


Abstract
Estrogen receptors (ER) are activated by the steroid hormone 17β-estradiol. Estrogen receptor alpha (ER-α) forms a regulatory network in mammary epithelial cells and in breast cancer with the transcription factors FOXA1 and GATA3. GATA3 is one of the most frequently mutated genes in breast cancer and is capable of specifying chromatin localization of FOXA1 and ER-α. How GATA3 mutations found in breast cancer impact genomic localization of ER-α and the transcriptional network downstream of ER-α and FOXA1 remains unclear. Here, we investigate the function of a recurrent patient-derived GATA3 mutation (R330fs) on this regulatory network. Genomic analysis indicates that the R330fs mutant can disrupt localization of ER-α and FOXA1. Loci co-bound by all three factors are enriched for genes integral to mammary gland development as well as epithelial cell biology. This gene set is differentially regulated in GATA3 mutant cells in culture and in tumors bearing similar mutations in vivo. The altered distribution of ER-α and FOXA1 in GATA3-mutant cells is associated with altered chromatin architecture, which leads to differential gene expression. These results suggest an active role for GATA3 zinc finger 2 mutants in ER-α positive breast tumors.
INTRODUCTION
Cooperative action of transcription factors creates complex gene regulatory networks that fine tune gene expression to meet physiologic demands. Disruption of these regulatory networks is often associated with human diseases including cancer. Estrogen receptor alpha (hereafter ER-α), FOXA1 and GATA3 are essential transcription factors for mammary gland development and physiology, and in breast cancers, the expression levels of these three genes are highly correlated (1–7). High level expression of these three transcription factors is a defining characteristic of luminal subtypes of breast tumors. Recent large-scale molecular profiling of breast tumors identified frequent mutations in GATA3 and FOXA1 (8–11). More than 10% of breast tumors carry GATA3 mutations, and these mutations are frequently observed in invasive ductal carcinoma. In contrast, FOXA1 mutations are frequently detected (∼7%) in invasive lobular carcinoma. In both cases, cells were found to be heterozygous for most mutations, with many located within sequences coding for their DNA-binding domains. In the case of GATA3, >70% of the cases are small nucleotide deletions or insertions, while less than 30% are missense mutations. These indel mutations induce frame-shifts, leading to protein truncation or extension. The functional consequences of these mutations are underexplored (7).
GATA3 and FOXA1 are known to act as pioneer transcription factors in the context of breast cancer (12–15). These pioneer factors are capable of binding closed chromatin and inducing chromatin opening. Chromatin opening by pioneer factors leads to recruitment of other chromatin binding proteins (including other transcription factors, remodeling factors, and histone modifiers) ultimately followed by gene activation (12,15–18). Precise mechanistic description of these events is incomplete. In luminal breast cancer, ER-α forms a transcriptional regulatory network with FOXA1 and GATA3, which is critical to maintenance of epithelial cell identity (14,19,20). Importantly, the chromatin binding activities of GATA3 and FOXA1 are largely independent from estrogen action, and, therefore, GATA3 and FOXA1 can mark epithelial genes and for downstream estrogen regulation (13,19). While chromatin localization of FOXA1 and ER-α is influenced by GATA3 silencing (13), the impact of cancer-specific mutations on this process is unknown.
Cancer-related GATA3 mutations are focused in the C-terminal region of the protein, predominantly exons 5 and 6 (8,9,11). We previously classified those mutations, based on the location and predictive protein products, and found that mutations in the second zinc-finger domain (ZnFn2) are associated with poor patient outcomes (21). We also experimentally demonstrated that one of these mutations (R330fs: a heterozygous frame-shift mutation at arginine 330) induces alterations of GATA3 chromatin binding activity that results in crippling of the response to progesterone through loss of expression of progesterone receptor (21). These molecular alterations lead to more aggressive tumor phenotypes in vitro and in vivo (21). Other groups also observed similar aggressive phenotypes in breast cancer cells that express a second ZnFn2 mutation (D336fs: a heterozygous frame-shift mutation at aspartic acid 336) (22–24). The phenotypic alterations are partially explained by the action of the mutant protein (9,21,25). Here, we investigate the impact(s) of the R330fs GATA3 mutation on the localization and activity of ER-α and FOXA1. Our genomic analysis indicates that the GATA3 mutation interrupts the regulatory network of these transcription factors by inducing redistribution of FOXA1 and ER-α.
MATERIALS AND METHODS
Cell line and cell culture
The T47D cell line was originally purchased from ATCC. The CR3 GATA3 mutant cell clone (T47D R330fs/wild-type) was previously established by using CRISPR-Cas9 based gene editing technique (21). T47D cells that express FLP recombinase was used as GATA3 wild-type T47D control. All cells were grown in DMEM high-glucose medium supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C with 10% CO2.
ChIP-seq/ChIP-qPCR
The details of the procedures were previously described (12,21). After removing medium, cells were fixed with formaldehyde (1% in PBS) at room temperature for 10 min, and stored at −80 °C. Fixed cells were thawed on ice and treated with hypotonic buffer containing 10 mM HEPES–NaOH pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 340 mM sucrose, 10% glycerol, 0.5% Triton X-100 and Halt Protease & Phosphatase Single-Use Inhibitor Cocktail (Thermo Fisher Scientific) for 5 min. The cells were further resuspended in lysis buffer containing 20 mM Tris–HCl pH 8.0, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM PMSF, 5 mM sodium butyrate, 0.1% SDS and protease inhibitor cocktail. Chromatin was sonicated using a Covaris S220. Immunoprecipitation was performed with anti-ER-α (Santa Cruz, HC-20), anti-FOXA1 (abcam, ab5089), anti-H3K4me1 (abcam ab8895), anti-H3K27ac (abcam ab4729), or anti-H3K27me3 (abcam ab192985) antibody. Antibodies were capture by Dynabeads Protein A and G (Thermo Fisher Scientific). DNA was purified by AMPure XP (Beckman Coulter). Purified DNAs were used for qPCR or sequencing library preparation. ChIP-qPCR assay was conducted with three biological replicates, and ChIP-seq analysis was conducted with two biological replicates. The primers were designed based on the results from the ChIP-seq data. The primer sequences were listed in Supplementary Table S1. The specificity of PCR amplification was validated by melt curve analysis.
The sequencing libraries were prepared by the NEXTflex Rapid DNA-seq kit (Bioo Scientific Corporation). All sequencing was conducted at the NIEHS Epigenomics Core Facility (NextSeq 500 or NovaSeq 6000 NGS system, Illumina). Reads were filtered based on a mean base quality score >20, and mapped to hg19 genome by Bowtie 0.12.8 (with the following parameters: -I 0 -X 1000 -m 1) allowing only uniquely mapped hits (26). Duplicate reads were removed using MarkDuplicates.jar from picard-tools-1.107 package, and non-duplicate reads were used for the analysis. The details of the sequencing quality were summarized in Supplementary Table S1.
Peak and motif analysis
ChIP-seq peaks were identified by the HOMER v4.1 software using the default parameters for transcription factor datasets (27). Overlap between peak sets was determined by BEDtools (v2.25.0) intersectBed (28). Differential ER-α or FOXA1 binding events between GATA3 wild-type and mutant T47D cells were identified by EdgeR with FDR < 0.05 and |fold change| > 1.5 as described in (21). Read counts within a 400 bp window centered on each peak were collected from ChIP-seq data in wild-type T47D cells or CR3 cells.
For the motif frequency calculation, the HOMER tool perl script (findMotifs.pl) was used to find each motif within the peaks (400 bp window). FOXA1(Forkhead)/MCF7-FOXA1-ChIP-Seq(GSE26831)/Homer (Motif 88), ERE(NR), IR3/MCF7-ERa-ChIP-Seq(Unpublished)/Homer (Motif 71), or WGATAR was used to detect FOXA1, ER-α or GATA3 motif respectively. The strict WGATAR motif matrix file was generated by the HOMER tool perl script (seq2profile.pl).
Heatmap views were generated in R with heatmap.2 for regions ±1 kb relative to peak midpoints. Read counts were collected in 20 bp bins then normalized to 30 million total uniquely-mapped non-duplicate fragments per dataset. Peaks were organized into subgroups (as specified in individual figure panels); sort order within subgroups was specified by descending TF signal over the region ±200 bp relative to peak midpoints.
Assessment of TF colocalization
For a peak set of interest, each query region was redefined as a 400 mer centered on the called peak midpoint. The read density for a given ChIP-seq at a given peak was determined as the number of uniquely-mapped non-duplicate fragments for which the fragment midpoint was within the defined peak range (400 bp). To determine the significance of the read density per peak, we apply the method described by Jothi et al. (29) in which the probability of the observed signal is given by the sum of Poisson probabilities. Peaks are assigned as positive for a given ChIP-seq at FDR < 0.001, where the FDR is defined as the ratio of expected peaks with the given read density to observed peaks with the given read density. The defined peaks were summarized in Supplementary Table S2.
Transcriptome analysis
For the GSEA and Oncomine (Thermo Fisher Scientific) analysis, ChIP-seq peaks were associated with closest genes within 50 kb from the nearest TSS, based on a RefSeq annotation downloaded on 17 October 2016. The gene expression data (FPKM) used for GSEA analysis was calculated previously (21). The same gene lists were used to conduct IPA pathway analysis. Functional analysis of ER-α or FOXA1 peak associated genes was performed in Bioconductor (version 3.8) using package clusterProfiler (version 3.5) for the enrichment of ontology related to the biological process (30). P-value was adjusted using the BH (Benjamini-Hochberg) method. Enriched pathways with an adjusted P-value <0.01 and q-value <0.05 were reported. Top 12 Enriched ontology terms are shown as Bar plot. For the Oncomine analysis, ER-α or FOXA1 differential peaks are ranked by fold changes in gene expression, and the top 100 up- or down-regulated genes in CR3 cells were used for the Oncomine Concept analysis.
METABRIC analysis
Gene expression data was queried from the R-based API of the Cancer Genomics Data Server (R library cgdsr) for data type ‘brca_metabric_mrna’ from the study ‘brca_metabric’ (11). Data was collected for genes associated with the EFG peaks in all available samples, then filtered to remove all genes and samples without reported scores. Row-scaled Z-scores were calculated then averaged across samples per GATA3 mutation group (wild-type, ZnFn2 mutations or other mutations).
RESULTS
Colocalization of ER-α, FOXA1 and GATA3 is associated with active chromatin architectures
Colocalization of ER-α, FOXA1 and GATA3 was previously reported as integral to the biological program of luminal breast cancer and of a subset of normal mammary epithelial cells (13,19). However, prevalent models have important limitations including mutation in GATA3 itself (9,21). To gain insight into the ER-α /FOXA1/GATA3 network, we analyzed their genomic localization in a breast cancer cell line, T47D, which lacks known mutations in these transcription factors. We conducted chromatin immunoprecipitation coupled with DNA sequencing (ChIP-seq) for ER-α and FOXA1 under estrogenic condition (complete medium containing 10% FBS) and defined peaks by HOMER (31). We observed 16 872 ER-α -peaks and 39 041 FOXA1-peaks. We integrated our recently published GATA3 ChIP-seq data which describes 34 382 GATA3-peaks (21). To explore colocalization of these three factors, we first conducted peak overlap analysis. Consistent with previous studies, the peaks of these three transcription factors overlapped at a subset of binding sites. (Figure 1A and B) (13,19). To dissect the functionality of the observed colocalization, we also assessed chromatin accessibility, enhancer histone marks (H3K4me1 and H3K27ac), and a repressive chromatin mark (H3K27me3). Since peak-calling applies a restrictive threshold, and overlap frequencies are potentially underestimated, we re-classified ER-α peaks into four groups based on the significance (FDR < 0.001) of observed read counts in ER-α, FOXA1 and GATA3 ChIP-seq data at each peak (see Methods) (29): EFG: ER-α +, FOXA1+, GATA3+; EF: ER-α +, FOXA1+, GATA3−; EG: ER-α +, FOXA1−, GATA3+; ER alone: ER-α +, FOXA1−, GATA3−. This classification method showed more frequent colocalization of three factors (Supplementary Table S2). About 50% of ER-α peaks are categorized as EFG, indicating ER-α is frequently colocalized with both FOXA1 and GATA3 in T47D cells when ligand is present (Figure 1C). HOMER motif analysis confirmed co-occurrence of all three motifs at EFG peaks (Supplementary Table S3). An additional 25% of ER-α peaks colocalize with either FOXA1 or GATA3, indicating that the majority of ER-α co-binds with one or both pioneer factors. These peak groups were also confirmed by ChIP qPCR (Supplementary Figure S1A). Among these peak groups, EFG and EF showed strong ATAC-seq, H3K4me1 and H3K27ac signals, while EG and ER-α alone peaks showed less robust signals of those active chromatin marks. In all groups, ATAC-seq signals were detectable, suggesting ER-α localizes at open chromatin regions.
Figure 1.

Chromatin architecture at ER-α -FOXA1-GATA3 colocalization sites. (A) Venn diagram showing overlap between ER-α, FOXA1 and GATA3 peaks in T47D cells. (B) Representative genome browser tracks at an exemplar ER-α -FOXA1-GATA3 co-localized locus. (C, D) Heatmap showing H3K27ac, H3K27med, H3K4me1, ATAC, ER-α, FOXA1 and GATA3 ChIP-seq in each ER-α (C) or GATA3 (D) peak category. (E) Biological pathway analysis of genes associated with EFG peaks. Top 12 significantly enriched pathways are shown.
FOXA1 and GATA3 peak classification exhibited different patterns of chromatin localization. By applying the same peak classification method, FOXA1 and GATA3 peaks were separated into 4 groups (Figure 1D, Supplementary Figure S1B). While ∼50% of ER-α peaks are found in the EFG category, roughly half as many FOXA1 (26%) and GATA3 peaks (24%) were classified as EFG. This finding suggests that the estrogen receptor is more reliant on this transcription factor network than either FOXA1 or GATA3. As was the case with the ER-α -centric analysis, both GATA3 and FOXA1 datasets had the highest ATAC-seq and active enhancer signals in the EFG cluster, perhaps reflecting the estrogenic conditions under which these cells were propagated. Like ER-α alone peaks, FOXA1 alone peaks displayed clear ATAC-seq signals but significantly lower H3K27ac and H3K4me1 levels. In contrast, many GATA3 alone peaks have low ATAC-seq signals and enhancer marks, which is consistent with the context-dependent pioneer action of GATA3 (12). Those FOXA1 alone and GATA3 alone peaks are still enriched for Forkhead and GATA consensus motifs respectively (Supplementary Table S3).
To further gain insight into the outcome associated with co-binding, we associated EFG peaks (GATA3-peak based) to the closest genes and conducted gene ontology analysis. The genes associated with EFG peaks were significantly enriched for cell junction, mammary gland development, and reproductive system development, suggesting that ER-α–FOXA1–GATA3 cooperative binding is enriched at loci integral to luminal cell identity (Figure 1E). The remaining categories (EF, EG, ER-α alone, FOXA1 alone and GATA3 alone) were associated only with a subset of these pathways, and many enriched pathways were not related to luminal or mammary cell identity (Supplementary Figure S1C–G).
GATA3 mutation induces redistribution of ER-α
To dissect the impact of GATA3 mutations on the gene regulatory network between ER-α, FOXA1 and GATA3, we utilized a previously established GATA3 mutant cell line (CR3) (21). This cell line endogenously expresses one of the recurrent ZnFn2 mutations (R330fs) found in multiple breast cancer cohorts (8,11,32,33). The second zinc-finger domain is essential for the recognition of the GATA3 consensus motif (WGATAR). A two-nucleotide deletion from one allele induces a frame-shift and premature protein truncation (Figure 2A). Most R330fs mutations (9 out of 10 R330fs mutant breast tumor cases) are found to be heterozygous. Therefore, this cell line mimics the situation of R330fs in human breast tumors. To investigate the impact(s) of the mutation on ER-α and FOXA1, we first assessed protein expression levels of ER-α and FOXA1 in wild-type and mutant T47D cells. While the ER-α mRNA level was decreased in CR3 cells (21), the protein level was similar to that in wild-type T47D cells (Figure 2A, Supplementary Figure S2A). The FOXA1 expression level in the mutant cells was equivalent to wild-type cells (Figure 2A, Supplementary Figure S2A).
Figure 2.

R330fs mutation induces global redistribution of ER-α. (A) Western blot showing ER-α, FOXA1 and GATA3 expression in T47D and CR3 cells. (B) Scatter plot showing differential binding of ER-α at ER-α peaks between wild-type and mutant T47D cells. CPM indicates counts per million in each peak. Increased, decreased, unchanged binding events are shown in red, blue, and yellow, respectively. (C) Heatmap analysis showing read density of ER-α ChIP-seq at ER-α peaks in T47D (left) or CR3 (right) cells. (D–F) Metaplot profiles of normalized ATAC-seq (D), H3K4me1 ChIP-seq (E) and H3K27ac (F) signals at ER-α ChIP-seq peaks in T47D (red) or CR3 (blue) cells.
To investigate the impact(s) of the GATA3 mutation on chromatin localization of these co-factors, we conducted ChIP-seq in the GATA3 mutant T47D cells and defined binding peaks by HOMER. The peak number for ER-α was decreased in CR3 cells by about one third (16 872 peaks in T47D cells; 11 337 peaks in CR3 cells). We then used EdgeR (31) to define differential ER-α chromatin enrichment (FDR < 0.05 and |fold change| > 1.5) (Figure 2B, Supplementary Figure S2B). The majority of ER-α peaks (11,582 peaks, ∼62%) were unchanged in CR3 cells, while ER-α ChIP-seq signals at 3698 (∼20%) peaks were increased, and 3,512 (∼19%) ER-α peaks were significantly decreased when compared to control cells with wild-type GATA3 (Figure 2C). Differential enrichment of ER-α was confirmed by ChIP qPCR analysis (Supplementary Figure S2C).
To measure the impact(s) of differential binding of ER-α on chromatin architecture, we investigated chromatin accessibility by ATAC-seq and enhancer histone marks (H3Kme1 and H3K27ac) by ChIP-seq. Metaplot analysis of the ATAC-seq data displayed differential impacts in each peak group (Figure 2D–F). Loci where ER-α abundance remained unchanged showed minimal change in K4me1, K27ac and ATAC sensitivity. Loci where ER-α levels increase display concomitant increases in ATAC sensitivity and K27Ac, with slight changes in K4me1, suggesting increased enhancer activity (Supplementary Table S4). Finally, sites where ER-α levels decline have a decrease in K4me1 and K27ac levels with a slight decrease in ATAC, suggesting that a subset of these sites were decommissioned.
GATA3 mutation redirects FOXA1 chromatin localization
We next analyzed the effects of the GATA3 frameshift on FOXA1. FOXA1 ChIP-seq data indicated a similar number of FOXA1 peaks in T47D (39 041 peaks) and CR3 cells (37 877 peaks). Differential peak analysis (Figure 3A, B, Supplementary Figure S2B) identified 4765 increased peaks (∼9%), 5441 decreased peaks (∼11%), and 40 338 unchanged peaks (∼80%), indicating that FOXA1 redistribution is less frequent than that of ER-α. Chromatin architecture was also changed at FOXA1 binding sites. At sites where FOXA1 accumulation is not impacted by GATA3 mutation, enhancer histone marks and ATAC sensitivity were unchanged. At sites where FOXA1 levels changed significantly, ATAC sensitivity positively correlated with changes in FOXA1 binding, consistent with its function as a pioneer transcription factor (Figure 3C). Similarly, both H3K4me1 and H3K27ac levels also correlated with differential FOXA1 binding (Figure 3D–E). FOXA1-increased binding sites showed increased levels of these active enhancer marks, while FOXA1-decreased peak regions showed decreased levels of these histone modifications. Finally, we assessed ER-α and FOXA1 binding at the GATA3 binding peaks. For this analysis, we utilized the previously defined 5161 increased and 5954 decreased GATA3 peaks (21). The scatter plots clearly showed positive correlation between GATA3 redistribution and ER-α or FOXA1 redistribution (Figure 4A–C). FOXA1 and GATA3 differential binding in CR3 cells was also confirmed by ChIP qPCR (Supplementary Figure S2D, E). These data indicate the R330fs mutant induces redistribution of all three transcription factors at a subset of GATA3-bound loci.
Figure 3.

R330fs mutation reshapes FOXA1 distribution and chromatin architectures. (A) Scatter plot showing differential binding of FOXA1 at FOXA1 peaks in wild-type versus mutant T47D cells. CPM indicates counts per million in each peak. Increased, decreased, unchanged binding events are shown in red, blue, and yellow, respectively. (B) Heatmap analysis displaying normalized read density of FOXA1 ChIP-seq at FOXA1 peaks in T47D (left) or CR3 (right) cells. (C–E) Metaplot profiles of normalized ATAC-seq (C), H3K4me1 ChIP-seq (D) and H3K27ac (E) signals at FOXA1 ChIP-seq peaks in T47D (red) or CR3 (blue) cells.
Figure 4.
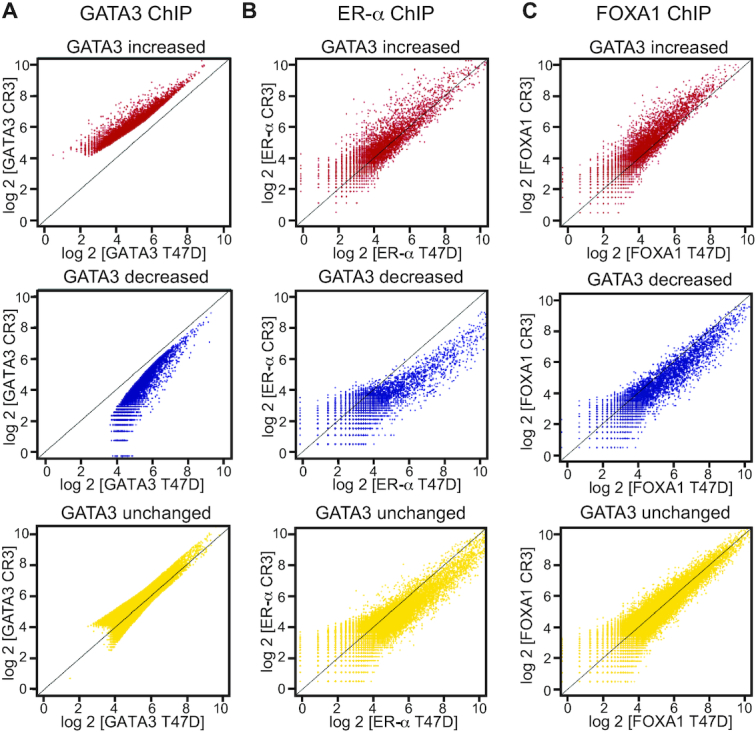
Relocalizations of ER-α and FOXA1 at GATA3 peaks. (A) Scatter plot showing GATA3 ChIP-seq signals in each differential GATA3 binding group. X-axis indicates GATA3 ChIP-seq in wild-type T47D cells, while Y-axis indicates GATA3 ChIP-seq in CR3 cells. (B) Scatter plot showing ER ChIP-seq signals in each GATA3 peak group. X-axis: ER ChIP-seq in wild-type T47D cells, Y-axis: ER ChIP-seq in CR3 cells. (C) Scatter plot showing FOXA1 ChIP-seq signals in each GATA3 peak group. X-axis: FOXA1 ChIP-seq in wild-type T47D cells, Y-axis: FOXA1 ChIP-seq in CR3 cells.
Redistribution of ER-α and FOXA1 reshapes transcriptional program
To measure the impact(s) of redistribution of ER-α and FOXA1 on gene expression, gene set enrichment analysis (GSEA) was conducted on data from cells bearing the GATA3 frame-shift mutant. We first assigned the ER-α and FOXA1 peaks to nearest genes (within 50 kb). The genes close to sites of ER-α -increased peaks in CR3 cells were frequently up-regulated when compared to wild-type T47D cells, whereas ER-α -decreased peaks are often observed near down-regulated genes (Figure 5A). Similar to the situation with ER-α, differential binding of FOXA1 exhibited strong positive correlation with gene expression changes in wild-type versus mutant T47D cells (Figure 5B). The gene ontology analysis of ER-α- or FOXA1-associated genes indicated that sites containing either ER-α or FOXA1 decreases in peaks were enriched for pathways related to gland development, epithelial cell development, and hormone signaling (Figure 5C, Supplementary Figures S3 and S4A, B). Increased binding of ER-α was associated with cell adhesion pathways, while FOXA1-increased sites were enriched in genes related to reproductive system development (Figure 5C, Supplementary Figure S4A, B). To dissect the upstream regulators of differentially expressed genes that are associated with ER-α or FOXA1 differential binding, IPA (Ingenuity Pathway Analysis, QIAGEN) pathway analysis was conducted. This upstream regulator analysis indicated that loss of ER-α and FOXA1 peaks were associated with down-regulation of signaling cascades regulated by ligand-dependent nuclear receptors such as PPARG (Figure 5D, Supplementary Figure S4C). TGFB1 pathway-related genes were also enriched in both cases. On the other hand, increased binding of ER-α was preferentially found at genes involved in AHR and CCL5, and these pathways were predicted to be activated (Figure 5E). Increased FOXA1 peaks were significantly associated with TP53, CD24 and TNF pathway activation (Supplementary Figure S4D).
Figure 5.

Relocalizations of ER-α and FOXA1 are associated with differential gene expression. (A) GSEA analysis indicating correlation between ER-α binding and gene expression in each ER-α peak group. (B) GSEA analysis indicating correlation between FOXA1 binding and gene expression in each FOXA1 peak group. (C) Biological pathway analysis of genes associated with ER-α decreased (top) or increased (bottom) peaks. Top 12 significantly enriched pathways are shown. (D) Upstream regulator analysis of genes associated with ER-α decreased binding. (E) Upstream regulator analysis of genes associated with ER-α increased peaks.
We then performed Oncomine analysis to explore the potential clinical relevance of the differential binding of ER-α and FOXA1 in breast cancer. The genes associated with decreased ER and FOXA1 peaks showed significant association with the genes that are down-regulated in highly aggressive breast cancer cohorts such as metastatic breast tumors and recurrent breast tumors (Supplementary Figure S5A-C). In contrast, the genes associated with increased ER peaks tend to be down-regulated in PR positive breast cancer. A subset of ER or FOXA1 increased peak-associated genes are also up-regulated in ERBB2 positive cancer (Supplementary Figure S5D, E). Taken together, these results suggest that the altered genomic distribution of ER-α and FOXA1 in the GATA3 R330fs mutant contributes to cellular reprogramming at the transcription level and to a more aggressive phenotype in the mutant cells.
GATA3 mutant disrupts ER-α -FOXA1-GATA3 network
Next, we examined the impact of the GATA3 mutation on colocalization of ER-α–FOXA1–GATA3. In the GATA3 mutant cells, peak overlap frequency of ER-α, FOXA1 and GATA3 was decreased, but colocalization was still detected (Figure 6A). Unlike the case with wild-type T47D, approximately 40% of all ER-α peaks were not found in association with either FOXA1 or GATA3 (Figure 6A). When we compared chromatin enrichment between GATA3 wild-type and mutant T47D cells in each peak group, binding of GATA3 was significantly decreased at EFG sites in the GATA3 mutant cells, while GATA3 alone peaks exhibited increased GATA3 binding (Figure 6B, C, Supplementary Figure S6A-B). In addition, binding of both ER-α and FOXA1, chromatin accessibility, and active enhancer marks were all reduced at EFG sites (Figure 6B, 6D, E), indicating that the GATA3 mutation preferentially impacts loci co-bound by all three factors.
Figure 6.

R330fs mutant interrupts ER-α -FOXA1-GATA3 network. (A) Venn diagram showing overlap between ER-α, FOXA1, and GATA3 peaks in CR3 cells. (B) Genome browser tracks at PR enhancer locus. ER-α, FOXA1, and GATA3 binding were drastically reduced. (C) Box plot showing normalized read of GATA3 ChIP-seq at EFG or GATA3 alone peaks in T47D (left) or CR3 (right) cells. Y-axis indicates normalized reads per peak. (D) Box plot showing normalized read of ER-α (left) or FOXA1 (right) ChIP-seq at EFG peaks. (E) Box plot showing normalized read of ATAC-seq, H3K4me1, and H3K27ac ChIP-seq signals at EFG peaks. (F, G) Bar graph showing the peak overlap frequency between ER-α or FOXA1 differential peaks and each peak group.
To interrogate the impact of GATA3 mutation on loci co-bound by all three factors, we examined ER-α and FOXA1 peak categories within differential binding peaks (defined above). The peak overlap analysis clearly indicated that both ER-α and FOXA1 reduction were more frequently observed at EFG peaks than any other category (Figure 6F, G). Metaplot analysis also confirmed that a significant reduction of GATA3 ChIP signals was detected at ER-α -decreased peaks in CR3 cells, while a moderate GATA3 increase was detected at ER-α -increased peaks in CR3 cells (Figure 7A). Similarly, GATA3 ChIP signals were also drastically reduced at FOXA1-decreased peaks in the mutant cells (Figure 7B). These results demonstrate that ER-α and FOXA1 chromatin binding mirror that of GATA3 at sites bound by all three factors.
Figure 7.

Motif analysis suggests decreased cooperation of ER-α -FOXA1-GATA3 complex in GATA3 mutant cells. (A) GATA3 binding profiles at ER-α differential binding peaks. GATA3 ChIP-seq data from T47D cells is shown in red, and the profile from CR3 cells is shown in blue. (B) GATA3 binding profiles at FOXA1 differential binding peaks. (C) GATA3, FOXA1, and ER-α motif frequency in ER-α increased or decreased binding sites. (D) GATA3, FOXA1, and ER-α motif frequency in FOXA1 increased or decreased binding sites. (E, F) HOMER de novo motif analysis of ER-α decreased (E) or increased (F) peaks. (G, H) HOMER de novo motif analysis of FOXA1 decreased (G) or increased (H) peaks. Top 5 motifs are indicated. (I) Heatmap showing EFG-associated gene expression in breast tumors. Gene expression data was extracted from METABRIC. The color scale indicates row Z-scores averaged across all samples per GATA3 mutation group.
To gain insights into the mechanisms underlying the GATA3 mutant-induced relocalization of FOXA1 and ER-α, we analyzed the motif frequency at differential binding sites. While the metaplot analysis displayed a weak increase of GATA3 ChIP-seq signals at peaks showing an increase in ER-α or FOXA1, the GATA3 consensus motif (WGATAR) was less frequently detected at these peaks as compared to ER-α and FOXA1 decreased peaks (Figure 7C, D). Similarly, peaks associated with increases in FOXA1 binding in CR3 cells carry less ER-α motifs than peaks associated with decreased FOXA1 binding, and ER-α increased peaks manifest less FOXA1 motifs than do ER-α decreased peaks. However, their own motifs are more frequently observed in increased peaks than decreased peaks. The de novo HOMER motif analysis is consistent with the model that ER-α and FOXA1 decreased binding is enriched at the sites that contain all three (ER-α, FOXA1 and GATA3) motifs, while increased peaks are enriched for motifs of other transcription factors (Figure 7E-H).
Finally, we assessed gene expression of EFG-peak associated genes in human breast tumors (METABRIC) (11). Breast tumors were previously categorized based on the types of GATA3 mutations (21). The breast tumors carrying the same type of ZnFn2 mutations as the mutant studied here showed altered expression of EFG-associated genes. In contrast, breast tumors that have intact GATA3 or other types of GATA3 mutations do not exhibit this correlation of gene expression, suggesting that GATA3 ZnFn2 mutations impact the ER-α–FOXA1–GATA3 regulated genes in vivo (Figure 7I). Collectively, these data suggest that GATA3 zinc finger 2 mutations disrupt the gene regulatory hub governed by ER-α, FOXA1 and GATA3.
DISCUSSION
Somatic mutations within transcription factors are a common phenomenon in cancer. One such example, mutation of the lineage-specifying transcription factor GATA3, occurs with high frequency in breast cancer. The biological significance of GATA3 mutations remains unclear. We previously established a GATA3 mutant cell line that endogenously expresses one of the recurrent GATA3 heterozygous mutants (R330fs) (21). This cell line exhibits more aggressive phenotypes at both cellular and transcriptome levels. The R330fs mutation alters distribution of wild-type GATA3 on chromatin leading to down-regulation of the progesterone receptor signaling pathway. R330fs mutant also shows a unique chromatin binding pattern as compared to wild-type. However, we observed more than a thousand differentially expressed genes between wild-type and mutant GATA3 cells, suggesting other potential mechanisms that may contribute to the drastic phenotypic changes observed in GATA3 mutant cells. Our present study extends analysis of this class of mutations to the estrogen receptor alpha/FOXA1/GATA3 transcriptional network. The resulting data and analysis demonstrate that the R330fs mutant is sufficient to interrupt and reshape chromatin localization of two GATA3 co-cofactors, ER-α and FOXA1 (Supplementary Figure S7). Redistribution of ER-α and FOXA1 is associated with changes in chromatin architecture and gene expression. Decreased binding of ER-α, FOXA1, and GATA3 was predominantly observed within those loci where all three factors are co-localized. These loci are enriched at sites adjacent to epithelial marker genes including CTSD (Figure 1B), KRT8 (Supplementary Figure S6A), CLND1 and OCLN. The R330fs mutant cells exhibit partial epithelial-to-mesenchymal transition (21). Therefore, impaired binding at the co-bound loci may be causal for the reduction in expression of key epithelial marker genes in mutant cells.
Silencing of GATA3 by siRNA changes localization of ER-α and FOXA1, and of enhancer activities that interact with ER-α -dependent genes (13). The R330fs mutation in the heterozygous state can induce both loss and gain of ER-α, FOXA1, and GATA3 binding with a net loss of all three factors in mutant cells at loci bound by all three transcription factors (Figure 6). At loci where either FOXA1 or ER-α peaks decrease in GATA3 mutant cells, GATA3 recognition motifs are enriched, suggesting that decreased levels of wild-type GATA3 at these sites results in decreased binding of partner transcription factors. In contrast, at loci where FOXA1 and ER-α binding increase in mutant cells, GATA3 motifs are not enriched and GATA3 binding is reduced (Figure 7), suggesting that ER-α and FOXA1 may be constrained in some manner by GATA3. These data suggest that the R330fs mutation selectively alters the correlation between GATA3, ER-α and FOXA1 observed in cancer and compromises the regulatory network governed by these three transcription factors.
Importantly, sites jointly bound by ER-α, FOXA1 and GATA3 showed strong open chromatin features as well as active enhancer status. Our GATA3 mutant cell line model suggests that intact binding of the three factors are essential to maintain a highly active chromatin state at these loci. The genes associated with these loci are strongly enriched in genes integral to mammary gland development and function as well as genes critical to the epithelial cell program. The impairment of this critical gene set likely contributes to the aggressive phenotype of mutant cells in culture and the poor outcome of tumors bearing similar mutations in vivo.
At a very simple level, transcription factors can be considered as a means of delivering regulatory information governing transcriptional output to specific loci within the genome. As a family, transcription factors are generally considered to be modular – they have various types of DNA interaction domains and diverse types of activation domains frequently interspersed with regulatory domains. In this work, we demonstrate that mutation in a DNA binding domain leads to aberrant transcription factor localization and downstream rewiring of a transcriptional network that controls cell identity. However, normal transcriptional regulation requires intact function of all domains of transcription factors. This is illustrated within the GATA transcription factor family where mutations that disrupt protein-protein interactions (34) or mutations that disrupt an activation domain (35) can also lead to aberrant localization of lineage-determining transcription factors and rewiring of transcriptional regulatory networks. As the relationship in cancer between transcription factor mutation and downstream function appear to be complex, interpretation of the clinical impact of the increasing number of such mutations identified in cancer will require careful investigation.
DATA AVAILABILITY
The RNA-seq, ChIP-seq, and ATAC-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible under accession number GSE99479 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99479) and GSE130703 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130703).
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge NIEHS/NIH core facilities (Epigenomics Core and Integrated Bioinformatics Core) for outstanding technical assistance. We thank Dr John D. Roberts and Dr Paula M. Vertino for insightful suggestions on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health [ES101965 to P.A.W., in part]; Division of Intramural Research Innovative Research Award from National Institute of Environmental Health Sciences, Division of Intramural Research [to M.T.]; National Institutes of Health [P20GM104360 to M.T.]; start-up fund provided by the University of North Dakota School of Medicine and Health Sciences, Department of Biomedical Sciences [to M.T.]. Funding for open access charge: National Institute of Environmental Health Sciences [ES101965].
Conflict of interest statement. None declared.
REFERENCES
- 1. Asselin-Labat M.L., Sutherland K.D., Barker H., Thomas R., Shackleton M., Forrest N.C., Hartley L., Robb L., Grosveld F.G., van der Wees J. et al.. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007; 9:201–209. [DOI] [PubMed] [Google Scholar]
- 2. Kouros-Mehr H., Bechis S.K., Slorach E.M., Littlepage L.E., Egeblad M., Ewald A.J., Pai S.Y., Ho I.C., Werb Z.. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell. 2008; 13:141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kouros-Mehr H., Slorach E.M., Sternlicht M.D., Werb Z.. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006; 127:1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mehra R., Varambally S., Ding L., Shen R., Sabel M.S., Ghosh D., Chinnaiyan A.M., Kleer C.G.. Identification of GATA3 as a breast cancer prognostic marker by global gene expression meta-analysis. Cancer Res. 2005; 65:11259–11264. [DOI] [PubMed] [Google Scholar]
- 5. Oh D.S., Troester M.A., Usary J., Hu Z., He X., Fan C., Wu J., Carey L.A., Perou C.M.. Estrogen-regulated genes predict survival in hormone receptor-positive breast cancers. J. Clin. Oncol. 2006; 24:1656–1664. [DOI] [PubMed] [Google Scholar]
- 6. Perou C.M., Sorlie T., Eisen M.B., van de Rijn M., Jeffrey S.S., Rees C.A., Pollack J.R., Ross D.T., Johnsen H., Akslen L.A. et al.. Molecular portraits of human breast tumours. Nature. 2000; 406:747–752. [DOI] [PubMed] [Google Scholar]
- 7. Takaku M., Grimm S.A., Wade P.A.. GATA3 in breast cancer: tumor suppressor or oncogene. Gene Expr. 2015; 16:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. The Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Usary J., Llaca V., Karaca G., Presswala S., Karaca M., He X., Langerod A., Karesen R., Oh D.S., Dressler L.G. et al.. Mutation of GATA3 in human breast tumors. Oncogene. 2004; 23:7669–7678. [DOI] [PubMed] [Google Scholar]
- 10. Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C. et al.. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015; 163:506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pereira B., Chin S.F., Rueda O.M., Vollan H.K., Provenzano E., Bardwell H.A., Pugh M., Jones L., Russell R., Sammut S.J. et al.. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016; 7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takaku M., Grimm S.A., Shimbo T., Perera L., Menafra R., Stunnenberg H.G., Archer T.K., Machida S., Kurumizaka H., Wade P.A.. GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome Biol. 2016; 17:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Theodorou V., Stark R., Menon S., Carroll J.S.. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013; 23:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carroll J.S., Liu X.S., Brodsky A.S., Li W., Meyer C.A., Szary A.J., Eeckhoute J., Shao W., Hestermann E.V., Geistlinger T.R. et al.. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005; 122:33–43. [DOI] [PubMed] [Google Scholar]
- 15. Zaret K.S., Carroll J.S.. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011; 25:2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iwafuchi-Doi M. The mechanistic basis for chromatin regulation by pioneer transcription factors. Wiley Interdiscipl. Rev. Syst. Biol. Med. 2019; 11:e1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. King H.W., Klose R.J.. The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife. 2017; 6:e22631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iwafuchi-Doi M., Zaret K.S.. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014; 28:2679–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kong S.L., Li G., Loh S.L., Sung W.K., Liu E.T.. Cellular reprogramming by the conjoint action of ERalpha, FOXA1, and GATA3 to a ligand-inducible growth state. Mol. Syst. Biol. 2011; 7:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin C.Y., Vega V.B., Thomsen J.S., Zhang T., Kong S.L., Xie M., Chiu K.P., Lipovich L., Barnett D.H., Stossi F. et al.. Whole-genome cartography of estrogen receptor alpha binding sites. PLos Genet. 2007; 3:e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takaku M., Grimm S.A., Roberts J.D., Chrysovergis K., Bennett B.D., Myers P., Perera L., Tucker C.J., Perou C.M., Wade P.A.. GATA3 zinc finger 2 mutations reprogram the breast cancer transcriptional network. Nat. Commun. 2018; 9:1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emmanuel N., Lofgren K.A., Peterson E.A., Meier D.R., Jung E.H., Kenny P.A.. Mutant GATA3 actively promotes the growth of normal and malignant mammary cells. Anticancer Res. 2018; 38:4435–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gustin J.P., Miller J., Farag M., Rosen D.M., Thomas M., Scharpf R.B., Lauring J.. GATA3 frameshift mutation promotes tumor growth in human luminal breast cancer cells and induces transcriptional changes seen in primary GATA3 mutant breast cancers. Oncotarget. 2017; 8:103415–103427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mair B., Konopka T., Kerzendorfer C., Sleiman K., Salic S., Serra V., Muellner M.K., Theodorou V., Nijman S.M.. Gain- and Loss-of-Function mutations in the breast cancer gene GATA3 result in differential drug sensitivity. PLoS Genet. 2016; 12:e1006279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adomas A.B., Grimm S.A., Malone C., Takaku M., Sims J.K., Wade P.A.. Breast tumor specific mutation in GATA3 affects physiological mechanisms regulating transcription factor turnover. BMC Cancer. 2014; 14:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jothi R., Cuddapah S., Barski A., Cui K., Zhao K.. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008; 36:5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nik-Zainal S., Davies H., Staaf J., Ramakrishna M., Glodzik D., Zou X., Martincorena I., Alexandrov L.B., Martin S., Wedge D.C. et al.. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang Y.Z., Yu K.D., Zuo W.J., Peng W.T., Shao Z.M.. GATA3 mutations define a unique subtype of luminal-like breast cancer with improved survival. Cancer. 2014; 120:1329–1337. [DOI] [PubMed] [Google Scholar]
- 34. Ang Y.S., Rivas R.N., Ribeiro A.J.S., Srivas R., Rivera J., Stone N.R., Pratt K., Mohamed T.M.A., Fu J.D., Spencer C.I. et al.. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 2016; 167:1734–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chlon T.M., McNulty M., Goldenson B., Rosinski A., Crispino J.D.. Global transcriptome and chromatin occupancy analysis reveal the short isoform of GATA1 is deficient for erythroid specification and gene expression. Haematologica. 2015; 100:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq, ChIP-seq, and ATAC-seq data have been deposited in NCBI’s Gene Expression Omnibus and are accessible under accession number GSE99479 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99479) and GSE130703 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130703).