

Abstract
The discovery that rare POT1 variants are associated with extremely long telomeres and increased cancer predisposition has provided a framework to revisit the relationship between telomere length and cancer development. Telomere shortening is linked with increased risk for cancer. However, over the past decade, there is increasing evidence to show that extremely long telomeres caused by mutations in shelterin components (POT1, TPP1, and RAP1) also display an increased risk of cancer. Here, we will review current knowledge on germline mutations of POT1 identified from cancer-prone families. In particular, we will discuss some common features presented by the mutations through structure-function studies. We will further provide an overview of how POT1 mutations affect telomere length regulation and tumorigenesis.
Keywords: telomere, POT1, cancer, germline mutation
Telomere length impacts tumorigenesis
Human telomere length is highly heterogeneous, typically ranging from 5 kb to 15 kb in the population, with considerable variability between individuals, among different tissues, and across different stages of a lifetime [1]. The importance of setting telomere length in a fine range has been well demonstrated. Critically short telomeres have been linked to degenerative diseases and increased cancer risk [2–6]. However, a growing body of epidemiological evidence has shown that individuals with extremely long telomeres have an increased risk for cancers, including gliomas, melanomas, and pulmonary and pancreatic cancers [7–12]. The molecular mechanisms of how defective telomere length maintenance contributes to tumorigenesis are not well understood. The genetic association of telomere length abnormalities with cancer has led to the identification of multiple population-linked SNPs and rare variants found in cancer patients, many of which are telomere-related. The first connection was revealed in 2013 with the identification of a gain-of-function mutation in the TERT promoter of tumor samples derived from familial melanoma cases across five generations [13]. This mutation resulted in an up to two-fold increase in TERT transcription. Since then, a number of germline mutations have been revealed in telomeric shelterin components, including POT1, TPP1, and RAP1 [14–25]. Among them, POT1 mutations are the most common and have been reported in a broad range of cancer types. In this review, we focus on emerging evidence from cancer-prone families as well as structure-function mechanistic studies linking germline mutations of POT1 to cancer susceptibility.
Deleterious germline POT1 variants identified in cancers
Approximately 10% of cutaneous malignant melanoma (CMM) cases occur in a familial setting, and 20–40% of familial melanomas exhibit mutations in the CDKN2A gene. In an attempt to identify new high-penetrance susceptibility genes for familial CMM, Shi et al. and Robles-Espinoza et al. performed whole-exome and genome sequencing on tumors derived from more than 100 cases of familial melanomas and identified POT1 as a second major susceptibility gene for CMM in several populations [14,15]. One missense POT1 variant (g.124493086C>T; p.Ser270Asn) was further traced back to approximately 10 generations ago and identified as a founder mutation in five unrelated melanoma-prone families in Italy [15]. To date, the tumor spectrum has been expanded to gliomas, cardiac angiosarcomas, chronic lymphocytic leukemia (CLL), colorectal cancer, and Hodgkin’s lymphoma [14–25].
POT1 directly binds single-stranded telomeric DNA sequences through its two N-terminal oligonucleotide/oligosaccharide-binding (OB) fold domains [26]. Yet due to its low abundancy inside the cell, POT1 telomeric localization requires interaction with TPP1 through its C-terminal half, which contains a third OB fold domain and a Holliday junction resolvase-like domain (Figure 1) [27–30]. This POT1/TPP1 heterodimer further stabilizes the protein complex and maintains the overall POT1 level in the cell. Once bound to the 3’ telomeric overhang, POT1 has two major functions. First, it inhibits the recruitment of ataxia telangiectasia and Rad3-related (ATR) to replication protein A (RPA) coated single-stranded DNA (ssDNA) by competitively binding RPA. Therefore, it represses the subsequent ATR-dependent DNA damage response (DDR) [31,32]. Second, it blocks access of telomerase to its substrate in late S phase by direct occupation of the G-strand overhang, but also acts as a telomerase processivity factor together with TPP1 [33,34]. As expected, any structural disruption caused by POT1 mutation may affect its telomere localization, protein stability, ssDNA binding, telomerase recruitment and/or processivity, and eventually, telomere length and function. Early somatic mutations of POT1 were found enriched in the OB folds in patients with chronic lymphocytic leukemia (CLL) [35]. However, current sequencing data has indicated that the positions of POT1 germline mutations may be located throughout the entire protein. Based on their distribution and structural consequences, these mutations can be separated into three groups:
Figure 1.
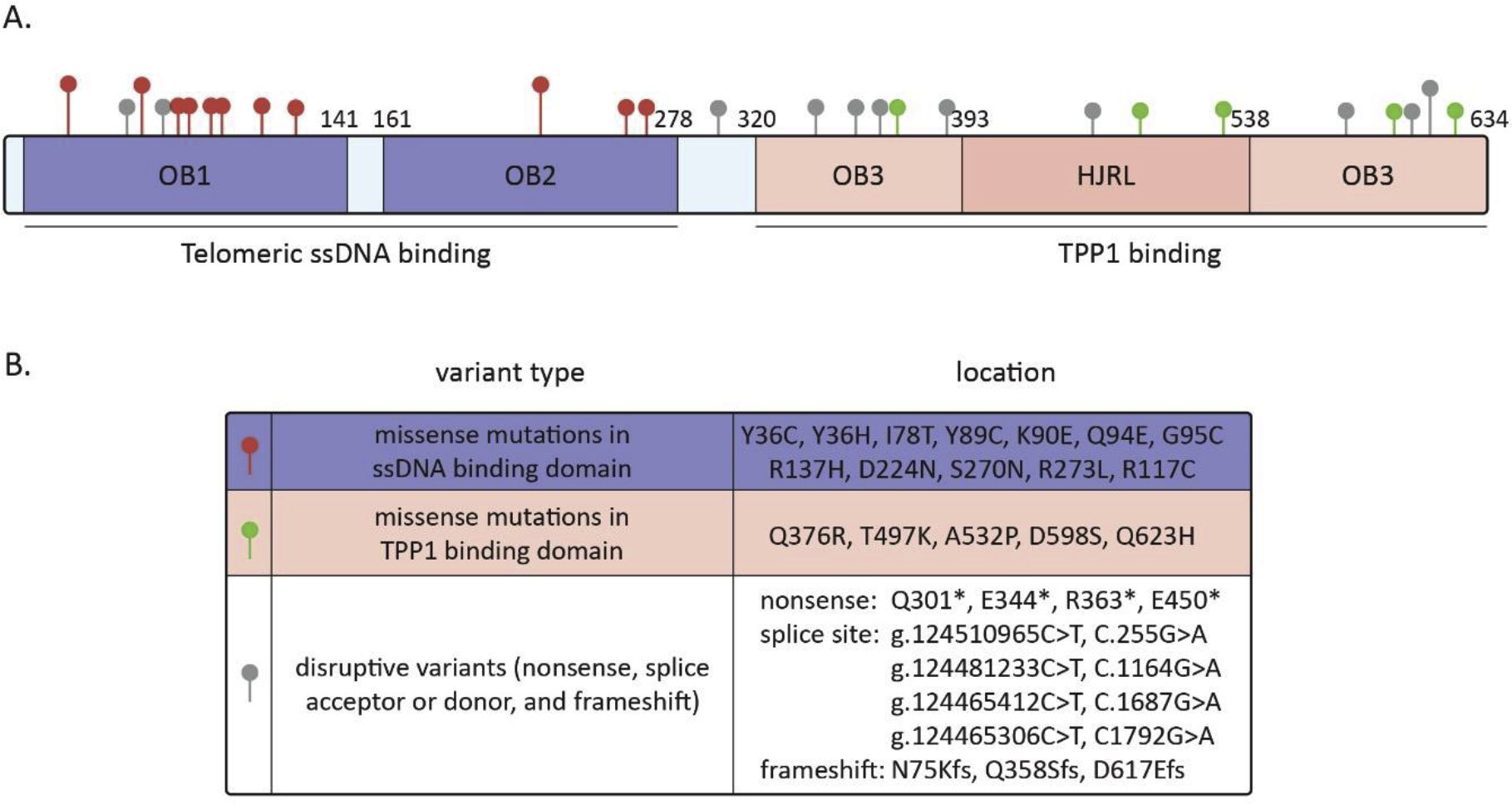
Rare germline variants in POT1 identified in familial cancers. (A) Schematic structure of human POT1 protein and conserved domains. OB1 and OB2 folds of POT1 mediating telomeric ss-DNA binding are colored in blue. The C-terminal OB3 and embedded HJRL domain mediating TPP1 binding are colored in red. The positions of variants identified in familial cancers are shown as pins on top of the protein. The taller pins represent the mutations that have been identified more than once. (B) Table of deleterious germline mutations identified in the POT1 gene.
Missense mutations in POT1 ssDNA binding OB folds
OB folds are important functional modules that mediate various protein-DNA, -RNA, and -protein interactions [36]. Structural analysis of the human POT1-ssDNA complex has revealed that the telomeric ssDNA adopts an irregular and extended conformation and binds both OB folds, where 24 protein residues of POT1 are used to make protein-ssDNA contact. A total of 12 missense mutations have been identified, whereas Tyr36Cys/Tyr36His, Ile78Thr, and Asp224Asn were found twice in two separate studies (Figure 1). Seven of them are predicted to be deleterious, either by directly affecting one of the 24 residues found at the POT1-telomeric polynucleotide interface (Tyr36Cys/Tyr36His, Tyr89Cys, Gln94Glu, Asp224Asn, Ser270Asn, Arg273Leu), or by affecting an α-helix structure involved in OB folding (Arg137His). Reduction or loss of POT1-DNA complex formation was confirmed in most of these mutations by in vitro telomeric ssDNA binding assay. So far, only Lys90Glu was reported to have the same DNA binding affinity as wild-type(wt) POT1 [37]. Functional characterization of the mutants was conducted in cells and mice exogenously expressing individual substitutions [37,38]. These studies demonstrated that all mutant alleles were capable of localizing to telomeres as expected, confirming that the mutations did not fully abolish binding to TPP1. As the POT1 variants identified from the patients were heterozygous, with carrier cells still retaining one wt allele, limited characterization of peripheral blood mononuclear cells (PBMCs) harboring Asp224Asn and Ser270Asn showed no presence of telomere dysfunction-induced foci (TIF), suggesting that the wt allele of POT1 is sufficient for ATR repression. Only when Tyr89Cys, Lys90Glu, Gln94Glu, and Ser270Asn are overexpressed exogenously, are TIF levels increased, possibly due to titration of wt POT1 present at single-stranded ends. All carriers of these variants, including Lys90Glu, have increased telomere lengths and numbers of fragile telomeres, a feature indicative of telomere replication-dependent defects.
One outlier of this group is the Arg117Cys mutation. Residue 117 is located within the OB1 fold, yet the single amino acid change greatly disrupts the overall protein conformation. As a result, this mutation was predicted to affect the interaction between OB1 and OB2 as well as the binding of POT1 to TPP1. Carriers of this variant demonstrated a severe phenotype, exemplified by reduced telomere bound POT1, increased TIF levels, and abnormally long/fragile telomeres [17].
Missense mutations in the POT1-TPP1 binding region
POT1 and TPP1 form a stable heterodimer that caps the telomere ends and regulates the access and processivity of telomerase [33,34]. As stated earlier, POT1 sequesters the 3’-overhang, thereby preventing telomerase access. In contrast to the negative regulation of telomerase by POT1, TPP1 positively mediates telomere lengthening by recruiting TERT through the seven amino acids, known as the TEL patch [39]. Recent structural work has revealed POT1 C- terminal binding to TPP1 spans a large region and makes extensive interactions [29,30]. POT1 mutations occur in this region, which involve residues 376, 497, 532, 598, and 623 (Figure 1). POT1 Ala532Pro and Gln623His mutants have been characterized in detail at the cellular level. Residue 623 is located in the binding pocket of POT1, which directly engages TPP1. The Gln623His mutant protein displayed a 4–5-fold decreased binding affinity to TPP1 and was less stable compared to the wt protein. When the Gln623His mutant was overexpressed in U2OS cells, it was able to localize to telomeres, though with decreased efficiency. However, this mutant was unable to completely repress DDR activation at telomeres. The Gln623His mutant displayed low to medium levels of TIF formation. This was a surprising result, since the Gln623His mutant did not significantly impact the ability of POT1 to interact with telomeric ssDNA, based on in vitro ssDNA binding results. Thus, either the overall expression of POT1 was too low to protect the telomere ends, or the TIFs arose from a capping-independent mechanism, such as defective telomere replication. Finally, cells carrying the Gln623His mutation showed a significant increase in telomere length and fragility after long term culture [29,30].
Unlike the Gln623His mutant, the phenotype of the Ala532Pro mutant is less severe. Structurally, residue 532 is located away from the TPP1 binding site and does not appear to have a significant impact on POT1/TPP1 complex formation. As expected, the mutant protein displayed almost the same binding affinity to TPP1 as wt POT1. However, in vitro protein purification demonstrated decreased expression of the Ala532Pro protein, possibly due to unfolded protein response-mediated degradation. When overexpressed in cells, the mutant still localized to telomeres and did not significantly impact telomere length during prolonged culture. The only significant phenotype presented by the Ala532Pro mutant was an increased incidence of fragile telomeres [30].
Nonsense and splicing mutations
Human POT1 has 19 exons and is predicted to produce 5 splice variants; transcripts 2, 3 and 5 are nonsense mediated mRNA decay candidates. Variants 1, 2, 3, and 5 share the same N-terminal sequence (encoding OB1 and OB2 domains) and terminate through different splicing to generate truncated versions distinct from the widely studied POT1 variant 1 protein. Variant 4 encodes a 55 kDa protein with an in-frame start codon ATG in exon 9, thereby missing most of the N-terminal OB1 domain [40]. Similar to the naturally occurring POT1 splice variants, nonsense and splicing mutations are likely to result in truncated products. Indeed, one such mutant (g.124510965C>T, c.255G>A) is predicted to cause skipping of exon 7 to generate an isoform that corresponds to the natural POT1 variant 4 [21]. Disruptive mutations represent the second most common type of mutations identified besides OB folds mutants. 11 disruptive mutations have been identified, most of which are located in the C-terminal half of POT1 (Figure 1). The expression of the majority splice mutants has been confirmed by the presence of an aberrant transcript shown by RT-PCR. While little functional characterization has been conducted in this group, current structural information indicates that the mutants will be impacted in two major ways at the protein level. First, there is predicted to be decreased POT1 expression due to nonsense mediated decay (E450*), misfolded protein degradation, or complete/partial loss of TPP1 binding and stabilization. This would elicit a phenotype similar to that exhibited by genetic knockdown of POT1 [27,41]. Second, if the mutants are stably expressed, then the phenotype will be due to gaining extratelomeric function or will be reminiscent of natural POT1 variants. Earlier characterization demonstrated that POT1 variants are expressed in multiple tissues. Intriguingly, the variants displayed different DNA binding affinities and telomere elongation abilities [42,43].
Mechanistic basis of telomere overlengthening caused by POT1 mutation
The most consistent phenotypes from the structure-function analysis of the POT1 variants are longer telomere length and elevated levels of fragile telomeres, which can be explained by several possibilities:
Loss of negative suppression by POT1
Loss of functional OB folds: POT1 blocks telomerase access through direct occupation of the 3’ single-stranded G overhang. When the binding of OB folds to the 3’ terminus is weakened by mutation, exposure of ssDNA renders constitutive telomerase access and elongation. Earlier work demonstrated that deletion of POT1 OB1 fold resulted in a dramatic increase in telomere length [27,44]. The germline OB fold mutants described here reinforce the importance of this cis-inhibitory effect of POT1 on telomerase.
Loss of TPP1 interaction: As mentioned earlier, TPP1 is required for the POT1 localization to telomeres. In addition, in vitro studies have shown that TPP1 enhances the binding of POT1 to ssDNA up to 10 fold [33,34]. Finally, TPP1 binding to POT1 stabilizes the POT1 protein, as demonstrated in recent studies of POT1 mutants. Thus, any partial or complete disruption of the POT1/TPP1 complex would result in decreased levels of functional POT1 at telomere ends, which in turn, would lead to extensive telomere elongation.
Decreased expression of POT1 protein: POT1 is the least abundant shelterin component, estimated to be present at around 15,000 molecules per cell. Each telomere may contain only 50–100 molecules of POT1 and TPP1 [45]. With such limited expression of POT1, any further decrease in its protein level would have a profound impact on its function at telomeres. Accordingly, a previous report demonstrated that POT1 suppression with shRNA led to an increase in telomere length [27,44]. Decreased levels of POT1 protein in POT1 mutants could occur due to a destabilized POT1/TPP1 complex, destabilized POT1 transcript, or a misfolded protein response, all of which would cause POT1 haploinsufficiency.
Loss of telomerase inhibition from CST complex
In addition to persistent telomerase access, loss of inhibition of telomerase following telomeric repeat extension can also lead to telomere elongation. In mammalian cells, the CST (CTC1-STN1-TEN1) heterotrimer appears to play a key role in this step. CST, an RPA like OB containing complex, is found transiently associated with telomeres [46,47]. In late S/G2 phase, after telomerase extends the telomeric DNA, the CST complex is brought to telomeres by TPP1 and POT1, where it limits accumulation of telomerase and terminates telomerase action [48,49]. Recent studies have confirmed significant lengthening of telomeres in the context of CST mutation or inhibition [47,50,51]. Another function of CST at telomeres is to facilitate proper DNA replication. Due to the repetitive nature of telomeres and their tendency to form secondary structures, DNA replication experiences frequent fork stalling and/or collapse in telomeres, which is manifested as fragile telomeres in metaphase spreads [52–55]. It is unclear whether POT1 regulates CST during telomere replication. Since most POT1 mutants have elevated fragile telomere signals, it is tempting to speculate that CST function is compromised in this setting. Indeed, Pinzaru et al. observed that reduced interaction between STN1 and TEN1 was associated with the Lys90Glu mutation, suggesting that destabilized CST complex assembly might contribute to the phenotype [37].
Gain-of-function variants generated by POT1 mutation
In addition to the loss-of-function or haploinsufficiency mutants as discussed above, there are also possibilities of gain-of-function mutants. The truncation mutants generated by nonsense or splicing mutation are such candidates. Previous studies have shown that naturally occurring POT1 splice variants display different DNA binding and telomere elongation capacities [42,43]. Recent CRISPR knockout of individual POT1 isoforms further demonstrated differences in their ability to block ATR activation and to regulate telomere overhang length [56]. Notably, most of the truncation mutants are predicted to lose their TPP1 interaction domain partially or completely, and thus are not expected to be recruited to telomeres. If such proteins are stable, would they gain extratelomeric functions, find new binding partners, or be reminiscent of natural POT1 variants? Work on the POT1a paralog in Arabidopsis thaliana demonstrated that POT1a is part of the telomerase complex and acts as a positive regulator of telomere length by stimulating repeat addition processivity. Alternatively, could human POT1 splice variants gain more access to existing partners, for example, CST complex? Again, recent work from Arabidopsis thaliana indicates that POT1a is able to displace TEN1 (T) by competing for the same binding site on STN1 (S) and forming a complex with the remaining CST components-STN1 (S) and CTC1 (C). The interaction between POT1a-CS is proposed to promote a telomere extendable state [57,58]. Thus, it is possible that by switching or competing the subunit within the CST heterotrimer, the dynamic POT1-CST interaction can either destabilize/titrate away functional CST to mimic CST deficiency phenotype as described above, or lead to increased assess and/or utilization of its telomeric DNA substrate by telomerase.
Do POT1 mutations confer a selective advantage to promote tumorigenesis and replication stress?
Cancers with POT1 germline mutations are often described as early onset. Pedigree analysis from published cancer-prone families is consistent with a profile of genetic anticipation. That is, POT1 mutations are inherited across generations, and cancers are diagnosed at an earlier age in later generations, with early mortality observed in some cases. This pattern supports a general role for POT1 variants in cancer predisposition. However, the overall penetrance of POT1 alleles associated with cancer is modest or incomplete. Not all mutation carriers develop cancers in their lifetime. Other than CMM, in which POT1 mutations have been detected in 2–4% families, the overall frequency of POT1 mutation is low. In parallel, work from mouse models indicated that POT1a (mouse paralog) inactivation alone is insufficient to initiate tumorigenesis. Only when coupled with p53 deficiency, does loss of POT1 function result in formation of various cancers [37,38,59]. Collectively, it is likely that these POT1 mutations provide a selective advantage to cancer cells and facilitate cancer development.
Recent large-scale studies on the elderly population indicate that telomere lengthening may offer modest benefits for health-span, but could concomitantly increase the risk of cancer [60]. We consider that the increased telomere length evoked by POT1 mutations could provide a selective advantage to cancer cells through a variety of mechanisms. (I) delayed replicative senescence checkpoint: Telomere shortening and the associated replicative apoptosis/senescence is the first barrier to limit cell growth and block tumorigenesis. With excessively long telomeres, cells are capable of undergoing more replications and are likely to accumulate more mutations. If some of them are driving mutations, or each of them is associated with some fitness advantage, then ultimately the chance of having a clonal expansion and tumor formation is increased. From the cancer spectrum, POT1 mutations seem to have a preference to CMM. It is known that melanoma genomes have the highest mutation load of any cancer [61,62]. (II) increased genome instability: Telomeres are prone to replication stress due to their repetitive nature and tendency to form G-quadruplex-like structures. The excessively long telomere length associated with POT1 mutations would exacerbate the replication burden. In accord with this notion, characterization of different POT1 mutations have consistently shown elevated levels of fragile telomeres, which indicates telomere replication defects. Moreover, other chromosomal aberrations, including chromosomal breaks, sister telomere fusions, and chromosomal fusions are also prominent. (III) bypass the requirement for TERT in tumorigenesis: Enabling unlimited replicative potential is one of the hallmarks of cancer. This is achieved most commonly by telomerase reactivation, or less frequently, via an alternative homologous recombination-based telomere maintenance mechanism. It is estimated that more than 85% of cancer cells need to acquire telomerase reactivation to gain a growth advantage. However, Taboski et al. demonstrated that excessively long telomeres bypass the requirement for TERT in tumorigenesis [63]. Thus, bypassing telomerase activation may account for the accumulation of tumor promoting mutations and increase the risk of neoplasms.
Conclusion
Emerging evidence has shown that individuals with excessively long telomeres have an increased risk for cancers, which has led to the identification of a number of genomic rare variants in shelterin components, including POT1. In addition to the loss-of-function or haploinsufficiency variants, there are also possibilities of existence of gain-of-function variants. POT1 variants may confer a selective advantage to promote tumorigenesis through a variety of mechanisms. The study of rare POT1 variants offers a unique opportunity to dissect POT1 function in vivo. It is noteworthy that evolution naturally produces POT1 variants in other organisms, such as mice and Arabidopsis thaliana. For example, the Brassicaceae lineage of POT1a is under positive selection and the amino acids that were changed serve to enhance the interaction of POT1a with the STN1 component of CST [58]. Studies from these organisms may help our understanding of how rare POT1 variants in humans contribute to tumorigenesis. Finally, rapid advances in whole-exome and -genome sequencing technologies are transforming knowledge from genetic sequence to biological consequence. It is likely that more rare variants associated with shelterin and other telomere length maintenance genes will be identified in the future, and can render new insight into the role of extremely long telomeres in the susceptibility to cancer development.
Figure 2.
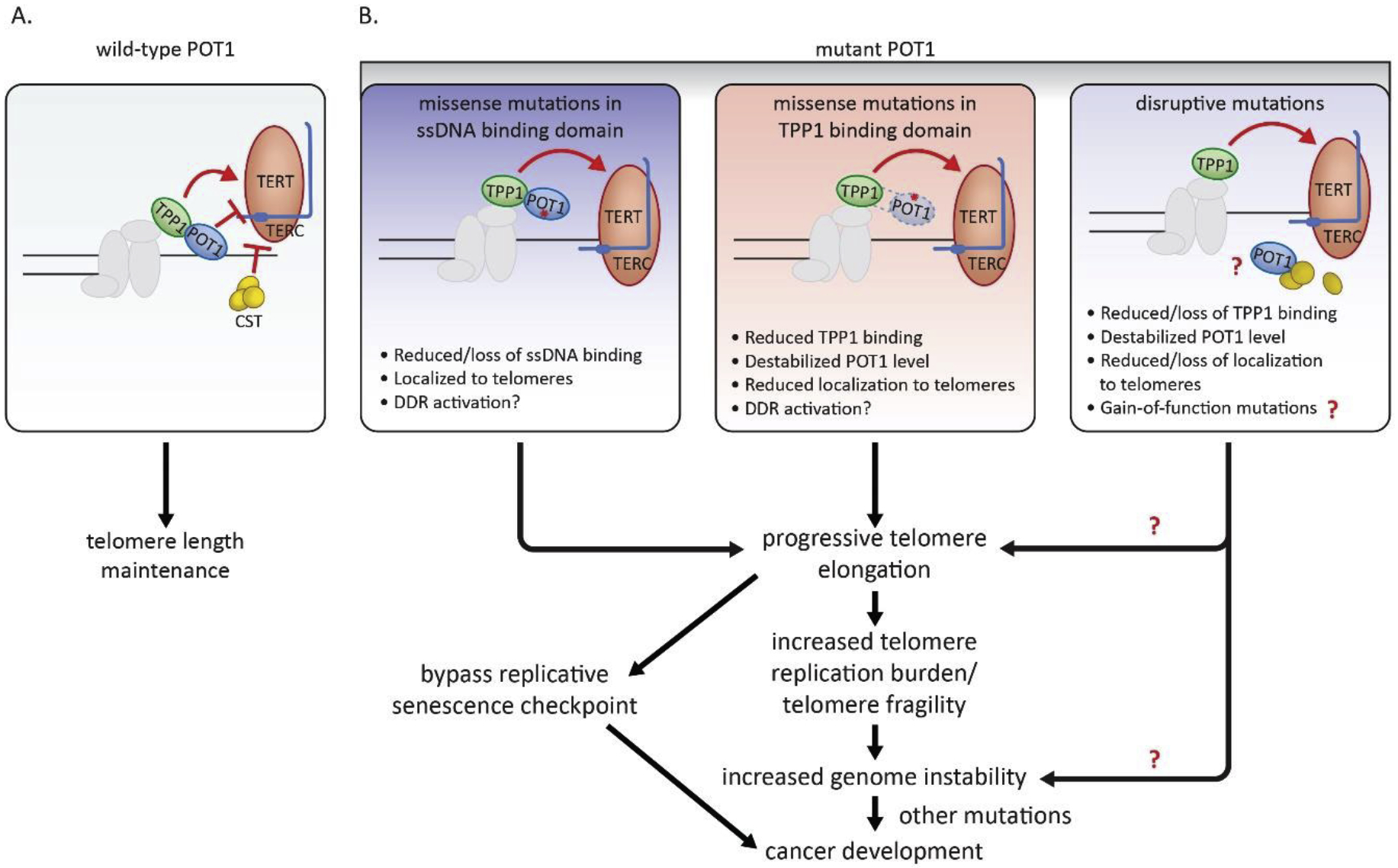
Impact of rare POT1 variants on telomere length, telomere DNA replication, genome integrity, and cancer. (A) Regulation of telomere length homeostasis by POT1. Wild-type POT1 together with TPP1 functions to protect telomere ends and to modulate telomerase access to 3’ ssDNA. The POT1-TPP1 complex both negatively and positively regulates telomerase recruitment and processivity. The CST complex facilitates telomere DNA replication and terminates telomerase action. (B) Rare POT1 variants lose the ability to protect telomeres through different mechanisms, shown in the boxes. Ultimately, cells undergo progressive telomere elongation, which further exacerbates replication burden and telomere fragility, consequently contributing to genome instability and cancer. Also, excessively long telomeres may allow bypass of the replicative senescence checkpoint and promote tumorigenesis.
ACKNOWLEDGEMENTS
We apologize to colleagues whose work could not be included owing to space constraints. We would like to thank Drs. Dorothy Shippen and Lea Harrington for the critical discussions on the manuscript, and Lauren Brick for assistance with the illustrations. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cleal K, Norris K, Baird D: Telomere Length Dynamics and the Evolution of Cancer Genome Architecture. Int J Mol Sci 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Savage SA: Beginning at the ends: telomeres and human disease. F1000Res 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aviv A, Anderson JJ, Shay JW: Mutations, Cancer and the Telomere Length Paradox. Trends Cancer 2017, 3:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, Varmus HE: Structure and variability of human chromosome ends. Mol Cell Biol 1990, 10:518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC: Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346:866–868. [DOI] [PubMed] [Google Scholar]
- 6.Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, Akdemir KC, Seth S, Song X, Wang Q, et al. : Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 2017, 49:349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Telomeres Mendelian Randomization C, Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, Wade KH, Timpson NJ, Evans DM, et al. : Association Between Telomere Length and Risk of Cancer and Non-Neoplastic Diseases: A Mendelian Randomization Study. JAMA Oncol 2017, 3:636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang C, Doherty JA, Burgess S, Hung RJ, Lindstrom S, Kraft P, Gong J, Amos CI, Sellers TA, Monteiro AN, et al. : Genetic determinants of telomere length and risk of common cancers: a Mendelian randomization study. Hum Mol Genet 2015, 24:5356–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh KM, Codd V, Rice T, Nelson CP, Smirnov IV, McCoy LS, Hansen HM, Elhauge E, Ojha J, Francis SS, et al. : Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 2015, 6:42468–42477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iles MM, Bishop DT, Taylor JC, Hayward NK, Brossard M, Cust AE, Dunning AM, Lee JE, Moses EK, Akslen LA, et al. : The effect on melanoma risk of genes previously associated with telomere length. J Natl Cancer Inst 2014, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luu HN, Huang JY, Wang R, Adams-Haduch J, Jin A, Koh WP, Yuan JM: Association between leukocyte telomere length and the risk of pancreatic cancer: Findings from a prospective study. PLoS One 2019, 14:e0221697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rode L, Nordestgaard BG, Bojesen SE: Long telomeres and cancer risk among 95 568 individuals from the general population. Int J Epidemiol 2016, 45:1634–1643. [DOI] [PubMed] [Google Scholar]
- 13.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. : TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339:959–961. [DOI] [PubMed] [Google Scholar]
- 14.Robles-Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, Pooley KA, Pritchard AL, Tiffen JC, Petljak M, et al. : POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 2014, 46:478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac-de Paillerets B, Nagore E, Avril MF, et al. : Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 2014, 46:482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]; • These are the first two studies documenting the link of POT1 as a major susceptibility gene for familial melanoma. The authors showed that patients carrying rare POT1 variants had excessively long telomere length, suggesting that perturbance of POT1 function and loss of telomere length homeostasis may have important roles in melanoma development.
- 16.Bainbridge MN, Armstrong GN, Gramatges MM, Bertuch AA, Jhangiani SN, Doddapaneni H, Lewis L, Tombrello J, Tsavachidis S, Liu Y, et al. : Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst 2015, 107:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calvete O, Martinez P, Garcia-Pavia P, Benitez-Buelga C, Paumard-Hernandez B, Fernandez V, Dominguez F, Salas C, Romero-Laorden N, Garcia-Donas J, et al. : A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li-Fraumeni-like families. Nat Commun 2015, 6:8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speedy HE, Kinnersley B, Chubb D, Broderick P, Law PJ, Litchfield K, Jayne S, Dyer MJS, Dearden C, Follows GA, et al. : Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood 2016, 128:2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson TL, Hattangady N, Lerario AM, Williams C, Koeppe E, Quinonez S, Osborne J, Cha KB, Else T: A new POT1 germline mutation-expanding the spectrum of POT1-associated cancers. Fam Cancer 2017, 16:561–566. [DOI] [PubMed] [Google Scholar]
- 20.McMaster ML, Sun C, Landi MT, Savage SA, Rotunno M, Yang XR, Jones K, Vogt A, Hutchinson A, Zhu B, et al. : Germline mutations in Protection of Telomeres 1 in two families with Hodgkin lymphoma. Br J Haematol 2018, 181:372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potrony M, Puig-Butille JA, Ribera-Sola M, Iyer V, Robles-Espinoza CD, Aguilera P, Carrera C, Malvehy J, Badenas C, Landi MT, et al. : POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol 2019, 181:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, Price A, Ma YP, Sherborne AL, Palles C, et al. : Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun 2016, 7:11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calvete O, Garcia-Pavia P, Dominguez F, Bougeard G, Kunze K, Braeuninger A, Teule A, Lasa A, Ramon YCT, Llort G, et al. : The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur J Hum Genet 2017, 25:1278–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong K, Robles-Espinoza CD, Rodriguez D, Rudat SS, Puig S, Potrony M, Wong CC, Hewinson J, Aguilera P, Puig-Butille JA, et al. : Association of the POT1 Germline Missense Variant p.I78T With Familial Melanoma. JAMA Dermatol 2019, 155:604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Lange T: Shelterin-Mediated Telomere Protection. Annu Rev Genet 2018, 52:223–247. [DOI] [PubMed] [Google Scholar]
- 26.Lei M, Podell ER, Cech TR: Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol 2004, 11:1223–1229. [DOI] [PubMed] [Google Scholar]
- 27.Ye JZ, Hockemeyer D, Krutchinsky AN, Loayza D, Hooper SM, Chait BT, de Lange T: POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev 2004, 18:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu D, Safari A, O’Connor MS, Chan DW, Laegeler A, Qin J, Songyang Z: PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 2004, 6:673–680. [DOI] [PubMed] [Google Scholar]
- 29.Chen C, Gu P, Wu J, Chen X, Niu S, Sun H, Wu L, Li N, Peng J, Shi S, et al. : Structural insights into POT1-TPP1 interaction and POT1 C-terminal mutations in human cancer. Nat Commun 2017, 8:14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rice C, Shastrula PK, Kossenkov AV, Hills R, Baird DM, Showe LC, Doukov T, Janicki S, Skordalakes E: Structural and functional analysis of the human POT1-TPP1 telomeric complex. Nat Commun 2017, 8:14928. [DOI] [PMC free article] [PubMed] [Google Scholar]; • These two studies described the crystal structure of POT1-TPP1 binding complex. The authors performed the structural and functional analysis of several TPP1-interaction disrupting POT1 mutants and showed a defective POT1-TPP1 complex results in POT1 instability and telomere elongation, which in turn promotes cancer development.
- 31.Gong Y, de Lange T: A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol Cell 2010, 40:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L: TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, Lei M: The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445:506–510. [DOI] [PubMed] [Google Scholar]
- 34.Xin H, Liu D, Wan M, Safari A, Kim H, Sun W, O’Connor MS, Songyang Z: TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 2007, 445:559–562. [DOI] [PubMed] [Google Scholar]
- 35.Ramsay AJ, Quesada V, Foronda M, Conde L, Martinez-Trillos A, Villamor N, Rodriguez D, Kwarciak A, Garabaya C, Gallardo M, et al. : POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet 2013, 45:526–530. [DOI] [PubMed] [Google Scholar]
- 36.Murzin AG: OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J 1993, 12:861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinzaru AM, Hom RA, Beal A, Phillips AF, Ni E, Cardozo T, Nair N, Choi J, Wuttke DS, Sfeir A, et al. : Telomere Replication Stress Induced by POT1 Inactivation Accelerates Tumorigenesis. Cell Rep 2016, 15:2170–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this study, the authors showed that POT1 inactivation promotes thymic lymphoma formation, and suggested a general role of defective telomere replication in tumorigenesis.
- 38.Gu P, Wang Y, Bisht KK, Wu L, Kukova L, Smith EM, Xiao Y, Bailey SM, Lei M, Nandakumar J, et al. : Pot1 OB-fold mutations unleash telomere instability to initiate tumorigenesis. Oncogene 2017, 36:1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this study, the authors showed that POT1 OB-fold mutations elicited a DNA damage response at telomeres, and the damage was later repaired by an error prone repair pathway. This increased genome instability combined with telomere overlengthening favor tumor initiation.
- 39.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR: The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 2012, 492:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T: POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J 2005, 24:2667–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veldman T, Etheridge KT, Counter CM: Loss of hPot1 function leads to telomere instability and a cut-like phenotype. Curr Biol 2004, 14:2264–2270. [DOI] [PubMed] [Google Scholar]
- 42.Baumann P, Podell E, Cech TR: Human Pot1 (protection of telomeres) protein: cytolocalization, gene structure, and alternative splicing. Mol Cell Biol 2002, 22:8079–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colgin LM, Baran K, Baumann P, Cech TR, Reddel RR: Human POT1 facilitates telomere elongation by telomerase. Curr Biol 2003, 13:942–946. [DOI] [PubMed] [Google Scholar]; • In this study, the authors showed that three human POT1 splice variants had different abilities to elongate telomeres, which did not correlate with their telomeric single-stand DNA binding affinity.
- 44.Loayza D, De Lange T: POT1 as a terminal transducer of TRF1 telomere length control. Nature 2003, 423:1013–1018. [DOI] [PubMed] [Google Scholar]
- 45.Takai KK, Hooper S, Blackwood S, Gandhi R, de Lange T: In vivo stoichiometry of shelterin components. J Biol Chem 2010, 285:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S, Saito M, Ishikawa F: RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell 2009, 36:193–206. [DOI] [PubMed] [Google Scholar]
- 47.Chen LY, Redon S, Lingner J: The human CST complex is a terminator of telomerase activity. Nature 2012, 488:540–544. [DOI] [PubMed] [Google Scholar]
- 48.Wu P, Takai H, de Lange T: Telomeric 3’ overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 2012, 150:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wan M, Qin J, Songyang Z, Liu D: OB fold-containing protein 1 (OBFC1), a human homolog of yeast Stn1, associates with TPP1 and is implicated in telomere length regulation. J Biol Chem 2009, 284:26725–26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen LY, Majerska J, Lingner J: Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev 2013, 27:2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryan C, Rice C, Harkisheimer M, Schultz DC, Skordalakes E: Structure of the human telomeric Stn1-Ten1 capping complex. PLoS One 2013, 8:e66756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu P, Min JN, Wang Y, Huang C, Peng T, Chai W, Chang S: CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 2012, 31:2309–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kasbek C, Wang F, Price CM: Human TEN1 maintains telomere integrity and functions in genome-wide replication restart. J Biol Chem 2013, 288:30139–30150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stewart JA, Wang F, Chaiken MF, Kasbek C, Chastain PD 2nd, Wright WE, Price CM: Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J 2012, 31:3537–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang F, Stewart JA, Kasbek C, Zhao Y, Wright WE, Price CM: Human CST has independent functions during telomere duplex replication and C-strand fill-in. Cell Rep 2012, 2:1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim H, Li F, He Q, Deng T, Xu J, Jin F, Coarfa C, Putluri N, Liu D, Songyang Z: Systematic analysis of human telomeric dysfunction using inducible telosome/shelterin CRISPR/Cas9 knockout cells. Cell Discov 2017, 3:17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Renfrew KB, Song X, Lee JR, Arora A, Shippen DE: POT1a and components of CST engage telomerase and regulate its activity in Arabidopsis. PLoS Genet 2014, 10:e1004738. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this study, the authors proposed a two-state model in which Arabidopsis thaliana POT1a and TEN1 compete for the same binding site on STN1, thus forming two dynamic complexes. When POT1a is engaged with STN1 and CTC1, the POT1a-CS complex associates with telomerase and extends telomere length. When POT1a is displaced by TEN1, the CST complex represses telomerase activity and terminates the telomerase action.
- 58.Beilstein MA, Renfrew KB, Song X, Shakirov EV, Zanis MJ, Shippen DE: Evolution of the Telomere-Associated Protein POT1a in Arabidopsis thaliana Is Characterized by Positive Selection to Reinforce Protein-Protein Interaction. Mol Biol Evol 2015, 32:1329–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Akbay EA, Pena CG, Ruder D, Michel JA, Nakada Y, Pathak S, Multani AS, Chang S, Castrillon DH: Cooperation between p53 and the telomere-protecting shelterin component Pot1a in endometrial carcinogenesis. Oncogene 2013, 32:2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kuo CL, Pilling LC, Kuchel GA, Ferrucci L, Melzer D: Telomere length and aging-related outcomes in humans: A Mendelian randomization study in 261,000 older participants. Aging Cell 2019:e13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. : Signatures of mutational processes in human cancer. Nature 2013, 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cancer Genome Atlas N: Genomic Classification of Cutaneous Melanoma. Cell 2015, 161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taboski MA, Sealey DC, Dorrens J, Tayade C, Betts DH, Harrington L: Long telomeres bypass the requirement for telomere maintenance in human tumorigenesis. Cell Rep 2012, 1:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In this study, the authors engineered a human tumor cell line containing loxP-flanked hTERT to enable the control of hTERT expression and telomere length. Using this system, the authors showed that when telomere reserves are long, tumor cells are capable of forming tumors robustly in the absence of hTERT expression (after loxP-hTERT cassette excision).