YFA0503900]; Science and Technology Program of Guangdong Province in China [2014A030308011, 2017B030301016, 2019A1515010472, 2019B030301009]; Shenzhen government [ZDSYS20190902093401689, KQJSCX20180328093403969, JCYJ20180507182044945]. Funding for open access charge: National Key R&D Program of China [2017YFA0503900].
Conflict of interest statement. None declared.
REFERENCES
- 1. Volkova M., Russell R. 3rd. Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011; 7:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferrans V.J., Clark J.R., Zhang J., Yu Z.X., Herman E.H.. Pathogenesis and prevention of doxorubicin cardiomyopathy. Tsitologiia. 1997; 39:928–937. [PubMed] [Google Scholar]
- 3. Aries A., Paradis P., Lefebvre C., Schwartz R.J., Nemer M.. Essential role of GATA-4 in cell survival and drug-induced cardiotoxicity. Proc. Natl Acad. Sci. U.S.A. 2004; 101:6975–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patient R.K., McGhee J.D.. The GATA family (vertebrates and invertebrates). Curr. Opin. Genet. Dev. 2002; 12:416–422. [DOI] [PubMed] [Google Scholar]
- 5. Grepin C., Nemer G., Nemer M.. Enhanced cardiogenesis in embryonic stem cells overexpressing the GATA-4 transcription factor. Development. 1997; 124:2387–2395. [DOI] [PubMed] [Google Scholar]
- 6. Charron F., Paradis P., Bronchain O., Nemer G., Nemer M.. Cooperative interaction between GATA-4 and GATA-6 regulates myocardial gene expression. Mol. Cell. Biol. 1999; 19:4355–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kobayashi S., Volden P., Timm D., Mao K., Xu X., Liang Q.. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010; 285:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kobayashi S., Lackey T., Huang Y., Bisping E., Pu W.T., Boxer L.M., Liang Q.. Transcription factor gata4 regulates cardiac BCL2 gene expression in vitro and in vivo. FASEB J. 2006; 20:800–802. [DOI] [PubMed] [Google Scholar]
- 9. Maharsy W., Aries A., Mansour O., Komati H., Nemer M.. Ageing is a risk factor in imatinib mesylate cardiotoxicity. Eur. J. Heart Fail. 2014; 16:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aries A., Whitcomb J., Shao W., Komati H., Saleh M., Nemer M.. Caspase-1 cleavage of transcription factor GATA4 and regulation of cardiac cell fate. Cell Death Dis. 2014; 5:e1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park A.M., Nagase H., Liu L., Vinod Kumar S., Szwergold N., Wong C.M., Suzuki Y.J.. Mechanism of anthracycline-mediated down-regulation of GATA4 in the heart. Cardiovasc. Res. 2011; 90:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneider J.G., Finck B.N., Ren J., Standley K.N., Takagi M., Maclean K.H., Bernal-Mizrachi C., Muslin A.J., Kastan M.B., Semenkovich C.F.. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006; 4:377–389. [DOI] [PubMed] [Google Scholar]
- 13. Liang Q., De Windt L.J., Witt S.A., Kimball T.R., Markham B.E., Molkentin J.D.. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J. Biol. Chem. 2001; 276:30245–30253. [DOI] [PubMed] [Google Scholar]
- 14. Kim Y., Ma A.G., Kitta K., Fitch S.N., Ikeda T., Ihara Y., Simon A.R., Evans T., Suzuki Y.J.. Anthracycline-induced suppression of GATA-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Mol. Pharmacol. 2003; 63:368–377. [DOI] [PubMed] [Google Scholar]
- 15. Kugel S., Mostoslavsky R.. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 2014; 39:72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tasselli L., Zheng W., Chua K.F.. SIRT6: novel mechanisms and links to aging and disease. Trends Endocrinol. Metab. 2017; 28:168–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haigis M.C., Guarente L.P.. Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006; 20:2913–2921. [DOI] [PubMed] [Google Scholar]
- 18. Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L., Barrett J.C. et al.. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008; 452:492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mostoslavsky R., Chua K.F., Lombard D.B., Pang W.W., Fischer M.R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M.M. et al.. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006; 124:315–329. [DOI] [PubMed] [Google Scholar]
- 20. Zhong L., D’Urso A., Toiber D., Sebastian C., Henry R.E., Vadysirisack D.D., Guimaraes A., Marinelli B., Wikstrom J.D., Nir T. et al.. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010; 140:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian M., Liu Z., Peng L., Tang X., Meng F., Ao Y., Zhou M., Wang M., Cao X., Qin B. et al.. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife. 2018; 7:e34836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu J., Sun D., Liu Z., Li M., Hong H., Liu C., Gao S., Li H., Cai Y., Chen S. et al.. SIRT6 suppresses isoproterenol-induced cardiac hypertrophy through activation of autophagy. Transl. Res. 2016; 172:96–112. [DOI] [PubMed] [Google Scholar]
- 23. Sundaresan N.R., Vasudevan P., Zhong L., Kim G., Samant S., Parekh V., Pillai V.B., Ravindra P.V., Gupta M., Jeevanandam V. et al.. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 2012; 18:1643–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cai Y., Yu S.S., Chen S.R., Pi R.B., Gao S., Li H., Ye J.T., Liu P.Q.. Nmnat2 protects cardiomyocytes from hypertrophy via activation of SIRT6. FEBS Lett. 2012; 586:866–874. [DOI] [PubMed] [Google Scholar]
- 25. Yu S.S., Cai Y., Ye J.T., Pi R.B., Chen S.R., Liu P.Q., Shen X.Y., Ji Y.. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. Br. J. Pharmacol. 2013; 168:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferrer C.M., Alders M., Postma A.V., Park S., Klein M.A., Cetinbas M., Pajkrt E., Glas A., van Koningsbruggen S., Christoffels V.M. et al.. An inactivating mutation in the histone deacetylase SIRT6 causes human perinatal lethality. Genes Dev. 2018; 32:373–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X.X., Wang X.L., Tong M.M., Gan L., Chen H., Wu S.S., Chen J.X., Li R.L., Wu Y., Zhang H.Y. et al.. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3alpha-dependent antioxidant defense mechanisms. Basic Res. Cardiol. 2016; 111:13. [DOI] [PubMed] [Google Scholar]
- 28. Wang S., Wang Y., Zhang Z., Liu Q., Gu J.. Cardioprotective effects of fibroblast growth factor 21 against doxorubicin-induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 2017; 8:e3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brito V.B., Nascimento L.V., Nunes R.B., Moura D.J., Lago P.D., Saffi J.. Exercise during pregnancy decreases doxorubicin-induced cardiotoxic effects on neonatal hearts. Toxicology. 2016; 368–369:46–57. [DOI] [PubMed] [Google Scholar]
- 30. Liu B., Wang J., Chan K.M., Tjia W.M., Deng W., Guan X., Huang J.D., Li K.M., Chau P.Y., Chen D.J. et al.. Genomic instability in laminopathy-based premature aging. Nat. Med. 2005; 11:780–785. [DOI] [PubMed] [Google Scholar]
- 31. Ehler E., Moore-Morris T., Lange S.. Isolation and culture of neonatal mouse cardiomyocytes. J. Vis. Exp. 2013; 79:e50154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qian M.X., Pang Y., Liu C.H., Haratake K., Du B.Y., Ji D.Y., Wang G.F., Zhu Q.Q., Song W., Yu Y. et al.. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell. 2013; 153:1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Etchegaray J.P., Zhong L., Li C., Henriques T., Ablondi E., Nakadai T., Van Rechem C., Ferrer C., Ross K.N., Choi J.E. et al.. The histone deacetylase SIRT6 restrains transcription elongation via promoter-proximal pausing. Mol. Cell. 2019; 75:683–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oda M., Kumaki Y., Shigeta M., Jakt L.M., Matsuoka C., Yamagiwa A., Niwa H., Okano M.. DNA methylation restricts lineage-specific functions of transcription factor Gata4 during embryonic stem cell differentiation. PLoS Genet. 2013; 9:e1003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arceci R.J., King A.A., Simon M.C., Orkin S.H., Wilson D.B.. Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol. Cell. Biol. 1993; 13:2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dai Y.S., Markham B.E.. p300 Functions as a coactivator of transcription factor GATA-4. J. Biol. Chem. 2001; 276:37178–37185. [DOI] [PubMed] [Google Scholar]
- 37. Pikkarainen S., Tokola H., Kerkela R., Ruskoaho H.. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004; 63:196–207. [DOI] [PubMed] [Google Scholar]
- 38. Liang Q., Wiese R.J., Bueno O.F., Dai Y.S., Markham B.E., Molkentin J.D.. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol. Cell. Biol. 2001; 21:7460–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tremblay J.J., Viger R.S.. Transcription factor GATA-4 is activated by phosphorylation of serine 261 via the cAMP/protein kinase a signaling pathway in gonadal cells. J. Biol. Chem. 2003; 278:22128–22135. [DOI] [PubMed] [Google Scholar]
- 40. Liszt G., Ford E., Kurtev M., Guarente L.. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005; 280:21313–21320. [DOI] [PubMed] [Google Scholar]
- 41. Jackson M.D., Schmidt M.T., Oppenheimer N.J., Denu J.M.. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003; 278:50985–50998. [DOI] [PubMed] [Google Scholar]
- 42. Yuan H., Marmorstein R.. Structural basis for sirtuin activity and inhibition. J. Biol. Chem. 2012; 287:42428–42435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bitterman K.J., Anderson R.M., Cohen H.Y., Latorre-Esteves M., Sinclair D.A.. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002; 277:45099–45107. [DOI] [PubMed] [Google Scholar]
- 44. Chimenti F., Bizzarri B., Maccioni E., Secci D., Bolasco A., Chimenti P., Fioravanti R., Granese A., Carradori S., Tosi F. et al.. A novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene-[4-(4′-chlorophenyl)thiazol-2-yl)hydrazone. J. Med. Chem. 2009; 52:530–536. [DOI] [PubMed] [Google Scholar]
- 45. Ghizzoni M., Wu J., Gao T., Haisma H.J., Dekker F.J., George Zheng Y.. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. Eur. J. Med. Chem. 2012; 47:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lasko L.M., Jakob C.G., Edalji R.P., Qiu W., Montgomery D., Digiammarino E.L., Hansen T.M., Risi R.M., Frey R., Manaves V. et al.. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017; 550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bowers E.M., Yan G., Mukherjee C., Orry A., Wang L., Holbert M.A., Crump N.T., Hazzalin C.A., Liszczak G., Yuan H. et al.. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 2010; 17:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fisher J.B., Horst A., Wan T., Kim M.S., Auchampach J., Lough J.. Depletion of Tip60 from in vivo cardiomyocytes increases myocyte density, followed by cardiac dysfunction, myocyte fallout and lethality. PLoS One. 2016; 11:e0164855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fazzio T.G., Huff J.T., Panning B.. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008; 134:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yanazume T., Hasegawa K., Morimoto T., Kawamura T., Wada H., Matsumori A., Kawase Y., Hirai M., Kita T.. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol. Cell. Biol. 2003; 23:3593–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang J., Chen J.. SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J. Biol. Chem. 2010; 285:11458–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kimura A., Horikoshi M.. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes Cells. 1998; 3:789–800. [DOI] [PubMed] [Google Scholar]
- 53. Karmodiya K., Krebs A.R., Oulad-Abdelghani M., Kimura H., Tora L.. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics. 2012; 13:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tennen R.I., Berber E., Chua K.F.. Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech. Ageing Dev. 2010; 131:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T.. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012; 18:1639–1642. [DOI] [PubMed] [Google Scholar]
- 56. Ueno M., Kakinuma Y., Yuhki K., Murakoshi N., Iemitsu M., Miyauchi T., Yamaguchi I.. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. J. Pharmacol. Sci. 2006; 101:151–158. [DOI] [PubMed] [Google Scholar]
- 57. Mao Z., Hine C., Tian X., Van Meter M., Au M., Vaidya A., Seluanov A., Gorbunova V.. SIRT6 promotes DNA repair under stress by activating PARP1. Science. 2011; 332:1443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang H., Khan S., Wang Y., Charron G., He B., Sebastian C., Du J., Kim R., Ge E., Mostoslavsky R. et al.. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013; 496:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kugel S., Feldman J.L., Klein M.A., Silberman D.M., Sebastian C., Mermel C., Dobersch S., Clark A.R., Getz G., Denu J.M. et al.. Identification of and molecular basis for SIRT6 Loss-of-Function point mutations in cancer. Cell Rep. 2015; 13:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ravi V., Jain A., Khan D., Ahamed F., Mishra S., Giri M., Inbaraj M., Krishna S., Sarikhani M., Maity S. et al.. SIRT6 transcriptionally regulates global protein synthesis through transcription factor Sp1 independent of its deacetylase activity. Nucleic Acids Res. 2019; 47:9115–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Suzuki S., Iben J.R., Coon S.L., Kino T.. SIRT1 is a transcriptional enhancer of the glucocorticoid receptor acting independently to its deacetylase activity. Mol. Cell. Endocrinol. 2018; 461:178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bates D.L., Chen Y., Kim G., Guo L., Chen L.. Crystal structures of multiple GATA zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by GATA. J. Mol. Biol. 2008; 381:1292–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.