


Abstract
The miRNA pathway has three segments—biogenesis, targeting and downstream regulatory effectors. We aimed to better understand their cellular control by exploring the miRNA-mRNA-targeting relationships. We first used human evolutionarily conserved sites. Strikingly, AGOs 1–3 are all among the top 14 mRNAs with the highest miRNA site counts, along with ANKRD52, the phosphatase regulatory subunit of the recently identified AGO phosphorylation cycle; and the AGO phosphorylation cycle mRNAs share much more than expected miRNA sites. The mRNAs for TNRC6, which acts with AGOs to channel miRNA-mediated regulatory actions onto specific mRNAs, are also heavily miRNA-targeted. In contrast, upstream miRNA biogenesis mRNAs are not, and neither are downstream regulatory effectors. In short, binding site enrichment in miRNA targeting machinery mRNAs, but neither upstream biogenesis nor downstream effector mRNAs, was observed, endowing a cellular capacity for intensive and specific feedback control of the targeting activity. The pattern was confirmed with experimentally determined miRNA-mRNA target relationships. Moreover, genetic experiments demonstrated cellular utilization of this capacity. Thus, we uncovered a capacity for intensive, and specific, feedback-regulation of miRNA targeting activity directly by miRNAs themselves, i.e. segment-specific feedback auto-regulation of miRNA pathway, complementing miRNAs pairing with transcription factors to form hybrid feedback-loop.
INTRODUCTION
MicroRNAs (miRNA), an evolutionarily conserved category of non-coding RNAs, are vital transcriptome regulators. Components and segments of the miRNA pathway—biogenesis, targeting and regulatory actions—are well understood. Aberrancies in the pathway are frequently associated with human diseases, such as cancer (1,2). However, how cells control the pathway is not clear yet, and the complexity of the miRNA–mRNA targeting relationship is a critical challenge.
Most miRNA biogenesis starts from RNA polymerase II production of pri-miRNA transcripts. The Drosha RNase III enzyme, with help from the RNA binding protein DGCR8, processes the pri-miRNA into pre-miRNAs, with one pri-miRNA producing up to 6 pre-miRNAs. Some pre-miRNAs are generated directly from mRNA introns and, thus, bypass the pri-miRNA and Drosha processing steps (3). The pre-miRNAs move, mainly via the Exportin-5 (XPO5) nucleocytoplasmic shuttle, out of nucleus into cytoplasm. The Dicer1 RNase III enzyme, in cooperation with the RNA binding protein TARBP2, processes the pre-miRNA into a mature 22-nucleotide long miRNA (4–6). The miRNA is then loaded onto the Argonaute (AGO) proteins to exert regulatory actions onto target mRNAs via base pairing between its seed sequence, which in human is only six to eight nucleotide long, and cognate binding sites.
The loaded AGOs, together with the p-body (processing body) scaffold protein TNRC6 (Trinucleotide Repeat Containing 6) they recruit, form the core of the miRNA targeting machinery, bridging upstream miRNA biogenesis to downstream regulatory effectors. The AGO PAZ domain binds to the 3′-end of loaded miRNA, and the PIWI domain to the 5′-end, orienting the miRNA to facilitate base pairing with target mRNAs. Meanwhile, loaded AGOs disassociate from TARBP2 and DICER1 (6), and recruit TNRC6A/B/C. The TNRC6s, in turn, recruit downstream effectors – general translation inhibition and/or mRNA destabilization machinery such as the CCR4-NOT and PAN2–PAN3 complexes. Thus, the AGOs, together with recruited TNRC6s, channel miRNA-mediated regulatory actions onto specific target mRNAs. Given such importance, it is not surprising that AGOs are regulated by many post-translational mechanisms (7). For instance, there are multiple phosphorylation sites and multiple cognate protein kinases (7–11).
Recently, Golden et al. discovered an AGO phosphorylation cycle (12), which was soon independently confirmed in both human cells and in Caenorhabditis elegans (13), revealing a new layer of regulation of miRNA targeting activity. Through iterative rounds of CRISPR/Cas9 library screening for regulators of miRNA pathway, they identified ANKRD52 and PPP6C—interacting components of protein phosphatase 6 (PPP6) complex—and cognate protein kinase CSNK1A1. Briefly, miRNA–mRNA binding triggers CSNK1A1 phosphorylation of AGO2 on multiple serine residues (S824–S834), which are evolutionarily conserved in all AGO proteins and are within a structurally unresolved loop of the PIWI domain near the miRNA–target interface (12,13). The phosphorylation disrupts AGO–miRNA binding to mRNAs (13). Meanwhile, PPP6 de-phosphorylates AGO2, presumably preparing it ready for the next target binding and phosphorylation cycle. Thus, the AGOs, ANKRD52, PPP6C and CSNK1A1 form a functional module within the miRNA targeting machinery (12,13).
The shortness of miRNA seed sequence leads to a complexity in the miRNA–mRNA target relationship. It enables individual miRNAs to target multiple, and sometimes a large number of, mRNAs; conversely, one mRNA can have binding sites for potentially a large number of unique miRNAs. Unfortunately, the shortness also means low signal-to-noise ratio in transcriptome-wide miRNA binding site identification efforts. Though somewhat mitigated by analyzing evolutionary conservation or combinatorial patterns of multiple miRNA binding sites (14–16), this technical difficulty is perhaps why the complexity remains largely under-appreciated. Current research focuses mostly on individual binding sites instead of the overall binding site distribution pattern, such as the study of miRNA functions in cell cycle regulation (17,18) and in cancers (1,2).
The miRNA binding site distribution is, however, likely a rich source for functional exploration. In transcription regulation, the notion of functionally related genes sharing common transcription factor (TF) binding sites has long been a fruitful assumption (19,20). It is conceivable to assume the same in miRNA-mediated transcriptome regulation. Moreover, TF binding site distribution often reveals feedback regulation. It can be a TF binding to its sites within its own genomic regulatory regions to feedback-control transcription (21). There are also numerous reports of a miRNA pairing with a TF in a feedback loop; the miRNA regulates the TF mRNA, and the latter regulates the miRNA gene's transcription (2,22,23). A computational study has shown enrichment of such loops in the human regulatory network (24). As for miRNA pathway itself, DICER1 is directly feedback-controlled by miR-103/107 and Let-7 in human (25,26). And, in C. elegans, the alg-1 AGO ortholog is directly regulated by mir-71 (27). However, it is unclear to which extent the pathway is directly feedback-controlled in this manner.
Thus, this study explored the distribution of evolutionarily conserved miRNA binding sites in the human transcriptome and, fortunately, shed new light onto cellular control of the miRNA pathway. Among heavily miRNA-targeted mRNAs, i.e. the mRNAs with highest predicted miRNA binding site counts, we detected significant enrichment of miRNA targeting machinery mRNAs, but neither upstream miRNA biogenesis nor downstream effector mRNAs. The AGO phosphorylation cycle mRNAs best exemplified the enrichment, and also share more than expected common miRNA binding sites. Thus, our analysis uncovered a cellular capacity for intensive and specific auto-feedback regulation of miRNA targeting activity by miRNAs themselves. Moreover, genetic experiments demonstrated cellular utilization of this capacity and strong repression of miRNA targeting activity mRNAs. The results also imply miRNA binding site distribution as a rich resource for further functional exploration.
MATERIALS AND METHODS
Evolutionarily conserved miRNA binding sites
To alleviate the high noise issue associated with computational miRNA binding site prediction, we restricted our analysis to evolutionarily conserved binding sites for conserved miRNA families in human and mouse. The set of sites and their cumulative weighted context++ scores were downloaded from the TargetScan database 7.2 in July 2018 (14). At the time of download, this was the most current version. For human, the dataset contains 120,702 evolutionarily conserved miRNA binding sites in the 3′-UTRs of 13,035 genes. We have previously used the dataset of TargetScan 7.1 (28), with which all observations of this study were originally made. During our updating to the new version, we noticed a severe shortening of the AGO2 3′-UTR to 895 base pairs in TargetScan 7.2, which is not consistent with the current AGO2 gene model. So, just for AGO2, we continued to use its information in TargetScan 7.1.
Wildtype and miRNA biogenesis deficient mutant RNA-seq datasets and analyses
We downloaded the wildtype and Dicer1−/− mouse embryonic stem cell Illumina HiSeq2000 RNA-seq dataset from the NCBI GEO database (accession number GSE55338). In the mutant Dicer1−/- cells, the exon coding for the majority of the second RNaseIII domain was removed. Consequently, the mutant cells express a mutant Dicer1 mRNA and a non-functional mutant Dicer1 protein with an internal in-frame 90-amino-acid deletion (29), and were nearly devoid of mature miRNAs (30). The dataset contains two wildtype, and three Dicer1−/− mutant, replicates. The paired-end sequencing reads were aligned to the mouse reference genome (mm10) with the STAR alignment software, followed by gene expression level determination with the HTSeq-count software. The counts were then converted to reads per kilobase of transcript, per million mapped reads (RPKM).
We also downloaded the wildtype and Drosha−/− HCT116 cell Illumina HiSeq2500 RNA-seq dataset from the NCBI GEO database (accession number GSE80258). The HCT116 cell has been a good choice for genomic-editing due to its near diploid. For this dataset, the Drosha−/− variant was generated by a TALEN-Cas9 induced frameshift mutation, resulting in non-functional Drosha protein and abolishment of canonical miRNA production (31). The sequencing reads in the comparative RNA-seq dataset were aligned to the human reference genome (hg38) with the STAR alignment software, followed by gene expression level determination with the HTSeq-count software. The counts were then converted to reads per kilobase of transcript, per million mapped reads (RPKM).
Public HCT116 cell miRNA-seq dataset and analysis
We downloaded the HCT116 miRNA-seq dataset from the NCBI GEO database (accession number GSE77989) (31). The dataset contains three replicates for wildtype HCT116 cells. The FASTQ sequences produced from an Illumina MiSeq sequencer were aligned to the human reference genome (hg38) by the Bowtie2 alignment software. By comparing with genomic coordinates of miRNAs (obtained from mirbase.org), miRNA reads were chosen and counted. The counts were then converted to read per million mapped reads (RPM), followed by average RPM calculation.
Polysome profiling analysis
Polysomes were isolated as previously described with the Biocomp Instruments Inc. gradient maker and fractionator equipment (32). Briefly, the cells were treated with 100 μg/ml cycloheximide for 15 min at 37°C, 5% CO2 and then washed twice with cold DPBS. The cells were lysed with lysis buffer (10 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.5% Triton X-100, 1× protease inhibitor cocktail (EDTA-free), 200 units/ml of RNase inhibitor). Collected cytoplasmic lysates were loaded on top of a 10–60% sucrose gradient, followed by centrifugation in a Beckman SW41 rotor at 390 000 g at 4°C for 2 h. The gradient was fractioned into 25 fractions. The heavy (10-mer or more) and light (2- to 9-mer) polysome fractions, identified based on the OD260 profile of the fractionation process, were collected for total RNA isolation.
The total RNA samples were processed for NGS sequencing analysis. The samples were first treated with DNase I to degrade any possible DNA contamination. Then the mRNA was enriched by using the oligo(dT) magnetic beads. The mRNA was fragmented into short fragments (∼200 bp). Then the first cDNA strand was synthesized by using random hexamer-primer. Buffer, dNTPs, RNase H and DNA polymerase I were then added to synthesize the second strand. The double stranded cDNA was purified with magnetic beads followed by end reparation and 3′-end single nucleotide A (adenine) addition. Finally, sequencing adaptors were ligated to the fragments, and the fragments were enriched by PCR amplification. Following qualify control and quantification, the libraries were analyzed on a BGI America DNBseq sequencer.
The raw sequencing reads were pre-processed to filter out low quality reads and to remove the multiplexing barcode sequences. The dataset has been deposited into the NCBI GEO database (access number GSE134818). In this study, the reads were analyzed in the same way as described above for downloaded public RNA-seq datasets.
Experimentally determined miRNA binding sites
We downloaded the miRTarBase release 7.0 (September 2017 release) human data from its websites in July 2018 (33,34). At the time of download, this was the most current version. In this study, only CLIP-seq generated data was used, in order to ensure the data was generated in a comprehensive and unbiased manner. The data was used as a list of experimentally determined miRNA–mRNA target relationship.
Computer software
The open source software package R (version 3.3) installed on a Mac Pro desktop computer was used for data analysis and plotting. For non-normal data, the Mann–Whitney–Wilcoxon tests were performed with the wilcox.test() method. For normal data, the t-tests were performed with the t.test() method. As for correlation analysis, the default Pearson correlation was used for normal data, and the Spearman correlation for non-normal data. For Loess regression, the loess() method was used.
Sliding window analysis
The mRNAs were sorted/ranked based on the specified metric. A window refers to a segment of the ranking, and the window size specifies the segment length, i.e. the number of mRNAs in a segment. The window slid from one end of the ranking, one position per step, to the other end of the ranking. At each step, the parameters of interest were calculated for the window.
RESULTS
The miRNA binding site distribution pattern
We recently studied miRNA binding site distribution in the human transcriptome (28). The distribution has long been known to be un-even; a small number of miRNAs target extra-ordinarily high numbers of mRNAs, and a small number of mRNAs contain extra-ordinarily high numbers of binding sites (35). Using evolutionally conserved miRNA–target relationships in the TargetScan database (14), we showed that the miRNA binding site distribution pattern can be described quantitatively by the so-called scale-free relationship (P(k) ∝ (K + a)−α, with P(K) as the number of mRNAs containing K unique binding sites; a and α positive constants) (28), which is commonly seen in many domains of biology such as regulatory networks and protein family size distribution (36–39). This is shown in Figure 1. The TargetScan 7.2 dataset identifies 13 035 mRNAs with at least one conserved 3′-UTR miRNA binding site. Overlapping sites were counted as one site in this study. A small number of mRNAs (24, <0.2%) are predicted to be extremely miRNA-targeted, each with 60 or more sites. The vast majority of the mRNAs have much fewer sites; >50% of the mRNAs (6926) have five or fewer sites (Figure 1).
Figure 1.
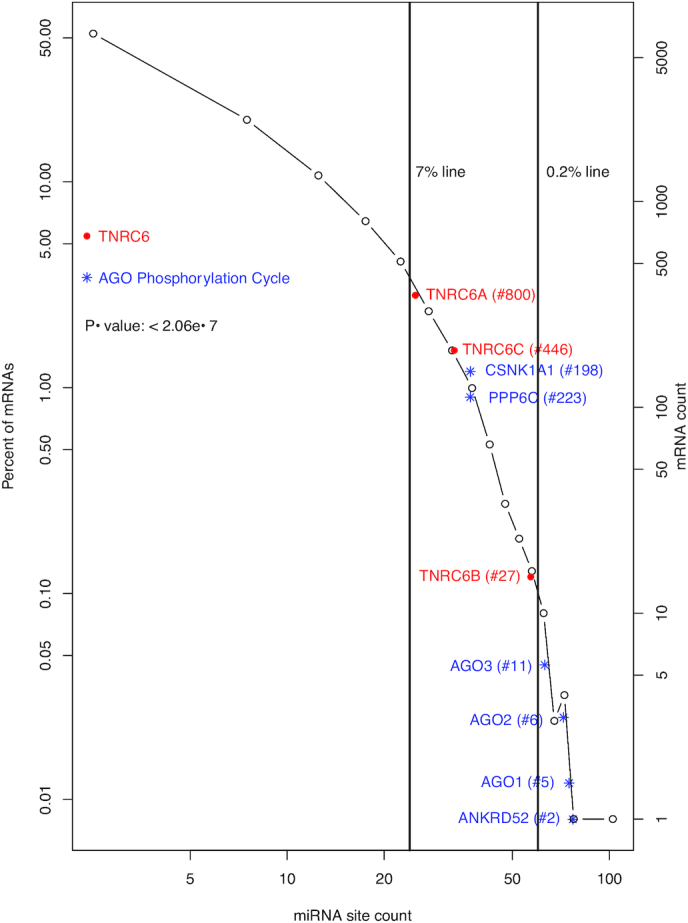
Distribution of miRNA binding sites in the human transcriptome and intensive targeting of AGO phosphorylation cycle and TNRC6 mRNAs by miRNAs. A histogram of human mRNAs based on their unique miRNA site counts is shown to illustrate the power-law relationship (P(K) ∝ (K+ a)−α). AGO phosphorylation cycle ( ) and the TNRC6 (
) and the TNRC6 ( ) mRNAs are plotted at their approximate positions to illustrate their high rankings, i.e. intensive targeting by miRNAs. The rankings are given inside the parentheses following gene symbols. The vertical lines denoting 0.2 and 7 percentile rankings are shown to further illustrate the high ranking; the 7-percentile ranking was chosen due to its proximity to the TNRC6A mRNA. The t-test P-value for comparing the whole transcriptome and the miRNA targeting machinery mRNAs (AGO phosphorylation cycle and TNRC6) is specified inside the plot.
) mRNAs are plotted at their approximate positions to illustrate their high rankings, i.e. intensive targeting by miRNAs. The rankings are given inside the parentheses following gene symbols. The vertical lines denoting 0.2 and 7 percentile rankings are shown to further illustrate the high ranking; the 7-percentile ranking was chosen due to its proximity to the TNRC6A mRNA. The t-test P-value for comparing the whole transcriptome and the miRNA targeting machinery mRNAs (AGO phosphorylation cycle and TNRC6) is specified inside the plot.
Herein, we explored two functional aspects of the distribution pattern: first, the cellular functions of the top predicted miRNA-targeted mRNAs; and second, the overall enhancement of cellular capacity for repression of the top mRNAs by the extreme enrichment of predicted binding sites and the utilization of this capacity for selected mRNA functional groups under specific experimental conditions. The latter is encouraged by the previously observed impact of miRNA site count on mRNA repression levels (40,41).
Enrichment of AGO phosphorylation cycle mRNAs among top miRNA-targeted mRNAs
We first explored the predicted top miRNA-targeted mRNAs. We speculated that, if a cellular function was controlled primarily by miRNAs, relevant mRNAs should be enriched among the top mRNAs, i.e. those with highest TargetScan conserved miRNA binding site counts. In the case of the miRNA pathway itself, such enrichment implies direct feedback control. Thus, the human mRNAs were ranked based on their binding site counts. We also used the histogram in Figure 1 for schematic interpretation of the site counts of individual mRNAs, i.e. how much a mRNA is predicted to be miRNA-targeted, relative to the whole human transcriptome.
The top 14 mRNAs with highest predicted miRNA binding site counts in their mRNA 3′-UTRs are listed in Table 1, section A. Consistent with previous analyses, TFs are in the list; for instance, the top ranked ZBTB20 is a zinc finger TF. This is not surprising since miRNAs, as discussed earlier, frequently pair with TFs to form feedback regulation loops. They also revealed an obvious enrichment of mRNAs for the AGO phosphorylation cycle proteins. Remarkably, AGO1, AGO2 and AGO3 are all in the list. AGO1 is the fifth ranked mRNA, AGO2 the 6th and AGO3 the 11th. Even more significantly, ANKRD52—the mRNA for regulatory subunit of the ANKRD52-PPP6C PPP6 phosphatase complex—is ranked the second. This enrichment of AGO1-3 and ANKRD52 mRNAs is illustrated schematically with blue symbol and text in Figure 1. They are all located to the right of the 0.2% line and, thus, ranked within the top 0.2%, This result suggests that the miRNA regulatory system likely targets the AGO phosphorylation cycle directly, constituting a major feedback auto-regulatory loop.
Table 1.
List of the top 14 mRNAs with highest counts of unique conserved 3′-UTR miRNA binding sites (A), other AGO phosphorylation cycle and the TNRC6 mRNAs (B) and miRNA biogenesis mRNAs (C). NCBI gene symbol, description, miRNA site count and descending rank are shown. The AGO phosphorylation cycle mRNAs are highlighted by bold font
| Gene symbol | Gene description | Site count | Rank | |
|---|---|---|---|---|
| A | ZBTB20 | Zinc finger and BTB domain containing 20 | 105 | 1 |
| ANKRD52 | Ankyrin repeat domain 52 | 77 | 2 | |
| NUFIP2 | FMR1 interacting protein 2 | 75 | 3–4 | |
| NFIB | Nuclear factor I B | 75 | 3–4 | |
| AGO1 | Argonaute 1 | 74 | 5 | |
| AGO2 | Argonaute 2 | 72 | 6 | |
| KLF7 | Kruppel like factor 7 | 68 | 7 | |
| LCOR | Ligand dependent nuclear receptor corepressor | 67 | 8–9 | |
| TAOK1 | TAO kinase 1 | 67 | 8–9 | |
| AAK1 | AP2 associated kinase 1 | 66 | 10 | |
| AGO3 | Argonaute 3 | 63 | 11–14 | |
| NFIA | Nuclear factor I A | 63 | 11–14 | |
| PURB | KH domain containing RNA binding | 63 | 11–14 | |
| CELF2 | CUGBP Elav-like family member 2 | 63 | 11–14 | |
| B | TNRC6B | Trinucleotide repeat containing 6B | 58 | 27 |
| CSNK1A1 | Casein kinase 1 alpha 1 | 39 | 198 | |
| PPP6C | Protein phosphatase 6 catalytic subunit | 38 | 223 | |
| TNRC6C | Trinucleotide repeat containing 6C | 31 | 446 | |
| AGO4 | Argonaute 4 | 27 | 670 | |
| TNRC6A | Trinucleotide repeat containing 6A | 25 | 800 | |
| C | DICER1 | Dicer 1, ribonuclease III | 25 | 800 |
| TARBP2 | TARBP2, RISC loading complex RNA binding subunit | 2 | ||
| XPO5 | Exportin 5 | 11 | 3208–3554 | |
| DGCR8 | DGCR8, microprocessor complex subunit | 9 | 3956–4391 | |
| DROSHA | Drosha ribonuclease III | 3 | 7739–8907 |
Next, we examined CSNK1A1 and PPP6C, the respective protein kinase and phosphatase in the AGO phosphorylation cycle. Consistent with our hypothesis, CSNK1A1 and PPP6C are both heavily miRNA-targeted, with 39 and 38 predicted miRNA binding sites, respectively. CSNK1A1 was ranked at the 198th, and PPP6C at the 223rd, both within the top 2% among the 13 035 genes with at least one conserved 3′-UTR miRNA binding sites. Their high ranks are shown schematically with blue symbol and text in Figure 1 as well. Thus, six AGO phosphorylation cycle mRNAs (AGO1–3, ANKRD52, CSNK1A1 and PPP6C) are likely heavily miRNA-targeted, all ranked within the top 2%. It is statistically significant, with a P-value of 1.3e–5 based on a Mann–Whitney–Wilcoxon test. Additionally, though not as heavily targeted as AGO1-3, the AGO4 mRNA has 27 unique 3′-UTR miRNA sites, and ranked at the 670th. Thus, the AGO phosphorylation cycle seems under direct feedback regulation by miRNAs.
The six mRNAs share significant numbers of common miRNA binding sites
It is usually assumed that functionally related genes share similarly gene expression regulation patterns and mechanisms, e.g. regulation by similar sets of transcription factors. In this case, we speculated that the six AGO phosphorylation cycle mRNAs share more common miRNA binding sites than expected by random chances.
This was indeed the case. The mRNAs for the six proteins share five common miRNA binding sites (miR-137, miR-30-5p, miR-302-3p/372-3p/373-3p/520-3p, miR-17-5p/20-5p/93-5p/106-5p/519-3p and miR-103-3p/107). This count was significant based on the following analysis. We randomly selected four mRNAs from the top 14 mRNAs in Table 1, one from the 25 genes with 39 miRNA binding sites and one from the 22 genes with 30 sites in their 3′-UTRs. We then counted common sites shared by mRNAs of the six randomly selected genes. In each computational experiment, this random-selection and calculation process was repeated 1000 times, followed by calculating the proportion of the 1000 attempts that gave a five or higher count. Repeated performance of this computational experiment never generated a proportion higher than 0.05. The proportion fluctuated in a tight range centered at 0.04, suggesting a P-value of 0.04. The significance is also illustrated by a box-plot in Figure 2. The majority of the randomly selected mRNA sets share no or just one common miRNA site.
Figure 2.
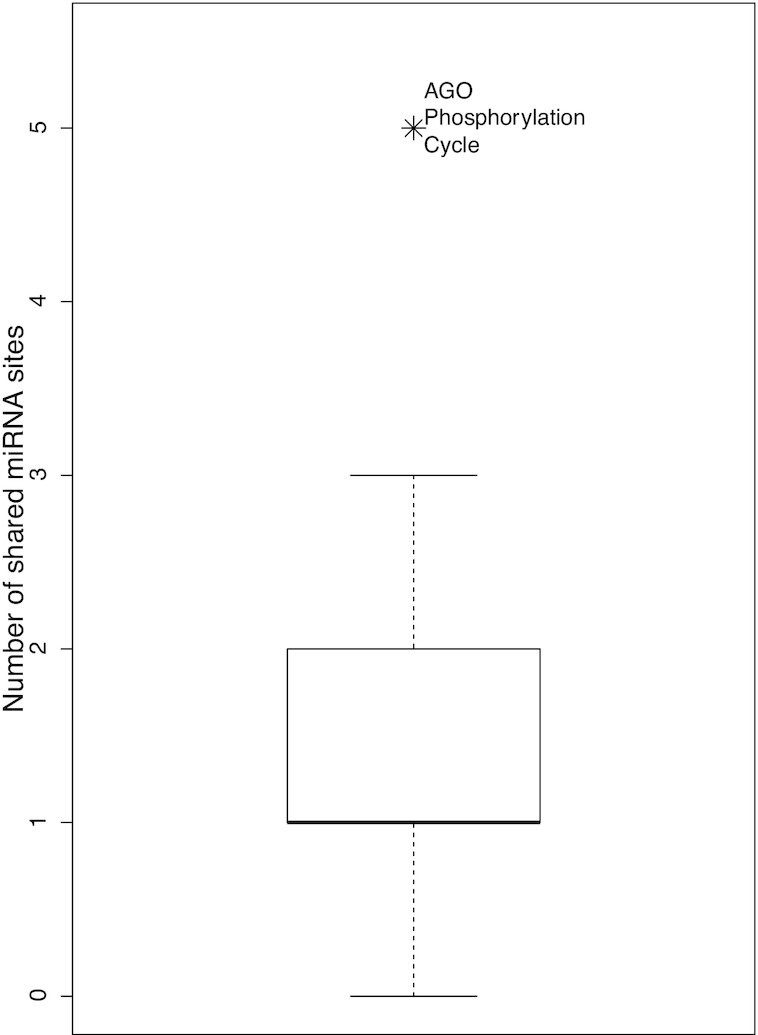
The six AGO phosphorylation cycle mRNAs share more than expected miRNA binding sites. A boxplot of numbers of shared miRNA sites of 1000 sets of randomly selected mRNAs (see text for details) is shown. A symbol is added to illustrate the significance of the observation that the six AGO phosphorylation cycle mRNAs share five sites.
All miRNA targeting machinery mRNAs are heavily targeted by miRNAs
As discussed in introduction, TNRC6s and AGOs act together to channel miRNA-mediated regulatory actions onto specific mRNAs; AGOs host and position miRNAs for target binding; TNRC6s bridge the loaded AGO proteins to downstream general regulatory effector proteins. Thus, we examined the three TNRC6 mRNAs. Indeed, the TNRC6B mRNA provided clear added evidence for intensive direct feedback regulation of miRNA targeting activity. It has 58 unique predicted 3′-UTR miRNA sites, and was ranked the 27th highest among the 13 035 genes. TNRC6A and TNRC6C were also targeted, with 25 and 31 conserved sites, respectively (Table 1). Moreover, the miRNA site counts of the TNRC6s and other mRNAs in section B of Table 1 should be interpreted in the context of the scale-free power-law distribution. In this distribution, the site count decreases precipitously along the rank, making the miRNA site counts of even highly ranked mRNAs seemingly less impressive. Thus, the high ranks of TNRC6s, and those of other mRNAs in section B of Table 1, are also shown schematically with red symbol and text in Figure 1. They are all ranked in the top 7% among all miRNA-targeted mRNAs.
Thus, in terms of the predicted miRNA binding site count, both AGO phosphorylation cycle and TNRC6 mRNAs support a cellular capacity for intensive auto-feedback regulation of miRNA targeting activity. Statistically, the enrichment has a P-value less than 2.06e–7. All of them, including AGO4, are ranked within the top 7% of miRNA–targeted mRNAs, all located to the right of the 7% line (Figure 1). More significantly, AGO1-3, ANKRD52 and TNRC6B are all among the top 30 mRNAs (Table 1, sections A and B). Interestingly, the TNRC6 mRNAs do not share the five common miRNAs targeting the AGO phosphorylation cycle mRNAs, suggesting differential regulation, i.e. by a different set of miRNAs.
Specificity of the binding site enrichment for miRNA targeting activity
We also examined other segments of the miRNA pathway – the upstream biogenesis and the downstream regulatory effectors. No enrichment of miRNA biogenesis mRNAs was detected among the heavily miRNA-targeted mRNAs. The miRNA biogenesis mRNAs (DGCR8, DROSHA, XPO5, TARBP2 and DICER1) are shown in Figure 3A, with red • symbol and text, in contrast to the miRNA targeting machinery mRNAs shown in blue * symbol. None of them had more than 25 miRNA sites, though it has been reported that DICER1 mRNA is directly regulated by miRNAs. Regulation of their expression might be dominated by indirect miRNA feedback control of cognate transcription factor mRNAs as well as other mechanisms (2,22,23,42–44). To some degree, DICER1, the last step of miRNA biogenesis before loading onto the AGO proteins, is a transition point. Its mRNA had 25 unique miRNA sites (ranked at the top 800th)—more than its upstream and partner proteins’ mRNAs (DROSHA, DGCR8, XPO5 and TARBP2) but less than miRNA targeting machinery mRNAs (Table 1).
Figure 3.

Lower levels of miRNA-targeting of upstream miRNA biogenesis and downstream regulatory effectors. The histogram in Figure 1 is used to show the contrast in the levels of targeting by miRNAs between two groups of mRNAs. The miRNA biogenesis () and miRNA targeting machinery () are shown in (A), and the downstream regulatory effectors () and miRNA targeting machinery () in (B). In (B), CNOTs 3 and 9–11 share one data point, as they have the same number of miRNA sites. The Mann–Whitney–Wilcoxon test P-values of the contrast between two groups of mRNAs are specified inside both plots.
Downstream of the targeting machinery, the general regulatory effector mRNAs also seem less miRNA-targeted. This is shown by the CNOTs (CNOT1-4, CNOT6, CNOT6 and CNOT7-11), PAN2 and PAN3 mRNAs schematically in Figure 3B. The CNOT and PAN mRNAs are shown with the red • symbol and text. Compared to the miRNA targeting machinery mRNAs (in blue * symbol), CNOT and PAN mRNAs have much lower miRNA binding site counts.
Thus, the intensive miRNA binding site enrichment seems specific for the mRNA targeting segment of the miRNA pathway. The specificity is supported by statistical analyses. The miRNA site counts of miRNA biogenesis mRNAs are not significantly different from the whole human transcriptome (P-value 0.33), and neither are those of the effector mRNAs (P-value 0.09). Both the miRNA biogenesis and the effector mRNAs have significantly lower miRNA binding site counts than the targeting machinery mRNAs, with P-values of 1.66e–3 and 2.71e–4, respectively (Figure 3A and B). The specificity for miRNA targeting activity is also illustrated schematically. In Figure 4, the miRNA pathway is divided into six steps—DROSHA processing (i), XPO5 nucleus export (ii), DICER1 processing (iii), the AGO phosphorylation cycle (iv), TNRC6 recruitment (v) and downstream regulatory effectors (vi). The human biogenesis mRNAs (steps i–iii) are shown with black symbols and text; the targeting step mRNAs (steps iv and v) in red symbols and text; and regulatory effector mRNAs (step vi), as exemplified by the CCR4–NOT and PAN2–PAN3 complexes, in blue symbol and text. The black line connects the median miRNA site count for each step, showing that overall miRNA site count increases from the first step of miRNA biogenesis (DROSHA) towards AGO phosphorylation cycle. Subsequently, it decreases towards the TNRC6s, and then towards downstream general effectors. Overall, the counts are higher in the targeting steps (steps 4–5) than in either the biogenesis steps or the downstream effector step (Figure 4A).
Figure 4.
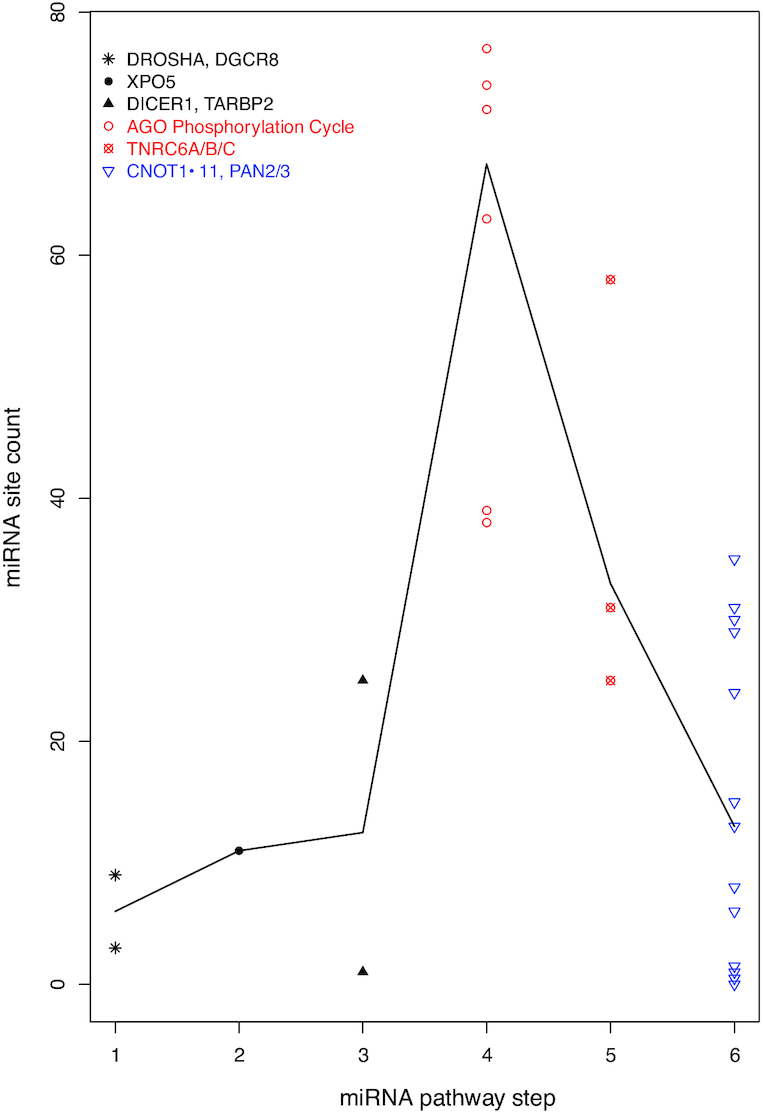
Specificity of auto-feedback regulation for the targeting segment of miRNA pathway. A scatter plot of mRNA miRNA binding site count is shown, with the mRNAs grouped into six steps of the miRNA pathway. The biogenesis steps are plotted in black, the targeting steps in red and the downstream effector in blue. The black line connects the median site counts of the six steps.
In short, our computational analysis of the miRNA binding site distribution uncovered a pattern of intensive enrichment of miRNA binding sites among the miRNA targeting activity mRNAs, but neither the upstream biogenesis nor the downstream regulatory effector mRNAs. Next, as mentioned at the beginning of Results section, we attempted to quantify the overall impact of the varying miRNA binding site counts on mRNA repression across the transcriptome, that is, to reveal the enhanced cellular capacity for miRNA-mediated repression of those mRNAs with the extremely high binding site counts. On the other hand, miRNA repression of target mRNAs is dynamically regulated; e.g. by the RNA binding proteins such as PUM1, HNRNPD/AUF1 and ELAVL1/HuR (45). For the miRNA targeting activity mRNAs, it became interesting to determine how much this capacity is utilized to enhance their repression under specific experimental conditions.
Functional confirmation of the auto-feedback regulation with genetic experimental results
Thus, we looked for opportunities to answer two questions: first, how much miRNA site enrichment correlates overall with higher levels of miRNA-mediated mRNA repression; and second, whether the cells utilize this capacity to auto-feedback-control miRNA targeting activity mRNAs. However, the miRNA binding site alone is not sufficient to answer the questions.
Confirmation in embryonic stem cells
Fortunately, as described in Materials and Methods, the results of a comparative RNA-seq analysis of the transcriptomes of wildtype and Dicer1 knockout mouse stem cells were publicly available (NCBI GEO database accession number GSE55338) (29,30,46), providing a perfect opportunity for functional assessment of miRNA regulatory actions and the potential auto-feedback regulation of miRNA targeting activity. To take advantage of this dataset, we downloaded the TargetScan mouse dataset and calculated counts of conserved miRNA binding sites for individual mRNAs. As expected, orthologous human and mouse mRNAs have highly similar miRNA binding site counts, with a spearman correlation coefficient of 0.92.
Then, we explored this dataset for answers to the two questions. First, the depletion of miRNAs in Dicer1−/− mutant cells relieved miRNA-mediated degradation, and thus should lead to higher expression levels, of miRNA–targeted mRNAs. How much individual mRNA expression levels increase upon Dicer1 knockout should reflect the degree to which the mRNAs are miRNA-regulated. This parameter can be used to answer the first question and to test our usage of miRNA site count as a metric of the potential for individual mRNAs to be miRNA-regulated. Thus, we calculated the log-ratio (log2(KO/WT)) of the expression levels in Dicer1−/− (KO) and wild type (WT) cells for each mRNA, and asked whether the log-ratios correlate with miRNA binding sites counts. This was indeed the case (Figure 5), despite the moderate nature of miRNA-mediated mRNA degradation enhancement, a myriad of non-miRNA-mediated regulatory mechanisms and various factors affecting the potency of miRNA regulatory actions such as relative expression levels of individual miRNA-mRNA pairs. The mRNAs were grouped based on the miRNA binding sites count, and the group mean log-ratios are plotted in Figure 5A versus the binding site count. The plot exhibits a clear positive correlation between the two parameters (with a Spearman correlation coefficient of 0.78 and a P-value smaller than 2.2e–16) (Figure 5A), thus validating miRNA site count as a metric of the potential for individual mRNAs to be miRNA-regulated. In the low binding site count ranges, where the mRNA groups are large, the correlation is nearly perfect. As the site count increases, due to the scale-free relationship (P(K) ∝ (K + a)−α), the mRNA groups become exponentially smaller and smaller; many groups containing just a few mRNAs, and some only one. The data points become, as expected statistically, more scattered. Yet, the positive relationship remains obvious.
Figure 5.

Overall positive correlation between miRNA site counts and the increases in mRNA expression levels in Dicer1−/− mutant cells (A–C) and the specificity of auto-feedback regulation for the targeting segment of miRNA pathway (D). (A) the mRNAs were grouped based on their binding site counts. The mean log-ratio (log2(KO/WT)) of the expression levels in Dicer1−/− (KO) and wild type (WT) cells was calculated for each group. A scatter plot of the mean log-ratios vs. the corresponding binding site counts is shown. Spearman correlation coefficient is also shown. For group sizes and the scale-free relationship, please see Figure 1. (B) a scatter plot of the log-ratio vs. miRNA binding site density. The miRNA binding site density is calculated as the log-ratio of binding site count to 3′UTR length. The mRNAs were sorted based this density. Then, a sliding-window analysis, with a window size of 100, was performed. The mean window log-ratio and miRNA site density were used to create the plot. The Pearson correlation and P-value are also shown. (C) a scatter plot of the log-ratios vs. relative miRNA binding site density. The relative miRNA binding site density is calculated as the residual of a loess regression (log2(binding site count) versus log2(3′UTR length)). The mRNAs were sorted based this density. Then, a sliding-window approach, with a window size of 100, was used to create the plot. The Pearson correlation and P-value are also shown. (D) a scatter plot of mRNA log2(KO/WT) values, with the mRNAs grouped into 6 steps of the miRNA pathway. As in Figure 4, the biogenesis steps are plotted in black, the targeting steps in red and the downstream effector in blue. The black line connects the mean log-ratio values of the 6 steps. The DICER1 mRNA is annotated with the * symbol to indicate its in-frame internal deletion mutation in the mutant stem cells, though the deleted 90-amino-acid-coding-region is only ∼3% of the full-length mRNA.
Additional analyses were done to ensure that the observed correlation was due to loss of miRNA expression instead of non-specific secondary effect upon Dicer deletion. We identified mRNAs with at least 1000-bp 3′-UTRs. This way, the mRNAs are likely under tight post-transcriptional—either miRNA- or non-miRNA-mediated—regulations. We then calculated the following two metrics of miRNA binding site enrichment in individual 3′-UTR sequences:
3′-UTR miRNA binding site density (log2(miRNA_Site_Count/3′-UTR_Length)). Using the sliding window analysis (see Materials and Methods), positive correlation between mRNA repression levels and this parameter was observed, with a correlation coefficient of 0.84 (Figure 5B).
Relative 3′-UTR miRNA binding site density. We performed a loess regression analysis of miRNA binding site count versus 3′-UTR length. The residue of the loess model is another measure of miRNA binding site enrichment; the larger the residue, the more miRNA binding site is enriched relative to the 3′-UTR length and embedded non-miRNA regulatory mechanisms. Once again, using the sliding window analysis, positive correlation between mRNA repression levels and this parameter was observed, with a correlation coefficient of 0.87 (Figure 5C).
The two metrics measure miRNA binding site enrichment relative to the 3′-UTR length and, thus, the amount of cognate signals embedded in the 3′-UTR sequences for non-miRNA-mediated regulatory actions. Their correlation with the levels of mRNA repression levels strongly suggests that the observed correlation is due to, collectively, the loss of miRNA expression instead of secondary effects. Thus, these results support the notion that miRNA site enrichment leads to enhanced capacity for miRNA-mediated repression at the cell's disposal. Dynamic usage of this capacity should confer the cells a post-transcriptional regulatory mechanism to control individual mRNAs with high miRNA site counts.
Second, in Figure 5D, the log-ratios of mRNAs for individual miRNA pathway steps are directly compared to illustrate the answer to the second question, with the mRNAs grouped and colored in the same manner as in Figure 4. The miRNA targeting activity mRNAs, i.e. the AGO phosphorylation cycle mRNAs and the TNRC6 mRNAs, have obviously higher log-ratios than both the miRNA biogenesis mRNAs and the down-stream effector mRNAs, with respective t-test P-values of 0.0085 and 0.0092, which is consistent with their higher binding site counts. Thus, in the case of these mRNAs, the enhanced capacity for miRNA-mediated repression was utilized. Variability of depression of miRNA targeting machinery mRNAs was also observed, perhaps resulting from dynamic usage of the enhanced capacity. For instance, some miRNA sites might be inaccessible due to blockage by other RNA binding proteins, and/or nonfunctional due to lack of expression of cognate miRNAs, under this particular experimental condition.
Interestingly, DROSHA and DGCR8 seemed upregulated in Dicer1−/− cells (Figure 5D), suggesting an active compensatory regulatory mechanism as part of cellular survival of the Dicer1 knockout. Whether this is miRNA-mediated became an interesting future research topic. Both DROSHA and DGCR8 have relatively low miRNA binding site counts (Table 1), but their sites have high efficacies. The DROSHA sites have TargetScan weighted context++ scores of –0.16 or better, and percentiles of 92 or better; the DGCR8 sites have a median score of –0.164 and a median percentile of 88. On the other hand, DGCR8 regulation by non-miRNA-mediated regulatory actions has also been reported (47).
Confirmation in the human HCT116 cells
Moreover, we performed functional assessment of the auto-feedback regulation in the HCT116 human cell line. This cell line has been used, perhaps due to its near diploid and thus suitability for genome editing, for at least two highly cited miRNAome studies (31,48). Therefore, the cells should have normal miRNA expression levels. Indeed, this is confirmed by the expression levels of the cognate miRNAs to the 5 miRNA binding sites shared by the AGO phosphorylation cycle mRNAs (Table 2). According to the miRbase database (49), miRNA stem loops for cognate miRNAs to 3 of the common sites (miR-17-5p/20-5p/93-5p/106-5p/519-3p, miR-103-3p/107 and miR-30-5p) are highly expressed, but those for the other two sites (miR-137 and miR-302-3p/372-3p/373-3p/520-3p) are lowly or not expressed, in the miRbase dataset collection (Table 2). Consistently, according to our analysis of the HCT116 miRNA-seq dataset in Kim et al. (31) that identified 1097 expressed miRNAs (see Materials and Methods), the cells express cognate miRNAs to the same 3 sites (miR-17-5p/20-5p/93-5p/106-5p/519-3p, miR-103-3p/107 and miR-30-5p) at very high levels (Table 2). For the miR-17-5p/20-5p/93-5p/106-5p/519-3p site, the cells express high levels of miR-17-5p (874.28 RPM), miR-20a-5p (554.38 RPM), miR-93-5p (4017.82 RPM) and miR-106b-5p (974.99 RPM); for the miR-103-3p/107 sites, the cells express high levels of miR-103a-3p (5902.59 RPM) and miR-107 (748.65); and for the miR-30-5p site, high levels of miR-30a-5p (292.2 RPM), miR-30b-5p (1057.2 RPM), miR-30c-5p (3217 RPM), miR-30d-5p (16185.82 RPM) and miR-30e-5p (15738.3 RPM). Except miR-30a-5p, all these cognate miRNAs are ranked among the top 100 highest expressed miRNAs, with miR-30d-5p and miR-30e-5p ranked among the top 20. Not surprisingly, the cognate miRNAs to the miR-137 and the miR-302-3p/372-3p/373-3p/520-3p sites are expressed at low levels (<1 RPM or not detected). Thus, the expression levels are consistent with the miRBase miRNA stem loop expression level information (Table 2) and provide experimental support for miRNA-feedback-regulation of the AGO phosphorylation cycle mRNAs.
Table 2.
Expression levels of cognate miRNAs and stem loops to the five conserved binding sites shared by AGO phosphorylation cycle mRNAs. The miRNA expression levels in the HCT116 cells were computed, as described in Materials and Methods, based on the miRNA-seq dataset in Kim et al. (31). The ranks are also listed for the miRNA ranked among the top-100 highest expressed miRNAs. The stem loop expression levels were collected from the miRbase website (www.mirbase.org). ND: Not Detected
| Cognate miRNAs | Cognate stem loops | ||||
|---|---|---|---|---|---|
| Sites | Name | Expression level (RPM) | Expression level ranking | Name | Expression level (RPM) per miRBase |
| miR-17-5p/20-5p/93-5p/106- 5p/519-3p | miR-17-5p | 874.28 | 78 | miR-17 | 9.52e3 |
| miR-20a-5p | 554.38 | 98 | miR20a | 7.48e3 | |
| miR-20b-5p | 0.188 | miR-20b | 545 | ||
| miR-93-5p | 4017.82 | 42 | miR-93 | 2e3 | |
| miR-106a-5p | ND | miR-106a | 7.94e3 | ||
| miR-106b-5p | 974.99 | 72 | miR-106b | 1.84e3 | |
| miR-519d-3p | ND | miR-519d | 3.23 | ||
| miR-30-5p | miR-30a-5p | 292.2 | miR-30a | 1.51e4 | |
| miR-30b-5p | 1057.2 | miR-30b | 2.25e3 | ||
| miR-30c-5p | 3217 | 45 | miR-30c-1 | 3.21e3 | |
| miR-30c-2 | 3.22e3 | ||||
| miR-30d-5p | 16185.82 | 17 | miR-30d | 1.17e4 | |
| miR-30e-5p | 15738.3 | 18 | miR-30e | 1.07e4 | |
| miR-103-3p/107 | miR-103a-3p | 5902.59 | 30 | miR-103a-1 | 1.24e4 |
| miR-103a-2 | 1.24e4 | ||||
| miR-107 | 748.65 | 87 | miR-107 | 1.19e4 | |
| miR-137 | miR-137 | ND | miR-137 | 246 | |
| miR-302-3p/372-3p/373-3p/520-3p | miR-302a-3p | 0.19 | miR-302a | 42.9 | |
| miR-302b-3p | ND | miR-302b | 0 | ||
| miR-302c-3p | ND | miR-302c | 0 | ||
| miR-302d-3p | 0.87 | miR-302d | 10.3 | ||
| miR-302e | ND | miR-302e | 0 | ||
| miR-372-3p | 0.56 | miR-372 | 91.9 | ||
| miR-373-3p | 0.68 | miR-373 | 2.22 | ||
| miR-520a-3p | ND | miR-520a | 4.76 | ||
| miR-520b-3p | ND | miR-520b | 9.8 | ||
| miR-520c-3p | ND | miR-520c | 6.02 | ||
| miR-520d-3p | ND | miR-520d | 0 | ||
| miR-520e-3p | ND | miR-520e | 33.3 | ||
We were able to identify a comparative RNA-seq dataset (NCBI GEO accession GSE80258) of the human HCT116 cells and its Drosha knockout genetic variant (50), providing another opportunity for this study. Same analyses were performed with this dataset, and showed that the direct auto-feedback regulation loop was active in HCT116 cells as well (Figure 6). Again, the miRNA site counts correlate well with the log-ratios, with a Spearman correlation coefficient of 0.67 (Figure 6A). Both miRNA site density and relative density correlate with the log-ratios as well, with Pearson correlation coefficients of 0.87 (Figure 6B and C). And the AGO phosphorylation cycle and the TNRC6 mRNAs have significantly higher log-ratios than the downstream effector mRNAs (with a t-test P-value of 0.008) and, to a lesser extent, the upstream miRNA biogenesis mRNAs (Figure 6D).
Figure 6.

Overall positive correlation between miRNA site counts and the increases in mRNA expression levels in Drosha−/− mutant cells (A–C) and the specificity of auto-feedback regulation for the targeting segment of miRNA pathway (D). (A) the mRNAs were grouped based on their binding site counts. The mean log-ratio (log2(KO/WT)) of the expression levels in Drosha−/− (KO) and wild type (WT) cells was calculated for each group. A scatter plot of the mean log-ratios versus the corresponding binding site counts is shown. Spearman correlation coefficient is also shown. For group sizes and the scale-free relationship, please see Figure 1. (B) a scatter plot of the log-ratio versus miRNA binding site density, as in Figure 5B. The Pearson correlation and P-value are also shown. (C) a scatter plot of the log-ratios vs. relative miRNA binding site density, as in Figure 5C. The Pearson correlation and P-value are also shown. (D) a scatter plot of mRNA log2(KO/WT) values, with the mRNAs grouped into steps of the miRNA pathway. As in Figure 4, the biogenesis steps are plotted in black, the targeting steps in red and the downstream effector in blue. The black line connects the mean log-ratio values of the steps.
We also functionally tested this auto-feedback regulation through analysis of miRNA-mediated translational inhibition. The approach was to use, as described below, a modified polysome-profiling analysis for comparative analysis of the HCT116 human cell line and its Dicer1−/− genetic variant (48). Since miRNA-mediated translational inhibition is known to be moderate, the translational difference between HCT116 wildtype and Dicer1−/− cells is likely moderate as well. To better detect the moderate difference, we adopted a modified version of the polysome-profiling analysis (51,52). The polysome fraction was divided into light (2- to 9-mer) and heavy (10-mer or more) sub-fractions, followed by RNA-seq analyses to quantify mRNA abundance in the two sub-fractions. The log-ratio (log2(heavy/light)) of the mRNA abundance in the sub-fractions could then be calculated. For miRNA-regulated mRNAs, this log-ratio should increase in Dicer1−/− cells, due to the disruption of miRNA biogenesis and, in turn, miRNA-mediated translational inhibition. Thus, we calculated the log-ratio increase from wild type to Dicer1−/− cells and used it as another metric of the degree to which the mRNA is miRNA-regulated. Once again, this metric correlated well with the miRNA-binding site count (Figure 7A). As in Figures 5A and 6A, strong correlation was observed at low miRNA site count ranges, and scattering showed up as miRNA site counts increase and mRNA groups became exponentially smaller and smaller. Again, both miRNA site density and relative density correlate with the log-ratios increase as well, with respective Pearson correlation coefficients of 0.62 and 0.65 (Figure 7B and C). The correlations suggested that miRNAs often retain their target mRNAs in the basal, though not completely inactive, translational stages (the light polysome sub-fractions). The AGO phosphorylation cycle and the TNRC6 mRNAs exhibited obviously higher increases in the log-ratio in Dicer1−/− cells than the upstream miRNA biogenesis mRNAs, and minor increases than the downstream effector mRNAs (Figure 7D). Consistent with Figure 5D, DGCR8 displayed some increases in this dataset as well (Figure 7D).
Figure 7.

Overall positive correlation between miRNA site counts and the increases in mRNA translation activity in Dicer1−/− mutant cells (A–C) and the specificity of auto-feedback regulation for the targeting segment of miRNA pathway (D). (A) a scatter plot of miRNA binding site counts (x axis) versus the mean difference between Dicer1−/− (KO) and wild-type (WT) log-ratios of the mRNAs with the corresponding counts (y axis). Spearman correlation coefficient is also shown. For group sizes and the scale-free relationship, please see Figure 1. (B) a scatter plot of the log-ratio difference versus miRNA binding site density, as in Figure 5B. The Pearson correlation and P-value are also shown. (C) a scatter plot of the log-ratios difference versus relative miRNA binding site density, as in Figure 5C. The Pearson correlation and P-value are also shown. (D) a scatter plot of log-ratio difference, with the mRNAs grouped into six steps of the miRNA pathway. As in Figure 4, the biogenesis steps are plotted in black, the targeting steps in red and the downstream effector in blue. The black line connects the mean log-ratio value differences of the six steps. The DICER1 mRNA is annotated with the * symbol to indicate its mutation in the mutant cells.
Thus, genetic experimental results supported an overall trend that miRNA site enrichment leads to enhanced cellular capacity for miRNA-mediated repression. In the case of miRNA targeting activity mRNAs, we demonstrated utilization of this capacity in direct and specific auto-feedback regulation. This was supported by analyses of both miRNA regulatory functions – mRNA destabilization and translation inhibition.
Confirmation of miRNA site enrichment with experimentally determined miRNA binding sites
Another valuable resource for this study is the miRNA-mRNA target relationship identified experimentally with CLIP-seq-based high-throughput approaches in a comprehensive and unbiased manner (53–55). The popular miRTarBase database collects and annotates such datasets (33,34). Thus, the miRTarBase CLIP-seq dataset was downloaded. The dataset identified 10 276 mRNAs with at least one miRNA binding sites. We ranked the mRNAs based on their unique miRNA binding site counts in the dataset, and repeated our analysis.
The result confirmed the specific enrichment of miRNA targeting machinery among top miRNA–targeted mRNAs (Figures 8 and 9). Except for PPP6C mRNA, they all are located to the right of the 15% line and, thus, ranked within the top 15%, (Figure 8A). AGO2 and AGO3 are both highly ranked, at the 4th and 47th, respectively; TNRC6B and TNRC6A at the 40th and 43rd, respectively; and so is ANKRD52, ranked within the top 2 percentile. Not surprisingly, the miRNA biogenesis mRNAs are not as highly ranked (Figure 8A), and neither are the CNOTs, PAN2 and PAN3 mRNAs (Figure 8B). CNOT3, CNOT8–11 and PAN3 have no miRNA site in miRTarbase dataset (Figure 8B). Once again, DICER1 was intermediately ranked (Figure 8A), consistent with its role as a transition point from miRNA biogenesis to target-binding activity.
Figure 8.

Confirmation of higher levels of miRNA-targeting of the targeting machinery than other segments of the miRNA pathway with the miRTarBase experimental dataset. As in Figure 1, a histogram of human mRNAs based on their miRNA site counts in the miRTarBase dataset is shown, illustrating the power-law relationship. The vertical line denotes the position of 15th percentile ranking, which was chosen due to its proximity to the AGO1 mRNA, to help illustrate the high level of miRNA-targeting of the targeting machinery mRNAs. The histogram is also used to display the contrast in the levels of targeting by miRNAs between two groups of mRNAs. The miRNA biogenesis () and miRNA targeting machinery () are shown in (A), and the downstream regulatory effectors () and miRNA targeting machinery () in (B). CNOTs 3 and 8–11 and PAN3 have no miRNA site in the miRTarbase dataset. They are plotted with one arbitrary data point (B) to help to illustrate the overall lower level of CNOT1-11 and PAN2/3 targeting by miRNAs. The Mann–Whitney–Wilcoxon test P-value of the contrast between two groups of mRNAs are specified inside both plots.
Figure 9.
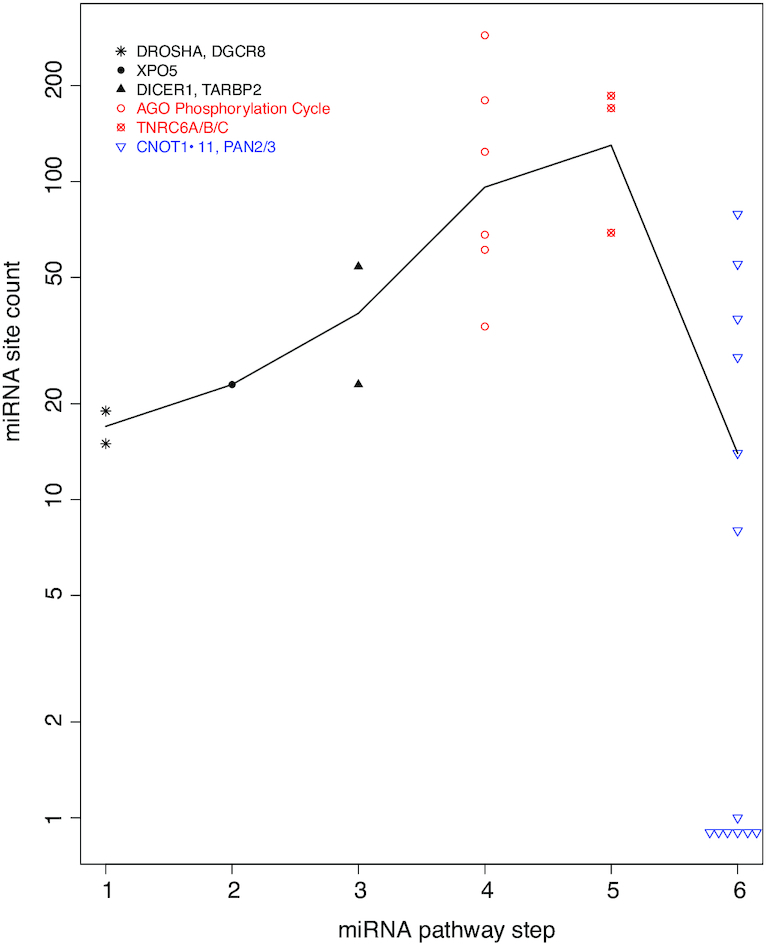
Specificity of auto-feedback regulation for the targeting segment of miRNA pathway based on the miRTarBase experimental dataset. As in Figure 4, a scatter plot of mRNA miRNA binding site count is shown, with the mRNAs grouped into six steps of the miRNA pathway. The biogenesis steps are plotted in black, the targeting steps in red and the downstream effectors in blue. The black line connects the median site counts of the six steps.
Additionally, the mRNAs are plotted in Figure 9, in the same manner as in Figures 4 and 5B, by steps of the miRNA pathway. Overall miRNA site count increases from the first step of miRNA biogenesis (DROSHA) towards the targeting steps (AGO phosphorylation cycle and TNRC6s), and then decreases towards downstream general effectors (Figure 9). The increase of miRNA site counts from miRNA biogenesis to targeting has a P-value of 2.5e–3 (Figure 8A); the decrease from targeting to downstream effector has a P-value of 2.6e–3 (Figure 8B).
However, differences were also observed. Overall, the enrichment became less dramatic; the P-value is very significant, but increases to 0.003 from 2.06e–7. AGO1 and ANKRD52 both ranked high, but not as much as in the ranking based on TargetScan dataset. TNRC6A, on the other hand, became much higher ranked at the 43rd. This might be a reflection of the different biases in the two datasets. The TargetScan dataset is based on computational analysis and, due to technical necessity, covers only orthologously conserved sites. The experimental approaches for the miRTarBase dataset do not have such limitation. Instead, they tend to bias toward miRNAs and mRNAs that are expressed at high levels in the studied cells and under the specific experimental conditions; miRNAs and mRNAs that are not or lowly expressed tend to be missed. To some degree, the biases in the two datasets complement each other. The exact causes for the discrepancy between the two datasets become an interesting topic for our future investigation.
DISCUSSION
MiRNAs are crucial components of cellular transcriptome regulatory machineries. Tremendous technical challenges remain to be solved. Among them is the low signal-to-noise ratio in miRNA binding site identification due to the shortness of the seed sequences—only six to eight nucleotides in human. This challenge has been partially alleviated by evolutionary conservation analysis and by detecting combinatorial patterns of multiple sites. Evolutionary conservation analysis is exemplified by the TargetScan dataset. This study took advantage of this dataset and revealed novel insights into how the miRNA pathway itself is regulated. Briefly, the AGO phosphorylation cycle and the TNRC6 mRNAs seems, in terms of their numbers of unique miRNA sites, under intensive direct auto-feedback regulation by miRNAs. This is consistent with frequent auto-feedback regulation of key regulators, such as transcription factors (21). On the other hand, the upstream miRNA biogenesis steps seem not under such regulation. As for the downstream effectors, this regulation seems, though somewhat active, much less dominant. Our computational observation was confirmed by experimental investigations, i.e. multiple experimental genetic analyses that compared wild type and miRNA-biogenesis-deficient mutant cells as well as the miRTarBase experimentally determined dataset.
Aberrancies in the miRNA pathway are frequently observed in human cancers, often with tissue-specific functions in the tumorigenic process. For instance, deleterious Dicer1 mutations are the genetic cause of the Dicer1 syndrome, a predisposition to distinctive inheritable tumors in the lungs, kidneys, ovaries, and thyroid (56). And Drosha mutations have been shown to be oncogenic in some tissues, and tumor suppressive in other tissues (57,58). Thus, it is interesting to investigate further whether the components of this direct auto-feedback-regulation are involved in tumor development and their potential as therapeutic targets.
Why the AGO phosphorylation cycle and the TNRC6s are targeted for this direct auto-feedback-regulation is an interesting issue. These proteins link the upstream miRNA biogenesis and the downstream general mRNA inhibition machinery. It is tempting to speculate that this link serves as the rate-limiting step of the miRNA regulatory activity, as such steps generally are under tighter control. However, there seems to be no data available to test this speculation.
Our computational analysis utilized the occurrence of miRNA sites in mRNAs. The sites confer the capacity for the mRNA to be repressed by the miRNA pathway. As discussed earlier, intuitively, this repression should not be constitutive, as the ability to dynamically relieve the repression is operationally advantageous for the cells, e.g. for rapid protein production in case of a need for quick adaptation to signals. Unfortunately, current computational analysis techniques are powerless in discerning such dynamics. To answer this issue, further experimental studies across different physiological conditions and/or transitions, and in multiple cell types, are needed.
These additional transcriptomic datasets will also enable computational mechanistic investigation into miRNA regulatory actions. While the miRNA sites and other regulatory signals embedded in mRNA sequences are the enabler of the regulatory actions, the datasets describe dynamic mRNA regulation patterns – the results of the regulatory actions. In other words, the datasets will serve as necessary inputs for computational reverse engineering efforts to decode the regulatory signals embedded in mRNA sequences. Among the many questions to be asked is whether and how specific combinatorial patterns of multiple regulatory signals dictate specific mRNA regulation patterns. The pattern can be a combination of multiple miRNA binding sites or a mix of miRNA sites with other regulatory elements, such as mRNA secondary structures.
These lines of future investigations will likely help to understand the functional advantages that the moderate regulatory actions of this regulatory system confer to the cells, i.e. why human cells maintain the seemingly uneconomical miRNA regulatory system. MiRNA-mediated mRNA degradation and translation inhibition are long known to be moderate. The results of our analysis of the public comparative RNA-seq datasets of wild type and miRNA-biogenesis-deficient mutant cells are consistent with this modesty. Our comparative polysome profiling of wild type and Dicer1−/− HCT116 cells revealed the retention of heavily miRNA-targeted mRNAs in the light polysome fractions. That is, miRNAs mediate moderate inhibition, instead of complete shut-off of, translation. Why the cell expends critical resources to express these mRNAs, but render them translationally inhibited and under enhanced degradation pressure, remains yet to be fully understood. While seemingly wasteful, similar strategies are implemented to minimize system latency in computer internal design. The computer step-by-step information retrieval process from the hard drive to memory, and then to CPU caches, is strikingly parallel to the gene expression process (59–62). Since the information retrieval process is much slower than CPU execution speed, CPU might potentially stay idle for extended periods of time waiting for the information. To minimize this latency, various strategies of speculative retrieval of information without CPU request are implemented (59–61). Similarly, the human gene expression process from transcription to mRNA, and then to protein production, is also time consuming. In case of a need for rapid response to signals, there must be regulatory mechanisms to bypass this slow process. Thus, it will be interesting to examine whether these computer optimization principles can be used as step stones toward a full understanding of the cellular functional advantages bestowed by the miRNA and other post-transcriptional regulatory systems in the context of efficient cellular operation.
It should be noted that miRNA-mediated mRNA inhibition exhibits technical similarities with transcription regulation by transcription factors. The complexity of regulator-target relationship applies to transcription regulation as well; a transcription factor usually regulates multiple genes, and a gene is always regulated by multiple transcription factors. And, in both cases, the consensus binding sites are short, leading to low signal-to-noise ratio in computational site predictions (63). Evolutionary conservation helped, in both cases, to alleviate this technical difficulty (64). Combinatorial patterns of multiple transcription factor binding sites and other genomic contextual information have provided additional means for improving the signal-to-noise ratios (65). It seems the combinatorial pattern of multiple sites also apply to miRNA-mediated mRNA inhibition (15,16). As our understanding of miRNA regulatory actions improves, it will be interesting to see whether additional similarities will be observed.
Finally, this study suggests that the overall miRNA binding site distribution pattern should be a rich source for further, technically more sophisticated, functional exploration. This distribution is currently relatively under-appreciated, perhaps due to the high levels of noise in such datasets. However, both computational and experimental approaches will improve, leading to more and more reliable datasets. It is our firm belief that the binding site distribution will play much more significant roles in our endeavor to a thorough understanding of miRNA-mediated transcriptome regulation.
DATA AVAILABILITY
The next generation sequencing (NGS) dataset has been submitted to the GEO database. The accession number is GSE134818.
ACKNOWLEDGEMENTS
We would like to acknowledge Dr Jordan Crago of Texas Tech University for proofreading this manuscript.
Notes
Present address: Audrey Gill, Phil and Penny Knight Campus Internship Program, University of Oregon, Eugene, OR 97403, USA.
FUNDING
National Institute of Health [R01LM010212, R15GM122006 to D.W., R01GM135167 to A.L.K.]. Funding for open access charge: Texas Tech University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hayes J., Peruzzi P.P., Lawler S.. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med. 2014; 20:460–469. [DOI] [PubMed] [Google Scholar]
- 2. Peng Y., Croce C.M.. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016; 1:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang J.S., Lai E.C.. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol. Cell. 2011; 43:892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chendrimada T.P., Gregory R.I., Kumaraswamy E., Norman J., Cooch N., Nishikura K., Shiekhattar R.. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005; 436:740–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haase A.D., Jaskiewicz L., Zhang H., Laine S., Sack R., Gatignol A., Filipowicz W.. TRBP, a regulator of cellular PKR and HIV-1 virus expression, interacts with Dicer and functions in RNA silencing. EMBO Rep. 2005; 6:961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacRae I.J., Ma E., Zhou M., Robinson C.V., Doudna J.A.. In vitro reconstitution of the human RISC-loading complex. Proc. Natl Acad. Sci. U.S.A. 2008; 105:512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jee D., Lai E.C.. Alteration of miRNA activity via context-specific modifications of Argonaute proteins. Trends Cell Biol. 2014; 24:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horman S.R., Janas M.M., Litterst C., Wang B., MacRae I.J., Sever M.J., Morrissey D.V., Graves P., Luo B., Umesalma S. et al.. Akt-mediated phosphorylation of argonaute 2 downregulates cleavage and upregulates translational repression of MicroRNA targets. Mol. Cell. 2013; 50:356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeng Y., Sankala H., Zhang X., Graves P.R.. Phosphorylation of Argonaute 2 at serine-387 facilitates its localization to processing bodies. Biochem. J. 2008; 413:429–436. [DOI] [PubMed] [Google Scholar]
- 10. Rudel S., Wang Y., Lenobel R., Korner R., Hsiao H.H., Urlaub H., Patel D., Meister G.. Phosphorylation of human Argonaute proteins affects small RNA binding. Nucleic Acids Res. 2011; 39:2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shen J., Xia W., Khotskaya Y.B., Huo L., Nakanishi K., Lim S.O., Du Y., Wang Y., Chang W.C., Chen C.H. et al.. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013; 497:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Golden R.J., Chen B., Li T., Braun J., Manjunath H., Chen X., Wu J., Schmid V., Chang T.C., Kopp F. et al.. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature. 2017; 542:197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quevillon Huberdeau M., Zeitler D.M., Hauptmann J., Bruckmann A., Fressigne L., Danner J., Piquet S., Strieder N., Engelmann J.C., Jannot G. et al.. Phosphorylation of Argonaute proteins affects mRNA binding and is essential for microRNA-guided gene silencing in vivo. EMBO J. 2017; 36:2088–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agarwal V., Bell G.W., Nam J.W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krek A., Grun D., Poy M.N., Wolf R., Rosenberg L., Epstein E.J., MacMenamin P., da Piedade I., Gunsalus K.C., Stoffel M. et al.. Combinatorial microRNA target predictions. Nat. Genet. 2005; 37:495–500. [DOI] [PubMed] [Google Scholar]
- 16. Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006; 38 (Suppl):S8–S13. [DOI] [PubMed] [Google Scholar]
- 17. Bueno M.J., Malumbres M.. MicroRNAs and the cell cycle. Biochim. Biophys. Acta. 2011; 1812:592–601. [DOI] [PubMed] [Google Scholar]
- 18. Chen J., Cai T., Zheng C., Lin X., Wang G., Liao S., Wang X., Gan H., Zhang D., Hu X. et al.. MicroRNA-202 maintains spermatogonial stem cells by inhibiting cell cycle regulators and RNA binding proteins. Nucleic Acids Res. 2017; 45:4142–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pan H., Qin K., Guo Z., Ma Y., April C., Gao X., Andrews T.G., Bokov A., Zhang J., Chen Y. et al.. Negative elongation factor controls energy homeostasis in cardiomyocytes. Cell Rep. 2014; 7:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou X., Geng L., Wang D., Yi H., Talmon G., Wang J.. R-Spondin1/LGR5 activates TGFbeta signaling and suppresses colon cancer metastasis. Cancer Res. 2017; 77:6589–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alon U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 2007; 8:450–461. [DOI] [PubMed] [Google Scholar]
- 22. Cheng C., Yan K.K., Hwang W., Qian J., Bhardwaj N., Rozowsky J., Lu Z.J., Niu W., Alves P., Kato M. et al.. Construction and analysis of an integrated regulatory network derived from high-throughput sequencing data. PLoS Comput. Biol. 2011; 7:e1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shalgi R., Lieber D., Oren M., Pilpel Y.. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 2007; 3:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsang J., Zhu J., van Oudenaarden A.. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell. 2007; 26:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martello G., Rosato A., Ferrari F., Manfrin A., Cordenonsi M., Dupont S., Enzo E., Guzzardo V., Rondina M., Spruce T. et al.. A MicroRNA targeting dicer for metastasis control. Cell. 2010; 141:1195–1207. [DOI] [PubMed] [Google Scholar]
- 26. Tokumaru S., Suzuki M., Yamada H., Nagino M., Takahashi T.. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis. 2008; 29:2073–2077. [DOI] [PubMed] [Google Scholar]
- 27. Inukai S., Pincus Z., de Lencastre A., Slack F.J.. A microRNA feedback loop regulates global microRNA abundance during aging. RNA. 2018; 24:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang F., Wang D.. The pattern of microRNA binding site distribution. Genes (Basel). 2017; 8:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harfe B.D., McManus M.T., Mansfield J.H., Hornstein E., Tabin C.J.. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc. Natl Acad. Sci. U.S.A. 2005; 102:10898–10903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Calabrese J.M., Seila A.C., Yeo G.W., Sharp P.A.. RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc. Natl Acad. Sci. U.S.A. 2007; 104:18097–18102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim Y.K., Kim B., Kim V.N.. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl Acad. Sci. U.S.A. 2016; 113:E1881–E1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karamysheva Z.N., Tikhonova E.B., Grozdanov P.N., Huffman J.C., Baca K.R., Karamyshev A., Denison R.B., MacDonald C.C., Zhang K., Karamyshev A.L.. Polysome profiling in Leishmania, human cells and mouse testis. J. Vis. Exp. 2018; 134:e57600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chou C.H., Chang N.W., Shrestha S., Hsu S.D., Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J. et al.. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–D247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chou C.H., Shrestha S., Yang C.D., Chang N.W., Lin Y.L., Liao K.W., Huang W.C., Sun T.H., Tu S.J., Lee W.H. et al.. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018; 46:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zare H., Khodursky A., Sartorelli V.. An evolutionarily biased distribution of miRNA sites toward regulatory genes with high promoter-driven intrinsic transcriptional noise. BMC Evol. Biol. 2014; 14:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barabasi A.L., Albert R.. Emergence of scaling in random networks. Science. 1999; 286:509–512. [DOI] [PubMed] [Google Scholar]
- 37. Jiang W., Guo Z., Lages N., Zheng W.J., Feliers D., Zhang F., Wang D.. A Multi-Parameter Analysis of Cellular Coordination of Major Transcriptome Regulation Mechanisms. Sci. Rep. 2018; 8:5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Padawer T., Leighty R.E., Wang D.. Duplicate gene enrichment and expression pattern diversification in multicellularity. Nucleic Acids Res. 2012; 40:7597–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo Z., Jiang W., Lages N., Borcherds W., Wang D.. Relationship between gene duplicability and diversifiability in the topology of biochemical networks. BMC Genomics. 2014; 15:577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grimson A., Farh K.K., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P.. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007; 27:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nielsen C.B., Shomron N., Sandberg R., Hornstein E., Kitzman J., Burge C.B.. Determinants of targeting by endogenous and exogenous microRNAs and siRNAs. RNA. 2007; 13:1894–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davis B.N., Hata A.. Regulation of MicroRNA biogenesis: a miRiad of mechanisms. Cell Commun Signal. 2009; 7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen J., Hung M.C.. Signaling-mediated regulation of MicroRNA processing. Cancer Res. 2015; 75:783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Slezak-Prochazka I., Durmus S., Kroesen B.J., van den Berg A.. MicroRNAs, macrocontrol: regulation of miRNA processing. RNA. 2010; 16:1087–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gebert L.F.R., MacRae I.J.. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019; 20:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng G.X., Do B.T., Webster D.E., Khavari P.A., Chang H.Y.. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat. Struct. Mol. Biol. 2014; 21:585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han J., Pedersen J.S., Kwon S.C., Belair C.D., Kim Y.K., Yeom K.H., Yang W.Y., Haussler D., Blelloch R., Kim V.N.. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009; 136:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cummins J.M., He Y., Leary R.J., Pagliarini R., Diaz L.A. Jr, Sjoblom T., Barad O., Bentwich Z., Szafranska A.E., Labourier E. et al.. The colorectal microRNAome. Proc. Natl Acad. Sci. U.S.A. 2006; 103:3687–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeong G., Lim Y.H., Kim Y.K.. Precise mapping of the transcription start sites of human microRNAs using DROSHA knockout cells. BMC Genomics. 2016; 17:908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Floor S.N., Doudna J.A.. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016; 5:e10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Freimer J.W., Hu T.J., Blelloch R.. Decoupling the impact of microRNAs on translational repression versus RNA degradation in embryonic stem cells. Elife. 2018; 7:e38014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darnell R.B. HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA. 2010; 1:266–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M. et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A., Haripal B., Zucker-Scharff I., Moore M.J., Park C.Y. et al.. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015; 29:2037–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Robertson J.C., Jorcyk C.L., Oxford J.T.. DICER1 syndrome: DICER1 mutations in rare cancers. Cancers (Basel). 2018; 10:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen K.S., Stroup E.K., Budhipramono A., Rakheja D., Nichols-Vinueza D., Xu L., Stuart S.H., Shukla A.A., Fraire C., Mendell J.T. et al.. Mutations in microRNA processing genes in Wilms tumors derepress the IGF2 regulator PLAG1. Genes Dev. 2018; 32:996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hata A., Kashima R.. Dysregulation of microRNA biogenesis machinery in cancer. Crit. Rev. Biochem. Mol. Biol. 2016; 51:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang D. Discrepancy between mRNA and protein abundance: insight from information retrieval process in computers. Comput. Biol. Chem. 2008; 32:462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang D., Gribskov M.. Examining the architecture of cellular computing through a comparative study with a computer. J. R. Soc. Interface. 2005; 2:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang D.G. ‘Molecular gene’: Interpretation in the right context. Biol. Philos. 2005; 20:453–464. [Google Scholar]
- 62. Condon A., Kirchner H., Lariviere D., Marshall W., Noireaux V., Tlusty T., Fourmentin E.. Will biologists become computer scientists? A truly interdisciplinary effort by computer scientists and biologists to understand how cells process information may yield new insights for both fields. EMBO Rep. 2018; 19:e46628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jayaram N., Usvyat D., AC R.M.. Evaluating tools for transcription factor binding site prediction. BMC Bioinformatics. 2016; 17:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dermitzakis E.T., Clark A.G.. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: conservation and turnover. Mol. Biol. Evol. 2002; 19:1114–1121. [DOI] [PubMed] [Google Scholar]
- 65. Berman B.P., Nibu Y., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M., Rubin G.M., Eisen M.B.. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl Acad. Sci. U.S.A. 2002; 99:757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The next generation sequencing (NGS) dataset has been submitted to the GEO database. The accession number is GSE134818.