


Abstract
The increasing use of CRISPR–Cas9 in medicine, agriculture, and synthetic biology has accelerated the drive to discover new CRISPR–Cas inhibitors as potential mechanisms of control for gene editing applications. Many anti-CRISPRs have been found that inhibit the CRISPR–Cas adaptive immune system. However, comparing all currently known anti-CRISPRs does not reveal a shared set of properties for facile bioinformatic identification of new anti-CRISPR families. Here, we describe AcRanker, a machine learning based method to aid direct identification of new potential anti-CRISPRs using only protein sequence information. Using a training set of known anti-CRISPRs, we built a model based on XGBoost ranking. We then applied AcRanker to predict candidate anti-CRISPRs from predicted prophage regions within self-targeting bacterial genomes and discovered two previously unknown anti-CRISPRs: AcrllA20 (ML1) and AcrIIA21 (ML8). We show that AcrIIA20 strongly inhibits Streptococcus iniae Cas9 (SinCas9) and weakly inhibits Streptococcus pyogenes Cas9 (SpyCas9). We also show that AcrIIA21 inhibits SpyCas9, Streptococcus aureus Cas9 (SauCas9) and SinCas9 with low potency. The addition of AcRanker to the anti-CRISPR discovery toolkit allows researchers to directly rank potential anti-CRISPR candidate genes for increased speed in testing and validation of new anti-CRISPRs. A web server implementation for AcRanker is available online at http://acranker.pythonanywhere.com/.
INTRODUCTION
CRISPR–Cas systems use a combination of genetic memory and highly specific nucleases to form a powerful adaptive defense mechanism in bacteria and archaea (1–4). Due to their high degree of sequence specificity, CRISPR–Cas systems have been adapted for use as programmable DNA or RNA editing tools with novel applications in biotechnology, diagnostics, medicine, agriculture, and more (5–9). In 2013, the first anti-CRISPR proteins (Acrs) were discovered in Pseudomonas aeruginosa phages able to inhibit the CRISPR–Cas system (10). Since then, Acrs able to inhibit a wide variety of different CRISPR subtypes have been found (10–28).
Multiple methods for identifying Acrs include screening for phages that escape CRISPR targeting (10,19–23), guilt-by-association studies (12,17,24,25,28), identification and screening of genomes containing self-targeting CRISPR arrays (11–13,24), and metagenome DNA screening for inhibition activity (26,27). Of these approaches, the ‘guilt-by-association’ search strategy is one of the most effective and direct, but it requires a known Acr to serve as a seed for the search. Thus, the discovery of one new validated Acr can lead to bioinformatic identification of others, as many Acrs have been discovered to be encoded in close physical proximity to each other, typically co-occurring in the same transcript with other Acrs or anti-CRISPR associated (aca) genes (12,17,28). Screening approaches are particularly useful in this regard, as they can potentially identify new Acr families.
Identification of self-targeting CRISPR arrays can also help in predicting new Acr families. Typically, a CRISPR array with a spacer targeting the host genome (self-targeting) is lethal to the cell (29). However, if a mobile genetic element (MGE) present in the cell carries acr genes, the CRISPR–Cas system could be inhibited, and this may allow a cell with a self-targeting array to survive. To find new Acrs, genomes containing self-targeting arrays are identified through bioinformatic methods, and the MGEs within are screened for anti-CRISPR activity, eventually narrowing down to individual proteins (11–13,24). Screens based on self-targeting also benefit from the knowledge of the exact CRISPR system that an inhibitor potentially exists for, as opposed to broad (meta-)genomic screens where a specific Cas protein has to be selected to screen against. Both types of screening additionally benefit from not requiring the prediction of a transcriptome or proteome that bioinformatic methods depend on, where incorrect annotations could lead to missed acr genes (24).
However, a weakness of all of these methods is that they are unable to predict a priori whether a gene may be an Acr, largely because Acr proteins do not share high sequence similarity or mechanisms of action (14,16,30–36). One theory to explain the high diversity of Acrs is the rapid mutation rate of the mobile genetic elements they are found in and the need to evolve with the co-evolving CRISPR–Cas systems trying to evade anti-CRISPR activity. Due to the relatively small size of most Acrs and their broad sequence diversity, simple sequence comparison methods for searching anti-CRISPR proteins are not expected to be effective. In this work, we report the development of AcRanker, a machine learning based method for direct identification of anti-CRISPR proteins. Using only amino acid composition features, AcRanker ranks a set of candidate proteins on their likelihood of being an anti-CRISPR protein. A rigorous cross-validation of the proposed scheme shows known Acrs are highly ranked out of proteomes. We then use AcRanker to predict 10 new candidate Acrs from proteomes of bacteria with self-targeting CRISPR arrays and biochemically validate three of them. Our machine learning approach presents a new tool to directly identify potential Acrs for biochemical validation using protein sequence alone.
MATERIALS AND METHODS
Data collection and preprocessing
To model the task of anti-CRISPR protein identification as a machine learning problem, a dataset consisting of examples from both positive (anti-CRISPR) and negative (non-anti-CRISPR) classes was needed. We collected anti-CRISPR information for proteins from the Anti-CRISPRdb (37). At the time the work was initiated, the database contained information for 432 anti-CRISPR proteins. In order to ensure that the machine learning model generalizes well to protein sequences that do not share high sequence similarity to known anti-CRISPR proteins, a 40% sequence identity threshold is used (38). The use of a 40% identity threshold represents a boundary where proteins above this threshold are likely to share the same structure and possibly function (39), thus providing a compromise between ensuring non-redundancy of the train and test datasets while retaining enough training examples for cross-validation. We used CD-HIT (40) to identify a non-redundant set (at the 40% sequence similarity threshold) of 20 experimentally verified Acrs (Supplementary Table S1). These proteins belong to different Acr classes: 12 of the proteins are active against subtype I-F CRISPR Cas systems, four against I-E, and four against II-A (10,13,17,20,22). This set constitutes the positive class of our dataset. We downloaded the complete proteomes of source species to which each of these proteins belong. Within these proteomes, any protein with 40% or higher sequence similarity with any protein in the set of known anti-CRISPR proteins was removed, and the remaining proteins were used to construct the negative dataset. For independent testing of the method, a dataset comprising 20 known Acrs separate from the training set (11–13,21,24,26,28,41) was used (Supplementary Table S2). The Acrs belonging to the test set were chosen to cover the wide variety of known Acr mechanisms and sequences (42), while mainly consisting of the three subtypes the model was trained on. Source proteomes for all these proteins were downloaded, based on open reading frame predictions on the NCBI database.
Feature extraction
In line with existing machine learning based protein function prediction techniques, we used sequence features (43) based on amino acid composition and grouped dimer and trimer frequency counts (44). For this purpose, amino acids are first grouped into seven classes based on their physicochemical properties (44) (Supplementary Table S3) and the frequency counts of all possible groups labeled as dimers and trimers in a given protein sequence are used in conjunction with amino acid composition. All three types of features (amino acid composition, di- and tri-meric frequency counts) are normalized to unit norm resulting in a 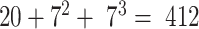 -dimensional feature vector representation for a given protein sequence (45,46).
-dimensional feature vector representation for a given protein sequence (45,46).
Machine learning model
The underlying machine learning model for AcRanker has been built using EXtreme Gradient Boosting (XGBoost) (47). In machine learning, boosting is a technique in which multiple weak classifiers are combined to produce a strong classifier. XGBoost is a tree-based method (47) that uses boosting in an end-to-end fashion, i.e., every next tree tries to minimize the error produced by its predecessor. XGBoost has been shown to be a fast and scalable learning algorithm and has been widely used in many machine learning applications (47).
In this work, we have used XGBoost as a pairwise ranking model to rank constituent proteins in a given proteome in descending order of their expected Acr behavior. The XGBoost model is trained in a proteome-specific manner to produce higher scores for known anti-CRISPR proteins as compared to non-anti-CRISPR proteins in a given proteome. In comparison to conventional XGBoost classification, the pairwise ranking model performed better in terms of correctly identifying known anti-CRISPR proteins in test proteomes in cross-validation (Supplementary Table S4). Specifically, given a set of training proteomes 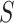 each with one or more known anti-CRISPR proteins, our objective is to obtain an XGBoost predictor
each with one or more known anti-CRISPR proteins, our objective is to obtain an XGBoost predictor 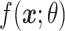 with learnable parameters
with learnable parameters 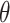 that generates a prediction score for a given protein sequence represented in terms of its feature vector
that generates a prediction score for a given protein sequence represented in terms of its feature vector 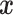 . In proteome-specific training, we require the model to learn optimal parameters
. In proteome-specific training, we require the model to learn optimal parameters 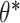 such that the score
such that the score 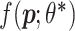 for a positive example
for a positive example 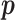 (known anti-CRISPR protein) should be higher than
(known anti-CRISPR protein) should be higher than 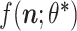 for all negative examples
for all negative examples  (non-Anti-CRISPR proteins) within the same proteome. The hyperparameters of the learning model are selected through cross-validation and optimal results are obtained with the number of estimators set at 120, a learning rate of 0.1, a subsampling of 0.6 and a maximum tree depth of 3.
(non-Anti-CRISPR proteins) within the same proteome. The hyperparameters of the learning model are selected through cross-validation and optimal results are obtained with the number of estimators set at 120, a learning rate of 0.1, a subsampling of 0.6 and a maximum tree depth of 3.
Performance evaluation
To evaluate the performance of the machine learning model, we have performed leave-one-out cross-validation as well as validation over an independent test set. In a single fold of leave-one-out cross-validation, we set aside the source proteome of a given anti-CRISPR protein for testing and train on all other proteomes. To ensure an unbiased evaluation, all sequences in the training set with a sequence identity of 40% or higher with any test protein or among themselves are removed from the training set. Furthermore, all proteins in the test set with >40% sequence identity with known anti-CRISPR proteins in the training set are also removed. This ensures that there is only one known anti-CRISPR protein in the test set in a single fold. The XGBoost ranking model is then trained and the prediction scores for all proteins in the test set are computed. Ideally, the known anti-CRISPR protein in the proteome should score the highest across all proteins in the given test proteome. This process is then repeated for all proteomes in our dataset. The rank of the known anti-CRISPR protein in its source proteome is used as a performance metric.
In bacteria, Acrs are usually located within prophage regions (13,48). Based on this premise, in another experiment for model evaluation, we passed only the proteins found within prophage regions to the model. To identify the prophage regions for a given bacterial proteome we used PHASTER (PHAge Search Tool Enhanced Release) web server (49) which accepts a bacterial genome and annotates prophage regions in it. The decision scores are computed for all phage proteins identified by PHASTER in the test proteome.
To help assess AcRanker's performance during leave-one-out cross-validation, BLAST (Basic Local Alignment Search Tool) (50) similarity was used to set a minimum performance expectation. For each protein in a given test proteome, we compute blastp scores (with default parameters) with the set of known Acrs (excluding the tested protein) and rank proteins in the increasing order of the respective e-values.
For independent validation, the ranking based XGBoost model trained over sequence features for all 20 source proteomes (Supplementary Table S1) has been tested for recently discovered Acrs (Supplementary Table S2) that are not part of our training set. The rank of a known Acr in its corresponding proteome was computed. Here again, we evaluated the model for both the complete proteome of the organism and the respective MGE subset identified by PHASTER.
AcRanker webserver
A webserver implementation of AcRanker is publicly available at http://acranker.pythonanywhere.com/. The webserver accepts a proteome file in FASTA format and returns a ranked list of proteins. The Python code for the webserver implementation is available at the URL: https://github.com/amina01/AcRanker.
Acr candidate selection
Self-Targeting Spacer Searcher (STSS; https://github.com/kew222/Self-Targeting-Spacer-Searcher) (11) was run with default parameters using ‘Streptococcus’ as a search term for the NCBI genomes database, which returned a list of all self-targets found in those genomes. Whether known acr genes were present in each of the self-targeting genomes was checked using a simple blastp search using default parameters with the Acr proteins stored within STSS. Twenty self-targeting genomes that contained at least one self-target with a 3′-NRG PAM were chosen for further analysis with AcRanker. Prophage regions with each genome were predicted using PHASTER (49). Then proteins found across all of the prophage regions predicted in a given genome were ranked with AcRanker.
To select individual gene candidates for synthesis and biochemical validation, the 10 highest ranked proteins from each genome were examined by visual inspection for a strong promoter, a strong ribosome binding site, and an intrinsic terminator. Promoters were searched for manually by looking for sequences closely matching the strong consensus promoter sequence TTGACA-17(±1)N-TATAAT upstream of the acr candidate gene, or any genes immediately preceding it. The presence of a strong ribosome binding site (resembling AGGAGG) near the start codon was similarly searched for and was required to be upstream of a gene candidate for selection. Last, given the nature of Acrs to be clustered together, genes neighboring the best candidates were also selected for further testing/validation and comprise part of the 10-member candidate test set.
Protein expression and purification
Each of the Acr candidates (Supplementary Table S5) were cloned into a custom vector (pET-based expression vector) such that each protein was N-terminally tagged with a 10xHis sequence, superfolder GFP, and a tobacco etch virus (TEV) protease cleavage site, available on Addgene (#140995–141004). Each Cas effector (Supplementary Table S6): Acidaminococcus sp. Cas12a (AsCas12a), Streptococcus pyogenes Cas9 (SpyCas9), Staphylococcus aureus Cas9 (SauCas9) and Streptococcus iniae Cas9 (SinCas9, Addgene #141076), were expressed as N-terminal MBP fusions. Proteins were produced and purified as previously described (33). Briefly, Escherichia coli Rosetta2 (DE3) containing Acr or Cas9 expression plasmids were grown in Terrific Broth (100 μg/ml ampicillin) to an OD600 of 0.6–0.8, cooled on ice, induced with 0.5 mM isopropyl-b-d-thiogalactoside and incubated with shaking at 16°C for 16 h. Cells were harvested by centrifugation, resuspended in wash buffer (20 mM Tris–Cl (pH 7.5), 500 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 5% (v/v) glycerol) supplemented with 0.5 mM phenylmethanesulfonyl fluoride and cOmplete protease inhibitor (Roche), lysed by sonication, clarified by centrifugation and purified over Ni-NTA Superflow resin (Qiagen) in wash buffer supplemented with 10 mM (wash) or 300 mM imidazole (elution). Elution fractions were pooled and digested overnight with recombinantly expressed TEV protease while dialyzed against dialysis buffer (20 mM Tris–Cl (pH 7.5), 125 mM NaCl, 1 mM TCEP, 5% (v/v) glycerol) at 4°C. The cleaved proteins were loaded onto an MBP-Trap (GE Healthcare) upstream of a Heparin Hi-Trap (GE Healthcare) in the case of SpyCas9, SauCas9 and SinCas9. Depending on the pI, TEV digested Acrs were loaded onto a Q (ML1, ML2, ML3, ML6, ML8, and ML10), heparin (ML4 and ML5), or SP (ML7 and ML9) Hi-Trap column. Proteins were eluted over a salt gradient (20 mM Tris–Cl (pH 7.5), 1 mM TCEP, 5% (v/v) glycerol, 125 mM–1 M KCl). The eluted proteins were concentrated and loaded onto a Superdex S200 Increase 10/300 (GE Healthcare) for SpyCas9, SauCas9, SinCas9 or Superdex S75 Increase 10/300 (GE Healthcare) for all the Acr candidates and developed in gel filtration buffer (20 mM HEPES-K (pH 7.5), 200 mM KCl, 1 mM TCEP and 5% (v/v) glycerol). The absorbance at 280 nm was measured by Nanodrop and the concentration was determined using an extinction coefficient estimated based on the primary amino acid sequence of each protein. Purified proteins were concentrated to approximately 50 μM for Cas9 effectors and 100 μM for Acr candidates. Proteins were then snap-frozen in liquid nitrogen for storage at –80°C. Purity and integrity of proteins was assessed by 4–20% gradient SDS-PAGE (Coomassie blue staining, Supplementary Figure S2A) and LC–MS (Supplementary Figure S2B).
RNA preparation
All RNAs (Supplementary Table S7) were transcribed in vitro using recombinant T7 RNA polymerase and purified by gel extraction as described previously (51). Briefly, 100 μg/ml T7 polymerase, 1 μg/ml pyrophosphatase (Roche), 800 units RNase inhibitor, 5 mM ATP, 5 mM CTP, 5 mM GTP, 5 mM UTP, 10 mM DTT, were incubated with DNA target in transcription buffer (30 mM Tris–Cl pH 8.1, 25 mM MgCl2, 0.01% Triton X-100, 2 mM spermidine) and incubated overnight at 37°C. The reaction was quenched by adding 5 units RNase-free DNase (Promega). Transcription reactions were purified by 12.5% (v/v) urea-denaturing PAGE (0.5× Tris–borate–EDTA (TBE)) and ethanol precipitation.
In vitro cleavage assay
In vitro cleavage assays were performed at 37°C in 1× cleavage buffer (20 mM Tris–HCl pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM DTT and 5% glycerol (v/v)) targeting a PCR amplified fragment of double-stranded DNA (Supplementary Table S8). For all cleavage reactions, the sgRNA was first incubated at 95°C for 5 min and cooled down to room temperature. The Cas effectors (SpyCas9, SauCas9, AsCas12a at 100 nM and SinCas9 at 200 nM respectively) were incubated with each candidate Acr protein at 37°C for 10 min before the addition of sgRNA (SpyCas9, SauCas9, AsCas12a sgRNA at 160 nM and SinCas9 sgRNA at 320 nM respectively) to form the RNP at 37°C for 10 min. The DNA cleavage reaction was then initiated with the addition of DNA target and reactions incubated for 30 min at 37°C before quenching in 1× quench buffer (5% glycerol, 0.2% SDS, 50 mM EDTA). Samples were then directly loaded to a 1% (w/v) agarose gel stained with SYBRGold (ThermoFisher) and imaged with a BioRad ChemiDoc.
Competition binding experiment
The reconstitution of the SinCas9–sgRNA–ML1 and SinCas9–sgRNA–AcrIIA2 complex was carried out as previously described (52). Briefly, purified SinCas9 and in vitro transcribed sgRNA were incubated in a 1:1.6 molar ratio at 37°C for 10 min to form the RNP. To form the inhibitor bound complexes, a 10-fold molar excess of AcrIIA20 (ML1) or AcrIIA2 were added and incubated with the RNP complex at 37°C for 10 min. For the competition binding experiment, a 10-fold molar excess of AcrIIA20 was first incubated with the RNP complex at 37°C before incubation with a 10-fold molar excess of AcrIIA2 at 37°C for 10 min. Each complex was then purified by analytical size-exclusion chromatography (Superdex S200 Increase 10/300 GL column, GE Healthcare) pre-equilibrated with the gel filtration buffer (20 mM HEPES-K (pH 7.5), 200 mM KCl, 1 mM TCEP and 5% (v/v) glycerol) containing 1 mM MgCl2. The peak fractions were concentrated by spin concentration (3-kDa cutoff, Merck Millipore), quenched in 1X SDS-Loading dye (2% w/v SDS, 0.1% w/v bromophenol blue and 10% v/v glycerol) and boiled down to 20 μl before loading onto a 4–20% gradient SDS-PAGE.
Mass spectrometry
Protein samples were analyzed using a Synapt mass spectrometer as described elsewhere (53).
RESULTS
A machine learning model for anti-CRISPR prediction
A major challenge in the discovery of new anti-CRISPR proteins is the diversity of amino acid sequences that have been discovered so far, and the lack of predictable structural features between them (54,55). While some Acrs and aca genes are predicted to contain an HTH fold (13,24,54,56,57), there is no broadly unifying structural motif, making traditional searching methods (such as BLAST similarity searching (50) poorly equipped to identify new Acr families. To address this challenge, we have developed AcRanker, a machine learning model that accepts a proteome as input and ranks its constituent proteins in decreasing order of their expected Acr character.
To build the model, we used EXtreme Gradient Boosting (XGBoost) based ranking (47) with 1-, 2- and 3-mer amino acid composition as input features (43). Other features were considered, but did not improve model performance, or were impractical to include (e.g. requiring experimental data to determine transcription or translation rates). Additionally, the use of sequence features alone can indirectly capture information about the structure of the protein and other properties, such as the isoelectric point and physiochemical properties, while being minimally restrictive. The utility of sequence features has been demonstrated previously (58), including work to predict binding sites within calmodulin (59), where the target proteins sequences are diverse.
To train the model we created a dataset comprising 20 experimentally verified Acrs taken from the anti-CRISPRdb (37) (Supplementary Table S1) and their source proteomes. Testing was performed on an additional set of 20 known Acrs, with different predicted mechanisms, sequence composition, and source organisms (Supplementary Table S2).
Cross-validation by single proteome omission
To evaluate the performance of AcRanker, we performed leave-one-out cross-validation using the training dataset. Out of the 20 known Acr proteomes tested individually, we observed that the ranking-based model ranked seven Acrs higher than other proteins in their respective proteomes (Table 1). In total, 14 out of the 20 known Acrs are ranked within the top 5% in their respective proteomes (Table 1).
Table 1.
Results for leave-one-out cross-validation
| Complete proteome | Prophage subset | ||||||
|---|---|---|---|---|---|---|---|
| Accession No. | Anti-CRISPR family | Proteome size | BLAST rank | AcRanker rank | Proteome Size | BLAST rank | AcRanker rank |
| YP_007392738.1 | AcrIE1 | 57 | 33 | 1 | - | - | - |
| YP_007392439.1 | AcrIE2 | 54 | 18 | 2 | - | - | - |
| YP_950454.1 | AcrIE3 | 52 | 17 | 1 | - | - | - |
| NP_938238.1 | AcrIE4 | 54 | 11 | 1 | - | - | - |
| YP_007392342.1 | AcrIF1 | 56 | 21 | 11 | - | - | - |
| YP_002332454.1 | AcrIF2 | 51 | 34 | 1 | - | - | - |
| YP_007392440.1 | AcrIF3 | 54 | 5 | 1 | - | - | - |
| YP_007392799.1 | AcrIF4 | 57 | 36 | 3 | - | - | - |
| YP_007392740.1 | AcrIF5 | 57 | 26 | 19 | - | - | - |
| WP_043884810.1 | AcrIF6 | 6095 | 1 | 80 | 361 | 1 | 15 |
| WP_019933870.1 | AcrIF6 | 3045 | 1 | 13 | 72 | 1 | 1 |
| WP_014702809.1 | AcrIF6 | 2689 | 1 | 130 | 57 | - | - |
| ACD38920.1 | AcrIF7 | 57 | 20 | 1 | - | - | - |
| AFC22483.1 | AcrIF8 | 68 | 30 | 1 | - | - | - |
| WP_031500045.1 | AcrIF9 | 4928 | 198 | 333 | 37 | - | - |
| KEK29119.1 | AcrIF10 | 3552 | 189 | 17 | 70 | 23 | 2 |
| AEO04364.1 | AcrIIA1 | 2951 | 183 | 770 | 146 | 60 | 87 |
| AEO04363.1 | AcrIIA2 | 2951 | 210 | 16 | 146 | 34 | 3 |
| AEO04689.1 | AcrIIA4 | 2951 | 59 | 21 | 146 | 9 | 4 |
| ASD50988.1 | AcrIIA5 | 54 | 5 | 8 | - | - | - |
Each row of the table indicates which Acr was excluded from the training dataset and used as a test dataset, and each number displayed is the ranking of the known Acr received from the indicated test proteome using either the blastp search against all other known Acrs (BLAST) or AcRanker. The Acrs from bacterial proteomes—AcrIF6, AcrIF9, AcrIF10, AcrIIA1, AcrIIA2 and AcrIIA4—were also ranked using only the subset of proteins predicted to reside within prophages as predicted by PHASTER (49). Two Acrs from bacterial proteomes did not occur in the predicted prophages (WP_014702809.1 and WP_031500045.1) and are indicated by dash placeholders. All three prophage proteome subset fields have been left empty for Acrs from phage proteomes.
Generally, we observe that the machine learning rankings for Acrs contained in phage proteomes are much better than those contained in bacterial proteomes, likely due to their smaller size (Table 1). To test if the relative rankings of the known Acrs found within bacterial proteomes would improve in the context of only prophage-derived proteins, we identified which proteins in the bacterial proteomes were found within prophages using PHASTER (49) and used only that subset to test both models. With the prophage subsets we did observe a higher ranking for the known Acrs due to the removal of higher-ranking proteins not found in the predicted prophages (Table 1).
As a baseline, we also compared the rankings obtained from the machine learning model to a blastp (50) ranking (Table 1). For each excluded Acr in the leave-one-out train/test cycles, the excluded Acrs proteome was used as a query set to BLAST against the 19 other Acrs used for training and the resulting e-values ranked from lowest to highest. These blastp scores represent a naïve search strategy that AcRanker seeks to improve upon. The BLAST search method, however, only returned the highest rank for the AcrIF6 family because three distant homologs (using the <40% identity threshold) were included in the training dataset. Interestingly, we also observed that the BLAST method gave higher ranks than AcRanker for AcrIF9, AcrIIA5 and AcrIIA1 (13,17,20). However, with the exception of AcrIF6, the BLAST rankings of all the Acrs fell outside of the top 5%, demonstrating the diversity of Acr families, the difficulty of predicting new Acrs de novo, and improvement gained using AcRanker.
We next asked which of the features used in AcRanker had the biggest impact on Acr ranking to determine if any biological insight could be gained. Performing a SHAP (SHapley Additive exPlanations) (60) analysis on the constructed model (Supplementary Figure S1) revealed that the three highest impact features were the presence or absence of three single amino acids: proline, glutamine, and leucine. However, the ‘blackbox’ nature of machine learning models, the relative continuity of the top 20 impact values, and the lack of a clear relationship between them prevent any clear conclusions from being drawn.
Independent set validation
To validate AcRanker, we used an independent testing dataset of 20 recently discovered Acrs not part of the training dataset (Supplementary Table S2). Of these 20 Acrs, three are found in phage (AcrIF14, AcrIIA6, and AcrIIIB1) and 10 (AcrIE4-F7, AcrIF11, AcrIF11.1, AcrIF11.2, AcrIC1, AcrIIA3, AcrIIA13, AcrIIC5, AcrVA1 and AcrVA4) were predicted to be in a prophage region using PHASTER. For the proteins predicted to be in a prophage both the complete bacterial and phage proteome were ranked with AcRanker, otherwise only the complete proteome was ranked (Supplementary Table S9). The results from the complete bacterial proteomes did generally not perform well (Supplementary Table S9), with only four (AcrIE5, AcrIC1, AcrIIA3 and AcrIIC5) out of 16 receiving ranks within the top 10. However, of the 13 proteins found within a phage/prophage, AcRanker ranked eight within the top 10, including two with the highest rank (Table 2).
Table 2.
Independent testing set validation results
| Prophage subset | ||||
|---|---|---|---|---|
| Accession no. | Anti-CRISPR family | Proteome size | BLAST rank | AcRanker rank |
| WP_064584002.1 | AcrIE4-F7 | 111 | 1 | 4 |
| WP_038819808.1 | AcrIF11 | 64 | 38 | 3 |
| WP_033936089.1 | AcrIF11.1 | 92 | 38 | 1 |
| EGE18857.1 | AcrIF11.2 | 59 | 1 | 30 |
| AKI27193.1 | AcrIF14 | 68 | 5 | 14 |
| WP_046701304.1 | AcrIC1 | 72 | 15 | 1 |
| WP_014930691.1 | AcrIIA3 | 74 | 10 | 2 |
| WP_149028791.1 | AcrIIA6 | 40 | 21 | 23 |
| AKS70260.1 | AcrIIA13 | 145 | 29 | 3 |
| WP_002642161.1 | AcrIIC5 | 367 | 237 | 6 |
| NP_666582.1 | AcrIIIB3 | 54 | 25 | 44 |
| WP_046701302.1 | AcrVA1 | 72 | 18 | 10 |
| WP_046699156.1 | AcrVA4 | 293 | 181 | 220 |
Thirteen proteomes containing non-redundant (<40% sequence identity) Acrs from phage or bacterial prophage (as predicted by PHASTER) were ranked with either AcRanker or a blastp search against the training set of Acrs.
Within the 20 Acr independent test set, AcRanker returns a higher rank for the majority of (pro-)phage proteomes compared to blastp searching (Table 2). Of the six cases where blastp ranked the known Acr higher than AcRanker, three (AcrIIA6, AcrIIIB1, AcrVA4) were ranked outside of the top 40% by both blastp and AcRanker, and would be unlikely to be discovered using either method. In two of the remaining three cases where blastp returned the higher rank (AcrIE4-F7 and AcrIF11), AcRanker was able to rank at least one member of the family within the top 10 of its respective predicted prophage proteome. AcrIF14 was the only case where blastp was able to rank the known Acr in the top 10 and AcRanker was not (Table 2). Generally, we observe better performance of AcRanker relative to blastp to identify Acrs, although the appearance of highly ranking known Acrs using blastp suggests a possibility that direct BLAST searching, as opposed to guilt-by-association searching, may be beneficial to locating certain undiscovered Acrs, for which there is some related precedent where three Acr families shared a homologous N-terminus (24).
anti-CRISPR candidate selection
Encouraged by the number of highly ranked Acrs from the test dataset, we proceeded to apply AcRanker to predict novel anti-CRISPRs from self-targeting genomes. Given the ubiquity of Streptococcus pyogenes Cas9 (SpyCas9) in gene editing and our inclusion of known SpyCas9 Acrs in the machine learning training dataset (AcrIIA1, AcrIIA2, AcrIIA4, AcrIIA5), we chose to focus specifically on Streptococcus species containing Cas9 proteins homologous to SpyCas9.
We began by generating a list of Streptococcus genomes containing at least one self-targeting type II-A CRISPR system using Self-Target Spacer Searcher, which has been previously described (11). We found 385 instances of self-targeting from type II-A CRISPR arrays occurring within 241 Streptococcus genome assemblies, six of which contained known Acrs. Of these 241 self-targeting arrays, we looked for instances where the target sequence was flanked by the 3′ NRG protospacer adjacent motif (PAM) characteristic of SpyCas9 and observed that it was present in 20 genomes. These 20 self-targeting arrays would be expected to be lethal for close homologs of SpyCas9, suggesting that other factors, such as the presence of Acrs (11), are preventing CRISPR self-targeting and cell death (Supplementary Table S10). During our original search of these 20 genomes, S. iniae strain UEL-Si1 was the only one that contained a previously discovered Acr, AcrIIA3 (13), providing a large proteome space to search for novel acr genes.
To identify new acr gene candidates, we first used PHASTER (49) to predict all of the prophages residing within the 20 self-targeting Streptococcus genomes as well as an additional Listeria monocytogenes genome (strain R2-502) containing a type II-A self-targeting CRISPR system (with six self-targets) and three well-known AcrIIA genes (13). We included the Listeria strain to determine if the known Acrs within it were returned as the top ranked genes, and if not, test the higher-ranking genes as potential additional Acrs within a known Acr-harboring strain. We created lists of the annotated proteins found within each genome's set of prophages. These protein lists were then ranked with AcRanker to predict the 10 highest ranked genes most likely to be an acr (Supplementary Table S11). Of the ∼200 genes returned, a subset was selected for further biochemical testing. The selection was based on previous observations that many Acrs are typically short genes with transcripts driven by strong promoters and ribosome binding sites that frequently end with intrinsic terminator sequences (11,13,24) (Figure 1). We also looked for proteins encoded in operons with other acr or aca genes, although this was rare, highlighting a challenge of guilt-by-association approaches.
Figure 1.

Acr candidates selected for biochemical testing. Ten Acr candidates were selected from manual inspection for further biochemical testing (blue fill). Each candidate is shown in its genomic context with its assigned rank from AcRanker noted in red. Homologous proteins share the same color border (green, blue). Homologs of AcrIIA3 (orange border) and AcrIIA1 (red border) are indicated. While testing the ML candidates, ML3 (yellow fill) was identified as a specific inhibitor of LmoCas9 (25).
As with the previous testing dataset, we observed that the known acr genes were highly ranked within the test proteomes. Interestingly, a few proteins contained in the same, or overlapping, transcripts as the known Acrs ranked higher with AcRanker (ML1 and ML2). We took these candidates as well as eight others (ML3–ML10) containing the features described above (Figure 1).
Biochemical validation of novel Acrs identified by AcRanker
To determine if the identified proteins were inhibitors of SpyCas9, we purified each candidate and tested their ability to directly inhibit DNA targeting in vitro. Of the ten candidate inhibitors, nine were successfully cloned, expressed and purified (Supplementary Figures S2A and B). To assess inhibition of DNA targeting in vitro, we first assayed the ability of SpyCas9 to cleave double stranded DNA (dsDNA) when incubated in the presence of a 50-fold excess of each candidate Acr (Figure 2A). While SpyCas9 was capable of complete DNA target cleavage, the generation of DNA cleavage products was attenuated in the presence of the positive control inhibitor AcrIIA4 and the candidates ML1 or ML8. To determine the potency of inhibition, we tested the ability of SpyCas9 to cleave the DNA target in the presence of a dilution series of ML1 or ML8 (Figure 2B). In contrast to AcrIIA4, an established potent inhibitor of SpyCas9 (13), both ML1 and ML8 inhibited SpyCas9 with around a 10-fold lower potency. We wondered if the high concentration of ML1 or ML8 required to completely inhibit Cas9 might represent an in vitro concentration-dependent artifact. To explore this, we assayed SpyCas9 DNA cleavage against a titration series of either non-target DNA competitor, BSA, ML2 or ML3 and observed no significant inhibition of SpyCas9, even with a 100-fold excess (Supplementary Figure S3B–D). Taken together, these data indicated that both ML1 and ML8 weakly inhibit SpyCas9 DNA cleavage in vitro.
Figure 2.

Inhibition of SpyCas9 and SauCas9 by newly discovered Acr candidates. (A) In vitro cleavage of dsDNA by SpyCas9 in the absence or presence of a 50-fold excess of AcrIIA4 (positive control) and each Acr candidate. (B) In vitro cleavage of dsDNA by SpyCas9 in the presence of increasing concentrations of (left to right) BSA (negative control), AcrIIA4 (positive control), ML1 and ML8 (Acr:RNP 0.1-, 1-, 2-,10-, 50- and 100-fold excess from left to right). (C) In vitro cleavage of dsDNA by SauCas9 in the absence or presence of a 25-fold excess of each Acr candidate. (D) In vitro cleavage of dsDNA by SauCas9 in the presence of increasing concentrations of (left to right) BSA (negative control), AcrllA5 (positive control, Acr:RNP 0.1-, 1-, 2-, 4-, 8- and 10-fold excess from left to right), ML3 and ML8 (Acr:RNP 0.1-, 1-, 2-,10-, 50- and 100-fold excess from left to right). Uncropped gel images for panels B and D are shown in Supplementary Figures S3 and S4.
We next tested the ability of the AcRanker-generated candidates to inhibit Staphylococcus aureus (SauCas9), another Cas9 commonly used for gene editing (61,62) to determine whether any of the candidates identified from self-targeting Streptococcus genomes had broader Cas9 inhibition activity. At a 25-fold excess relative to the SauCas9 RNP complex, ML3 and ML8 were able to inhibit SauCas9 dsDNA cleavage (Figure 2C). To determine potency, we incubated a dilution series of either ML3 or ML8 with SauCas9 before the addition of the DNA target. However, in comparison to AcrIIA5, an established strong inhibitor of SauCas9 (20,24,63), both Acr candidates inhibited SauCas9 with approximately 50-fold lower potency (Figure 2D, Supplementary Figure S4A and S4B), an activity we confirmed was not due to a false positive from the high concentration of protein in the assay (Supplementary Figure S4A).
Given the relatively weak inhibition of both SpyCas9 and SauCas9, we next tested the specificity of ML1, ML3 and ML8 by assaying their ability to block DNA targeting by either AsCas12a or the restriction enzyme AlwNI. Neither AcrIIA4, ML1, ML3 nor ML8 were able to inhibit DNA targeting by AlwNI, suggesting that they all are specific inhibitors of CRISPR effectors (Supplementary Figures S5A and B). Consistent with this, inhibition of AsCas12a was only observed with ML1 and ML8 at a 100-fold excess (Supplementary Figure S5C). Taken together, our data show that ML1, ML3 and ML8 are low potency inhibitors of SpyCas9 (ML1 and ML8) or SauCas9 (ML3 and ML8). While testing ML1–ML10 for Acr activity, Osuna, et al. described AcrIIA12, a specific inhibitor of LmoCas9 in plaque assays, which shares the same sequence as ML3 (25).
ML1: a potent inhibitor of SinCas9
ML1 was identified in the Streptococcus iniae (Sin) genome. Previous studies have reported anti-CRISPRs can exhibit either selective or broad-spectrum inhibition of divergent Cas effectors (14,33). Given that SinCas9 is ∼70% identical to SpyCas9 and only ∼26% identical to SauCas9 we wondered whether ML1 is a more potent inhibitor of SinCas9. To explore this, we cloned, expressed, and purified SinCas9 protein for use in in vitro DNA targeting assays. Like SpyCas9, SinCas9 was capable of cleaving dsDNA targets proximal to an NGG PAM using a sgRNA derived from a fusion of the tracrRNA and crRNA (Figure 3A, Supplementary Figures S6 and S7). Similar to SpyCas9, both ML1 and ML8 inhibited DNA cleavage by SinCas9 (Figure 3A). Using a titration of ML1 and ML8, we again assayed the potency of SinCas9 inhibition (Figure 3B, Supplementary Figure S6B). Strikingly, in contrast to the weak inhibition of SpyCas9, ML1 was able to potently inhibit DNA cleavage by SinCas9 (Figure 3B). To investigate at which step ML1 inactivates SinCas9 function, we carried out in vitro cleavage assays where ML1 was incubated with SinCas9 before and after the addition of sgRNA (Supplementary Figure S6C). In both cases the DNA cleavage activity of SinCas9 was potently inhibited, suggesting that ML1 inhibits activity after sgRNA binding to Cas9.
Figure 3.
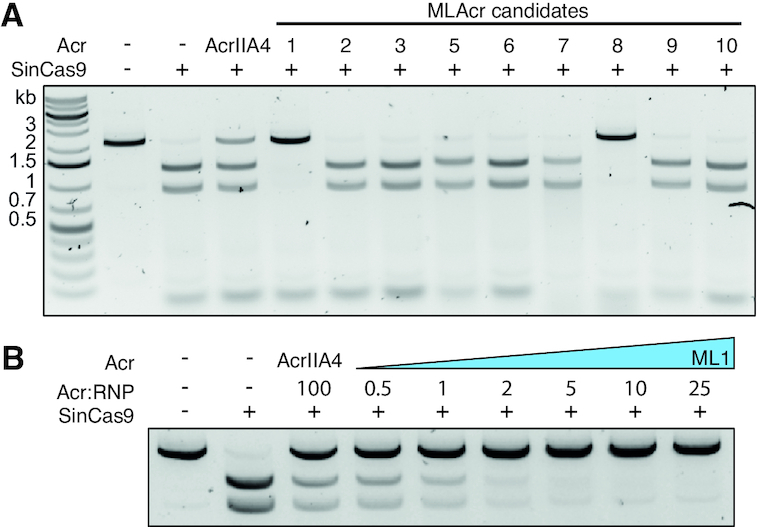
ML1 and ML8 inhibit SinCas9 with ML1 showing high potency. (A) In vitro cleavage of dsDNA by SinCas9 in the absence or presence of a 50-fold excess of each Acr candidate. (B) In vitro cleavage of dsDNA by SinCas9 in the presence of increasing concentrations of ML1. The uncropped gel image for panel B is shown in Supplementary Figure S6.
A number of reported type-IIA Acrs inhibit their cognate Cas9 by competing with target DNA through PAM mimicry (52,64). We noted that SinCas9 was susceptible to inhibition by AcrIIA4 at 100-fold excess (Figure 3A) and AcrIIA2 at 10-fold excess (Supplementary Figure S6D), both PAM mimics that inhibit PAM recognition by SpyCas9 (15,52). Like these established PAM mimics, ML1 is a small protein with a predicted negatively charged surface potential (isoelectric point of 4.3), suggesting that it too might compete with target DNA. To explore this idea, we developed a competition binding experiment to assay if the association of ML1 with SinCas9 might prevent the binding of AcrIIA2 (Figure 4A). First, we incubated either AcrIIA2 or ML1 with the SinCas9–sgRNA complex and observed a stable SinCas9–sgRNA–Acr complex on a gel filtration column (Figure 4B, Supplementary Figure S8A) with the complex components all resolvable on a protein gel (Figure 4C, Supplementary Figure S8B). To determine if ML1 binding to the SinCas9 RNP could prevent AcrIIA2 binding, we first formed the SinCas9–sgRNA–ML1 complex and then incubated with AcrIIA2 before resolving over a column. Incubating ML1 with the SinCas9 RNP before adding AcrIIA2 abolished AcrIIA2 co-elution with SinCas9–sgRNA (Figure 4C, Supplementary Figure S8B), suggesting that ML1 might occupy the same site on SinCas9. Collectively, these data are consistent with a model where ML1 directly binds to the SinCas9–sgRNA complex to form a complex that is incompatible with AcrIIA2’s ability to bind to the PAM interacting domain (52).
Figure 4.

ML1 competes with AcrIIA2 to bind to the SinCas9–sgRNA complex. (A) Flowchart for the competition binding experiment between ML1 and AcrIIA2. Binding of the Acr to the SinCas9–sgRNA RNP was reconstituted using size-exclusion chromatography (SEC). (B) Size-exclusion chromatogram of SinCas9-sgRNA in the presence of either ML1, AcrIIA2 or both Acrs with AcrIIA2 added after ML1. (C) Coomassie-stained polyacrylamide gel illustrating the components of the SinCas9-RNP fraction annotated (I), (II) and (III) in panel B.
DISCUSSION
With the growth of the anti-CRISPR field, there has been a need for improved tools to search the extensive proteomic space to find new anti-CRISPRs more efficiently. In this work we developed a machine learning method, AcRanker, as a first step toward the direct prediction of acr genes de novo with minimal knowledge a priori. We show that with only protein sequence features, AcRanker is able to highly rank Acrs from within prophage proteomes. Using a combination of AcRanker and self-targeting information from STSS (11), we were able to quickly reduce to a few top acr gene candidates for direct synthesis and testing of anti-CRISPR properties. From these candidates, we identified two novel Acrs: here named AcrIIA20 and AcrIIA21. AcrIIA20 (ML1) inhibits Streptococcus iniae Cas9 (SinCas9) with high potency and S. pyogenes Cas9 (SpyCas9) with low potency. With only 64 amino acids and a molecular weight of 7.3 kDa, to our knowledge it is the smallest type II Acr found to date. Based on the negative charge of AcrIIA20 and its competitive binding with AcrIIA2, we speculate that AcrIIA20 inhibits Cas9 dsDNA cleavage via a similar mechanism of PAM mimicry. In addition, we found AcrIIA21 (ML8), a broadly acting type II-A Acr, which is able to inhibit SpyCas9, SauCas9 as well as SinCas9, although with low potency.
The narrow and broader inhibition range of AcrIIA20 and AcrIIA21, respectively, is mirrored in their distribution in other genomes. Within the NCBI protein database, only a handful of homologs can be found for AcrIIA20 in closely related Streptococcus species (namely iniae, uberis and dysgalactiae). In contrast, sequences sharing homology with AcrIIA21 are found broadly in Lactobacillales and beyond, owing at least in part to its shared identity with replication initiator protein A, a single stranded DNA binding protein, suggesting nucleic acid binding as one potential mechanism of inhibition for AcrIIA21.
We also observe weak inhibition of SauCas9 with ML3 (AcrIIA12), which was shown to be a specific inhibitor of Listeria monocytogenes Cas9 (LmoCas9) while this study was being conducted (25). Because we were unable to test LmoCas9 (due to the difficulty of purifying it intact and active), we were unable to observe strong inhibition activity specific to its host Cas9. Similarly, we were unable to satisfactorily purify S. agalactiae Cas9 (SagCas9) to test ML4-ML10 against the Cas9 found in the same genomes in which they were found, leaving the door open for the possibility that they are specific against SagCas9.
AcRanker adds yet another tool to the anti-CRISPR hunter's toolbox by providing an alternative to BLAST and guilt-by-association searching to find new Acr families. In fact, we find that of the three candidates that we or others validated (ML1, ML3 and ML8), all had significantly higher rankings with AcRanker over BLAST (Supplementary Table S12). However, we do see some cases where BLAST ranks known Acrs higher than AcRanker (Tables 1 and 2), providing a potential complementary approach, although one we believe is less likely to lead to new Acrs.
The ability to identify potential new Acr candidates directly from protein sequence with AcRanker opens the door for testing many new proteins without the need for laborious screening efforts. Searching within prophages of genomes containing self-targeting CRISPR arrays promises to be particularly effective, as the potential inhibitors for a specific CRISPR system can be quickly ranked to make a short list of candidates to test. We expect that direct Acr prediction methods like AcRanker will continue to reveal many more Acrs distributed across many bacterial species, finding new Acrs with unique properties for yet unforeseen future biotechnology applications.
DATA AVAILABILITY
A webserver implementation of AcRanker is publicly available at http://acranker.pythonanywhere.com/. The Python code for the webserver implementation is available in the GitHub repository (https://github.com/amina01/AcRanker).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Blake McMahon for plasmid cloning and protein purification. We thank Haridha Shivram and Patrick Pausch for providing useful tips throughout the project. We also want to thank Dylan Smock for expressing proteins and Brittney W. Thornton for technical advice.
Author contributions: Conceptualization, F.A.A.M., K.E.W., J.A.D.; Methodology, A.A., K.E.W., S.E., G.J.K., F.A.A.M.; Software, A.A., F.A.A.M., K.E.W.; Investigation, A.A., K.E.W., S.E., F.A.A.; Biochemical Analysis, S.E., G.J.K., A.T.I.; Data Curation, A.A., K.E.W., S.E.; Writing, A.A., S.E., K.E.W., G.J.K., F.A.A.M.; Funding Acquisition, K.E.W., S.E., G.J.K., J.A.D., F.A.A.M., A.T.I.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This research was developed with funding from the Defense Advanced Research Projects Agency (DARPA) award HR0011-17-2-0043. The views, opinions, and/or findings expressed are those of the author and should not be interpreted as representing the official views or policies of the Department of Defense or the U.S. Government. Approved for public release; distribution is unlimited. This research was supported by the Allen Distinguished Investigator Program, through The Paul G. Allen Frontiers Group. We also acknowledge support from the National Science Foundation [MCB-1244557 to J.A.D.]; J.A.D. is an investigator of the Howard Hughes Medical Institute (HHMI). A mass spectrometer was purchased using National Institutes of Health support [1S10OD020062-01]; Amina Asif is funded via Information Technology and Telecommunication Endowment Fund at Pakistan Institute of Engineering and Applied Sciences. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. J.A.D. is a cofounder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, and Mammoth Biosciences. J.A.D. is a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, Metagenomi, and Inari. J.A.D. is a Director at Johnson & Johnson. J.A.D has research projects sponsored by Biogen, Pfizer, Apple Tree Partners, and Roche. The Regents of the University of California have patents pending for CRISPR related technologies on which the authors are inventors.
REFERENCES
- 1. Bolotin A., Quinquis B., Sorokin A., Dusko Ehrlich S.. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005; 151:2551–2561. [DOI] [PubMed] [Google Scholar]
- 2. Horvath P., Barrangou R.. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010; 327:167–170. [DOI] [PubMed] [Google Scholar]
- 3. Barrangou R. The roles of CRISPR–Cas systems in adaptive immunity and beyond. Curr. Opin. Immunol. 2015; 32:36–41. [DOI] [PubMed] [Google Scholar]
- 4. Knott G.J., Doudna J.A.. CRISPR–Cas guides the future of genetic engineering. Science. 2018; 361:866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song G., Jia M., Chen K., Kong X., Khattak B., Xie C., Li A., Mao L.. CRISPR/Cas9: a powerful tool for crop genome editing. Crop J. 2016; 4:75–82. [Google Scholar]
- 6. Ledford H. CRISPR: gene editing is just the beginning. Nature. 2016; 531:156–159. [DOI] [PubMed] [Google Scholar]
- 7. Zhang F., Wen Y., Guo X.. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum. Mol. Genet. 2014; 23:R40–R46. [DOI] [PubMed] [Google Scholar]
- 8. van Diemen F.R., Kruse E.M., Hooykaas M.J.G., Bruggeling C.E., Schürch A.C., van Ham P.M., Imhof S.M., Nijhuis M., Wiertz E.J.H.J., Lebbink R.J.. CRISPR/Cas9-mediated genome editing of Herpesviruses limits productive and latent infections. PLoS Pathogens. 2016; 12:e1005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Doudna J.A., Charpentier E.. The new frontier of genome engineering with CRISPR–Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 10. Bondy-Denomy J., Pawluk A., Maxwell K.L., Davidson A.R.. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013; 493:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watters K.E., Fellmann C., Bai H.B., Ren S.M., Doudna J.A.. Systematic discovery of natural CRISPR–Cas12a inhibitors. Science. 2018; 362:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marino N.D., Zhang J.Y., Borges A.L., Sousa A.A., Leon L.M., Rauch B.J., Walton R.T., Berry J.D., Joung J.K., Kleinstiver B.P. et al.. Discovery of widespread type I and type V CRISPR–Cas inhibitors. Science. 2018; 362:240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rauch B.J., Silvis M.R., Hultquist J.F., Waters C.S., McGregor M.J., Krogan N.J., Bondy-Denomy J.. Inhibition of CRISPR–Cas9 with Bacteriophage Proteins. Cell. 2017; 168:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harrington L.B., Doxzen K.W., Ma E., Liu J.J., Knott G.J., Edraki A., Garcia B., Amrani N., Chen J.S., Cofsky J.C. et al.. A broad-spectrum inhibitor of CRISPR–Cas9. Cell. 2017; 170:1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin J., Jiang F., Liu J.-J., Bray N.L., Rauch B.J., Baik S.H., Nogales E., Bondy-Denomy J., Corn J.E., Doudna J.A.. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv. 2017; 3:e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maxwell K.L. The anti-CRISPR story: a battle for survival. Mol. Cell. 2017; 68:8–14. [DOI] [PubMed] [Google Scholar]
- 17. Pawluk A., Staals R.H.J., Taylor C., Watson B.N.J., Saha S., Fineran P.C., Maxwell K.L., Davidson A.R.. Inactivation of CRISPR–Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat. Microbiol. 2016; 1:16085. [DOI] [PubMed] [Google Scholar]
- 18. Borges A.L., Davidson A.R., Bondy-Denomy J.. The discovery, mechanisms, and evolutionary impact of anti-CRISPRs. Annu. Rev. Virol. 2017; 4:37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He F., Bhoobalan-Chitty Y., Van L.B., Kjeldsen A.L., Dedola M., Makarova K.S., Koonin E.V., Brodersen D.E., Peng X.. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype ID immunity. Nat. Microbiol. 2018; 3:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hynes A.P., Rousseau G.M., Lemay M.L., Horvath P., Romero D.A., Fremaux C., Moineau S.. An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat. Microbiol. 2017; 2:1374–1380. [DOI] [PubMed] [Google Scholar]
- 21. Hynes A.P., Rousseau G.M., Agudelo D., Goulet A., Amigues B., Loehr J., Romero D.A., Fremaux C., Horvath P., Doyon Y. et al.. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat. Commun. 2018; 9:2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pawluk A., Bondy-Denomy J., Cheung V.H.W., Maxwell K.L., Davidson A.R.. A New Group of Phage Anti-CRISPR Genes Inhibits the Type I-E CRISPR–Cas System of Pseudomonas aeruginosa. mBio. 2014; 5:e00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pawluk A., Shah M., Mejdani M., Calmettes C., Moraes T.F., Davidson A.R., Maxwell K.L.. Disabling a Type I-E CRISPR–Cas Nuclease with a Bacteriophage-Encoded Anti-CRISPR Protein. MBio. 2017; 8:e01751-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watters K.E., Shivram H., Fellmann C., Lew R.J., McMahon B., Doudna J.A.. Potent CRISPR–Cas9 inhibitors from Staphylococcus genomes. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:doi:10.1073/pnas.1917668117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Osuna B.A., Karambelkar S., Mahendra C., Christie K.A, Garcia B., Davidson A.R., Kleinstiver B.P., Kilcher S., Bondy-Denomy J.. Listeria phages induce Cas9 degradation to protect lysogenic genomes. 2019; bioRxiv doi:30 September 2019, preprint: not peer reviewed 10.1101/787200. [DOI] [PMC free article] [PubMed]
- 26. Uribe R.V., van der Helm E., Misiakou M.A., Lee S.W., Kol S., Sommer M.O.A.. Discovery and Characterization of Cas9 Inhibitors Disseminated across Seven Bacterial Phyla. Cell Host Microbe. 2019; 25:233–241. [DOI] [PubMed] [Google Scholar]
- 27. Forsberg K.J., Bhatt I. V, Schmidtke D.T., Javanmardi K., Dillard K.E., Stoddard B.L., Finkelstein I.J., Kaiser B.K., Malik H.S.. Functional metagenomics-guided discovery of potent Cas9 inhibitors in the human microbiome. Elife. 2019; 8:e46540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee J., Mir A., Edraki A., Garcia B., Amrani N., Lou H.E., Gainetdinov I., Pawluk A., Ibraheim R., Gao X.D. et al.. Potent Cas9 inhibition in bacterial and human cells by AcrIIC4 and AcrIIC5 anti-CRISPR proteins. mBio. 2018; 9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heussler G.E., O’Toole G.A.. Friendly fire: Biological functions and consequences of chromosomal targeting by CRISPR-cas systems. J. Bacteriol. 2016; 198:1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pawluk A., Davidson A.R., Maxwell K.L.. Anti-CRISPR: discovery, mechanism and function. Nat. Rev. Microbiol. 2018; 16:12–17. [DOI] [PubMed] [Google Scholar]
- 31. Bondy-Denomy J., Garcia B., Strum S., Du M., Rollins M.F., Hidalgo-Reyes Y., Wiedenheft B., Maxwell K.L., Davidson A.R.. Multiple mechanisms for CRISPR–Cas inhibition by anti-CRISPR proteins. Nature. 2015; 526:136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maxwell K.L. Phages fight back: inactivation of the CRISPR–Cas bacterial immune system by anti-CRISPR proteins. PLoS Pathog. 2016; 12:e1005282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knott G.J., Thornton B.W., Lobba M.J., Liu J., Al-Shayeb B., Watters K.E., Doudna J.A.. Broad-spectrum enzymatic inhibition of CRISPR–Cas12a. Nat. Struct. Mol. Biol. 2019; 26:315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dong L., Guan X., Li N., Zhang F., Zhu Y., Ren K., Yu L., Zhou F., Han Z., Gao N. et al.. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 2019; 26:308–314. [DOI] [PubMed] [Google Scholar]
- 35. Zhang H., Li Z., Daczkowski C.M., Gabel C., Mesecar A.D., Chang L.. Structural basis for the inhibition of CRISPR–Cas12a by anti-CRISPR proteins. Cell Host Microbe. 2019; 25:815–826. [DOI] [PubMed] [Google Scholar]
- 36. Knott G.J., Cress B.F., Liu J.-J., Thornton B.W., Lew R.J., Al-Shayeb B., Rosenberg D.J., Hammel M., Adler B.A., Lobba M.J. et al.. Structural basis for AcrVA4 inhibition of specific CRISPR–Cas12a. eLife. 2019; 8:e49110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dong C., Hao G.F., Hua H.L., Liu S., Labena A.A., Chai G., Huang J., Rao N., Guo F.B.. Anti-CRISPRdb: a comprehensive online resource for anti-CRISPR proteins. Nucleic Acids Res. 2018; 46:D393–D398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Walsh I., Pollastri G., Tosatto S.C.E.. Correct machine learning on protein sequences: A peer-reviewing perspective. Brief. Bioinformatics. 2016; 17:831–840. [DOI] [PubMed] [Google Scholar]
- 39. Rost B. Twilight zone of protein sequence alignments. Protein Eng. 1999; 12:85–94. [DOI] [PubMed] [Google Scholar]
- 40. Huang Y., Niu B., Gao Y., Fu L., Li W.. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010; 26:680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bhoobalan-Chitty Y., Johansen T.B., Di Cianni N., Peng X.. Inhibition of Type III CRISPR–Cas immunity by an Archaeal virus encoded anti-CRISPR protein. Cell. 2019; 179:448–458. [DOI] [PubMed] [Google Scholar]
- 42. Hwang S., Maxwell K.L.. Meet the anti-CRISPRs: widespread protein inhibitors of CRISPR–Cas systems. CRISPR J. 2019; 2:23–30. [DOI] [PubMed] [Google Scholar]
- 43. Saidi R., Maddouri M., Mephu Nguifo E.. Protein sequences classification by means of feature extraction with substitution matrices. BMC Bioinformatics. 2010; 11:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shen J., Zhang J., Luo X., Zhu W., Yu K., Chen K., Li Y., Jiang H.. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:4337–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leslie C., Eskin E., Noble W.S.. The spectrum kernel: a string kernel for SVM protein classification. Proc. Pacific Symp. Biocomputing. 2002; 564–575. [PubMed] [Google Scholar]
- 46. Ben-Hur A., Weston J.. A user's guide to support vector machines. Methods Mol. Biol. 2010; 609:223–239. [DOI] [PubMed] [Google Scholar]
- 47. Chen T., Guestrin C.. XGBoost: a scalable tree boosting system. Proc. of the 22nd ACM SIGKDD Int. Conf. 2016; 785–794. [Google Scholar]
- 48. Koonin E. V., Makarova K.S.. Anti-CRISPRs on the march. Science. 2018; 362:156–157. [DOI] [PubMed] [Google Scholar]
- 49. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 51. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jiang F., Liu J.J., Osuna B.A., Xu M., Berry J.D., Rauch B.J., Nogales E., Bondy-Denomy J., Doudna J.A.. Temperature-responsive competitive inhibition of CRISPR–Cas9. Mol. Cell. 2019; 73:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Light S.H., Su L., Rivera-Lugo R., Cornejo J.A., Louie A., Iavarone A.T., Ajo-Franklin C.M., Portnoy D.A.. A flavin-based extracellular electron transfer mechanism in diverse Gram-positive bacteria. Nature. 2018; 562:140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang F., Song G., Tian Y.. Anti‐CRISPRs: the natural inhibitors for CRISPR‐Cas systems. Anim. Models Exp. Med. 2019; 2:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bondy-Denomy J., Davidson A.R., Doudna J.A., C.fineran Peter, Maxwell K.L., Moineau S., Peng X., Sontheimer E.J., Wiedenheft B.. A unified resource for tracking anti-CRISPR names. CRISPR J. 2018; 1:304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ka D., An S.Y., Suh J.Y., Bae E.. Crystal structure of an anti-CRISPR protein, AcrIIA1. Nucleic Acids Res. 2018; 46:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu Y., Zhang F., Huang Z.. Structural insights into the inactivation of CRISPR–Cas systems by diverse anti-CRISPR proteins. BMC Biol. 2018; 16:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Al-Shahib A., Breitling R., Gilbert D.R.. Predicting protein function by machine learning on amino acid sequences - a critical evaluation. BMC Genomics. 2007; 8:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Minhas F.U.A.A., Ben-Hur A.. Multiple instance learning of Calmodulin binding sites. Bioinformatics. 2012; 28:416–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lundberg S.M., Lee S.-I.. A unified approach to interpreting model predictions. Adv. Neural Inform. Process. Syst. 2017; 30:4765–4774. [Google Scholar]
- 61. Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. et al.. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015; 520:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yourik P., Fuchs R.T., Mabuchi M., Curcuru J.L., Robb G.B.. Staphylococcus aureus Cas9 is a multiple-turnover enzyme. RNA. 2019; 25:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Garcia B., Lee J., Edraki A., Hidalgo-Reyes Y., Erwood S., Mir A., Trost C.N., Seroussi U., Stanley S.Y., Cohn R.D. et al.. Anti-CRISPR AcrIIA5 potently inhibits all Cas9 homologs used for genome editing. Cell Rep. 2019; 29:1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang H., Patel D.J.. Inhibition mechanism of an anti-CRISPR suppressor AcrIIA4 targeting SpyCas9. Mol. Cell. 2017; 67:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A webserver implementation of AcRanker is publicly available at http://acranker.pythonanywhere.com/. The Python code for the webserver implementation is available in the GitHub repository (https://github.com/amina01/AcRanker).