

Abstract
Chronic hypoxia (CH) produces a time-dependent increase of resting ventilation and the hypoxic ventilatory response (HVR) that is called ventilatory acclimatization to hypoxia (VAH). VAH involves plasticity in arterial chemoreceptors and the central nervous system (e.g. nucleus tractus solitarius, NTS) but signals for this plasticity are not known. We hypothesized hypoxia inducible factor 1-α (HIF-1α), an O2-sensitive transcription factor, is necessary in the NTS for normal VAH. We tested this in two mouse models using loxP-Cre gene deletion. First, HIF-1α was constitutively deleted in CNS neurons (CNS-HIF-1α−/−) by breeding HIF-1α floxed mice with mice expressing Cre-recombinase driven by CaM Kinase II promoter. Second, HIF-1α was deleted in NTS neurons in adult mice (NTS-HIF-1α−/−) by microinjecting adeno-associated virus that expressed Cre-recombinase in HIF-1α floxed mice. In normoxic control mice, HIF-1α deletion in the CNS or NTS did not affect ventilation nor the acute HVR (10–15 min hypoxic exposure). In mice acclimatized to CH for one week, ventilation in hypoxia was blunted in CNS-HIF-1α−/− and significantly decreased in NTS-HIF-1α−/− compared to control mice (p < 0.0001). These changes were not explained by differences in metabolic rate or CO2. Immunofluorescence showed that HIF-1α deletion in NTS-HIF-1α−/− was restricted to glutamatergic neurons. The results support that HIF-1α is a necessary signal for VAH and the previously described plasticity in glutamatergic neurotransmission in the NTS with CH. HIF-1α deletion had no effect on the increase in normoxic ventilation with acclimatization to CH indicating this is a distinct mechanism from the increased HVR with VAH.
Keywords: Hypoxia, HIF-1α, Control of Breathing, Neuroplasticity, Hypoxic Ventilatory Response, Nucleus Tractus Solitarii
I. Introduction.
The body’s first line of defense against a decrease in blood oxygen (O2) levels is the hypoxic ventilatory response (HVR), which is an increase in ventilation due to reflex response triggered by arterial chemoreceptors. The activation of this chemoreflex occurs when decreased arterial oxygen partial pressure (PaO2) stimulates carotid body chemoreceptors that send sensory information to respiratory centers in the brainstem, including the nucleus tractus solitarius (NTS). This increased afferent input leads to increased phrenic nerve output and other ventilatory motor neuron activity to produce hyperventilation (Teppema & Dahan, 2010). The HVR exhibits plasticity with different time domains of hypoxia, including further increases in ventilation when hypoxia is sustained for two days to two months, termed ventilatory acclimatization to hypoxia (VAH) (Pamenter & Powell, 2016). Other changes in ventilatory control, such as persistent increase in ventilation and decreased arterial partial pressure of CO2 during normoxia, also occur with VAH, suggesting multiple mechanisms may be involved. For example, increased O2 sensitivity in carotid body chemoreceptors contributes to VAH, as well as increased phrenic nerve activity for a given arterial chemoreceptor afferent input to the respiratory centers, termed an increased central nervous system (CNS) gain of the HVR (Dwinell & Powell, 1999).
Considering that two or more days are necessary to produce significant VAH in humans and most animal models (Huey et al., 2000; Powell et al., 2000), it is likely that changes in gene expression contribute to such plasticity. Under low O2 conditions, hypoxia inducible factors (HIFs) promote changes in gene expression to maintain O2 homeostasis (Semenza, 2011). The HIF pathway controls expression of hundreds of hypoxia-related genes including erythropoietin (EPO), vascular endothelial growth factor (VEGF), nitric oxide synthases (NOS) (Semenza, 1998, 1999; Shimoda & Semenza, 2011; Yuan et al., 2011), and has been related to diseases involving hypoxia (Semenza, 2003, 2013; Bishop & Ratcliffe, 2015; Vohwinkel et al., 2015) as well as adaptation to high altitude in human populations (reviewed in Bigham & Lee, 2014; Simonson 2015). In the presence of O2, the prolyl hydroxylases (PHDs) target HIF alpha subunits (e.g., HIF-1α) for proteasome degradation. In hypoxia, the activity of O2-sensing PHDs is reduced, and HIF-1α accumulates in the cytoplasm. The dimerization of HIF-1α with the constitutive unit HIF-1β and other cofactors allows the HIF complex to bind hypoxia response element (HREs) in the DNA, leading to expression of various HIF-targeted genes (Semenza, 2011; Palazon et al., 2014). The multiple mechanisms resulting from these genes may contribute to the multiple mechanisms of ventilatory acclimatization to hypoxia described above.
Previous work with whole body, lifelong heterozygous deletion of the HIF-1α subunit in mice showed a role for HIF-1α both in the carotid body response to acute hypoxia and the HVR after CH (Kline et al., 2002). More recently, work using an inducible whole body heterozygous prolyl hydroxylase domain 2 (PHD2) deletion in mice showed a role for HIF-2α but not HIF-1α, in the HVR after acclimatization to CH (Hodson et al., 2016). In this study, changes in ventilation are attributed to a proliferative response in the carotid body, yet the potential contributions of central nervous system respiratory areas were not explored.
In our study, we tested the hypothesis that HIF-1α in medullary respiratory centers is necessary for VAH using two mouse models. First, to study the effects of HIF-1α deletion in adult neurons throughout the brain, we bred constitutive central nervous system (CNS)-null HIF-1α mice from mice containing loxP flanked alleles surrounding the critical exon 2 of the HIF-1α gene (Ryan et al., 2000) and transgenic mice expressing Cre recombinase under the control of the calcium/calmodulin-dependent protein kinase IIα (CaM kinase IIα) promoter (Dragatsis & Zeitlin, 2000). Second, to determine the effects of HIF-1α specifically in neurons within the NTS, the site of the first synapse in the CNS from carotid body chemoreceptors and critical respiratory center in the medulla for the HVR (Finley & Katz, 1992), we generated conditional NTS-null HIF-1α mice. We used the same HIF-1α loxP mice described above (Ryan et al., 2000) but using NTS-targeted microinjection of adeno-associated virus (AAV) that expressed Cre-recombinase to delete HIF-1α in a spatially (i.e. in the NTS) and temporally (i.e. in adults during exposure to chronic hypoxia) specific manner.
We measured ventilation and metabolic rates and characterized details of the HIF-1α deletion in both HIF-1α knock-out models before and after exposure to CH to test the hypothesis that HIF-1α in the central nervous system, and specifically in the NTS, is contributing to VAH.
II. Methods
1. Ethical approval
All surgical procedures and protocols were performed in accordance with the relevant guidelines of The University of California San Diego Institutional Animal Care and Use Committee (Protocol #S02143M, UCSD, IACUC). The experiments adhered to national standards of care and use of experimental animals, the American Physiological Society’s “Guiding Principles in the Care and Use of Animals” and the animal ethics guidelines outlined in the editorial of The Journal of Physiology (Grundy, 2015).
2. Mouse models
We studied adult (10–12 weeks old) male mice purchased from The Jackson Laboratory (NV, USA). Animals were housed in standard mouse cages in a vivarium with a 12:12-hour light-dark cycle and fed a standard mouse diet ad libitum.
CNS Neuron HIF-1α Null Mice
To delete HIF-1α in central neurons, we bred a mouse strain with loxP flanking exon 2 of the HIF-1α locus (Ryan et al., 2000) with mice expressing Cre recombinase and a LacZ reporter gene under the control of the CaM kinase IIα (CaMKIIα). We used the R1ag#5 line, which expresses Cre in neurons expressing CaMKIIα throughout the brain (Dragatsis & Zeitlin, 2000).
We isolated DNA from the tail of 4-week-old mice, conducted polymerase chain reaction (PCR), and genotyped each sample based on amplification with Cre-specific primers: Cre forward (CCGGGCTCGCACGAGCAA) and Cre reverse (GGCGCGGCAACACCATTTTT) (Gibco). The amplified Cre band was resolved at 450 bp in 1.2% agarose gel. Mice without Cre amplification were categorized as ‘wild type’ (CNS-HIF-1α+/+ i.e., loxP/loxP; Cre negative) and those with the Cre amplification as CNS-HIF-1α null mice (CNS-HIF-1α−/− i.e., loxP/loxP; Cre positive).
To quantify deletion of HIF-1α in the CNS within this model, we used real-time PCR assays for HIF-1α. Adult male CNS-HIF-1α null mice (n=5) were anesthetized with pentobarbital (50mg/kg, i.p.) after 4 hours of continuous hypoxia at 6% O2. The brain was immediately removed and frozen in isopentane on dry ice. A rodent brain slicer was used to obtain 1 mm thick sections from the NTS rostral and caudal to calamus scriptorius. Sections were then frozen on the sectioning blades with liquid N2 and 20 gauge hypodermic tubing was used to obtain bilateral punches of the NTS. DNA was isolated from the biopsies and subjected to real-time PCR with primers spanning the targeted region as well as primers for an undeleted control gene (c-jun) for normalization (Cramer et al. 2003). The following primers were used for the analysis: HIF-1α forward (CTATGGAGGCCAGAAGAGGGTAT) and reverse (CCCACATCAGGTGGCTCATAA). C-jun forward (GACGGACCGTTCTATGACTGC) and reverse (GGAGGAACGAGGCGTTGAG). Specific mRNA levels were measured by relative quantitation by comparison to a standard curve and detected by real time fluorescence using SYBR Green I dye, which is specific for its 1000 fold increase in fluorescence intensity for double-stranded DNA versus single-stranded DNA. Reverse transcriptase and PCR were accomplished in one tube using the Brilliant qRT-PCR Core Reagent Kit (Stratagene).
To localize Cre expression in CNS neuron HIF-1α null mice, we used the LacZ reporter gene expressing β-galactosidase. CNS-HIF-1α−/− null mice were perfused transcardially with saline and 4% paraformaldehyde (PFA) in phosphate buffer solution (PBS, pH = 7.40). Both respiratory regulatory centers, the brainstem and carotid body, were removed by dissection. Tissues were fixed for 5 min in 0.2% glutaraldehyde in 0.1 M Na-phosphate buffer (pH 7.3) containing 1.25 mM CaCl2 and 2 mM MgCl2. The tissues were then block stained using Xgal [5-bromo-4-chloro-3-indolyl-β-galactosidase] and washed 3 times in the same buffer before incubating overnight in the buffer plus 5 mM K3[Fe(CN6)], 5 mM K4[Fe(CN6)], and 0.1% X-Gal at 37°C. Tissue was post-fixed in 2% PFA in PBS overnight, cryoprotected in 30% sucrose in PBS, frozen in isopentane (−140°C) and sectioned into 30 μm slices using a cryostat (Cryocut 1800, Leica Biosystems, Wetzlar Germany). The X-Gal stained sections were imaged using a slide scanning system (NanoZoomer 2.0 HT, Hamamatsu Japan).
To confirm that HIF-1α deletion was localized in the CNS, we also examined carotid bodies for LacZ reporter as described above for neurons. To look for alterations in carotid body structure, we stained for tyrosine hydroxylase (TH), which is a marker of carotid body glomus cells. Briefly, carotid body sections (5 μm) were blocked with normal goat serum for 1 hour and then incubated with primary antibody against TH (1:1000, # AB152, Millipore) overnight at 4°C. The immunoreactive staining was detected using a streptavidin-peroxidase method and revealed at 37°C in dark chamber with 3, 3’-diaminobenzidine tetrahydrochloride (DAB, Sigma).
To determine if lifelong HIF-1α deficiency in the CNS had any effects on brain gross morphology, we used magnetic resonance imaging (MRI). Adult male wild-type (CNS-HIF-1α+/+, n=2) and CNS-HIF-1α null mice (n=2) were anesthetized with pentobarbital (50mg/kg, i.p.) and transcardially perfused and fixed with phosphate-buffered saline (PBS) containing 4% paraformaldehyde. MRI images were acquired on WT CNS-HIF-1α null mice using a 7Telsa Varian animal scanner and 2 cm volume coil with the following acquisition parameters: Spin Echo TR/TE 1000/13.1 FOV 20×20 mm matrix 256×128, 20 averages, slice thickness 0.8mm.
NTS neuron HIF-1α null mice
To delete HIF-1α in NTS neurons only, we used the same HIF-1α loxP mice described above (HIF1atm3Rsjo, The Jackson Laboratory) (Ryan et al., 2000) and microinjected adeno-associated virus expressing Cre-recombinase (Vector Biolabs, PA, USA) into the NTS. Adeno-associated virus containing Cre-recombinase expression sequence coupled to green fluorescent protein (AAV-Cre-GFP) was also microinjected into the NTS. AAV encoding only the LacZ reporter gene (AAV-LacZ-GFP) was microinjected for sham treatment (NTS-HIF-1α+/+ or wild-type). AAV serotype 2 was used as a viral vector for the gene encoding the Cre or β-galactosidase (Vector Biolabs, Philadelphia PA). Both AAV-Cre and AAV-LacZ had titer levels of 1.3 × 1012 GC/ml.
For stereotaxic AAV microinjections, mice were anesthetized (40 mg/kg Ketamine and 3 mg/kg Xylazine i.p.), and the scalp was shaved and sterilized with Betadine and alcohol and placed in a stereotaxic holder (Kopf Instruments, Tujunga CA) at a 45° angle nose-down. We made a midline incision and the muscle was retracted to expose the base of the skull. The dura was cut at the point it connects to the skull to expose the dorsal brainstem. We used the calamus scriptorius as a visual landmark for injection sites in the NTS. Injections were made bilaterally 0.1mm caudal and 0.3mm and 0.7mm rostral to the calamus scriptorius at depths of ~ 0.2mm. A total of 200nl of AAV was injected through 10 microinjections (Pneumatic Picopump PV830, World Precision Instruments, Sarasota FL) using a glass micropipette with a 10 μm tip diameter. Following the surgery, the mice were housed in a normoxic isolation room and observed daily for 5 days to allow for AAV shedding. Analgesic (Buprenorphine, 0.1mg/Kg) was administered i.p. before the animal wakes up from anesthesia. Another dose of analgesic was given overnight, and thereafter as necessary for up to 48 hours. Antibiotic (Enrofloxacin, 4mg/Kg) was administered i.p. once at the end of surgery, and then twice a day for 2 more days.
For histology experiments, mice were anesthetized to a deep surgical plane (confirmed by lack of response to a toe pinch) with sodium pentobarbital (Fatal Plus, Vorthech, 150 mg/kg i.p.) and fixed via transcardial perfusion, first with 0.9% saline/0.004% heparin, followed by 4% PFA. The brainstem was removed and postfixed in 4% PFA overnight followed by immersion in 30% sucrose. The brainstem was mounted, frozen and sliced on a Leica cryostat in 30 μm floating sections. To determine if AAV injection procedure deleted HIF-1α in a specific type of neuron, we used immunohistochemistry on floating sections from the NTS incubated with 0.5% Triton X-100 (Sigma) in phosphate-buffered saline (PBS) for 2 hours. After blocking washes and blocking in normal goat serum (5% in PBS), slices were incubated with the following primary antibodies: mouse anti-Vglut2 (1/1000; UC Davis), mouse anti-GAD67 (1/1000; #MAB5406, Millipore) or rabbit anti-cFOS (1/1000; #sc-253, Santa Cruz). As secondary antibodies, Alexa (Molecular Probes) were used. We used confocal laser microscopy (Leica, SP5) to co-localize GFP coupled to Cre-recombinase (AAV-Cre-GFP, Vector Biolabs, PA, USA) with Vglut2, GAD67 and cFOS. Mice were exposed to hypoxia (10% O2) for 3 hours in these experiments to measure acute responses to hypoxia. This sub-chronic exposure was not long enough to induce plasticity of the HVR in the CNS (cf. Dwinell & Powell, 1999) but it was long enough to generate a cFOS signal indicating neuronal activation in the NTS, while 15 min of hypoxia was not.
3. Chronic hypoxia
Mice were acclimatized to chronic hypoxia (PIO2 = 70 Torr) in a hypobaric chamber for 7 days (CH group). Acute sustained hypoxia (3 hours of normobaric hypoxia, PIO2 = 70 Torr) was used to identify neurons in the NTS activated by hypoxia (e.g. carotid body chemoreceptor input) with cFOS. Normoxic control mice were housed in the same conditions in the room outside the chamber. The chamber was opened once daily for cage cleaning and replacement of food and water.
4. Ventilatory chemoreflexes
Ventilatory responses to hypoxia and hypercapnia were measured in unrestrained mice using a whole body barometric plethysmograph (500 ml) modified for continuous flow (Jacky, 1978; Pamenter et al. 2014). Flow was maintained through the chamber while a pressure transducer (mp45 with 2 cm H2O diaphragm, Validyne, Northridge CA) recorded the changes due to the warming and expansion of inhaled gases. On the experimental day, the mice were weighed and sealed into the plethysmograph chamber along with a temperature and humidity probe (Thermalert TH5, Physitemp, Clifton NJ). A constant gas flow (335 ml/min) was delivered with a rotameter (603, Matheson, Montgomeryville PA) and measured with a flow meter (S-110, McMillan Co., Georgetown TX) upstream of the chamber. Gases exited the chamber through a valve and into a vacuum pump (Model 25, Precision Scientific Co, Chicago IL) to isolate pressure changes from breathing in the chamber during constant flow with high input and output impedances. This also allowed us to maintain chamber pressure near-atmospheric pressure and reference pressure measurements (Validyne, Northridge CA) in the chamber to atmosphere. Inspired and expired O2 and carbon dioxide (CO2) fractions were measured in a mass spectrometer (MGA1100, Marquette Electronics, Milwaukee WI) sampling from the chamber.
Hypoxic Ventilatory Response (HVR) protocol
Ventilatory measurements were performed after the animals acclimated to the plethysmograph chamber for 40 min at their chronic (breathing 10% O2 for CH and 21% O2 for normoxic Control group). Animals were then exposed to a 5 min challenge of experimental gas (e.g. 21% O2 for CH; 10% for Control) to test responsiveness. We then measured the acute HVR with ventilatory responses measured between 10 and 15 minutes of a switch to normoxia (21% O2) and hypoxia (10% O2). Mice were returned to chronic baseline conditions (10% O2 for CH; 21% O2 for Controls) between acute tests.
We also calculated O2 consumption () and CO2 production () during ventilatory measurements by measuring inspired and expired O2 and CO2) fractions and chamber flow rate, correcting for evaporative water loss according to the method developed by Withers (1977).
After experiments mice were perfused for histology as mentioned before, or euthanized with overdose of sodium pentobarbital (Fatal Plus, Vortech).
5. Data acquisition and analysis
All ventilatory parameters were recorded on a digital acquisition program (Labview v2.5.0, National Instruments, Austin TX), sampling at a rate of 200 Hz. A customized Matlab-based program (Mathworks, Natick MA) was used to analyze a 20 second minimum region of interest between 10–15 min after changing gas concentrations. The respiratory frequency (fR, breaths/min) and tidal volume (VT, ml) was calculated from cyclic peaks in the plethysmograph pressure pulses and VT was measured using 0.02 ml calibration pulses and the equations developed by Drorbaugh & Fenn (1955). The product of fR and VT is inspired ventilation, which was normalized body mass ventilation (V̇I, ml/min*kg).
Data was expressed as mean ± SD. Statistical analysis was performed using multivariate ANOVA or two way ANOVA tests followed by Fisher’s post hoc analysis (StatView, 5.0, USA); p< 0.05 was set as the level of statistical significance.
III. Results
1. HIF-1α deletion in the CNS
The goal of our experiment was to preserve HIF-1α expression in the peripheral nervous system, and especially the carotid bodies, while deleting it in the respiratory centers in the brain. To determine the distribution of neurons lacking HIF-1α in CNS-HIF-1α null mice, we observed LacZ staining as an indication of Cre expression (in a ROSA mouse strain). LacZ staining was distributed uniformly throughout neurons in the brainstem (Fig. 1A), including the NTS (Fig. 1B) and absent in the carotid bodies (Fig. 1C).
Figure 1.
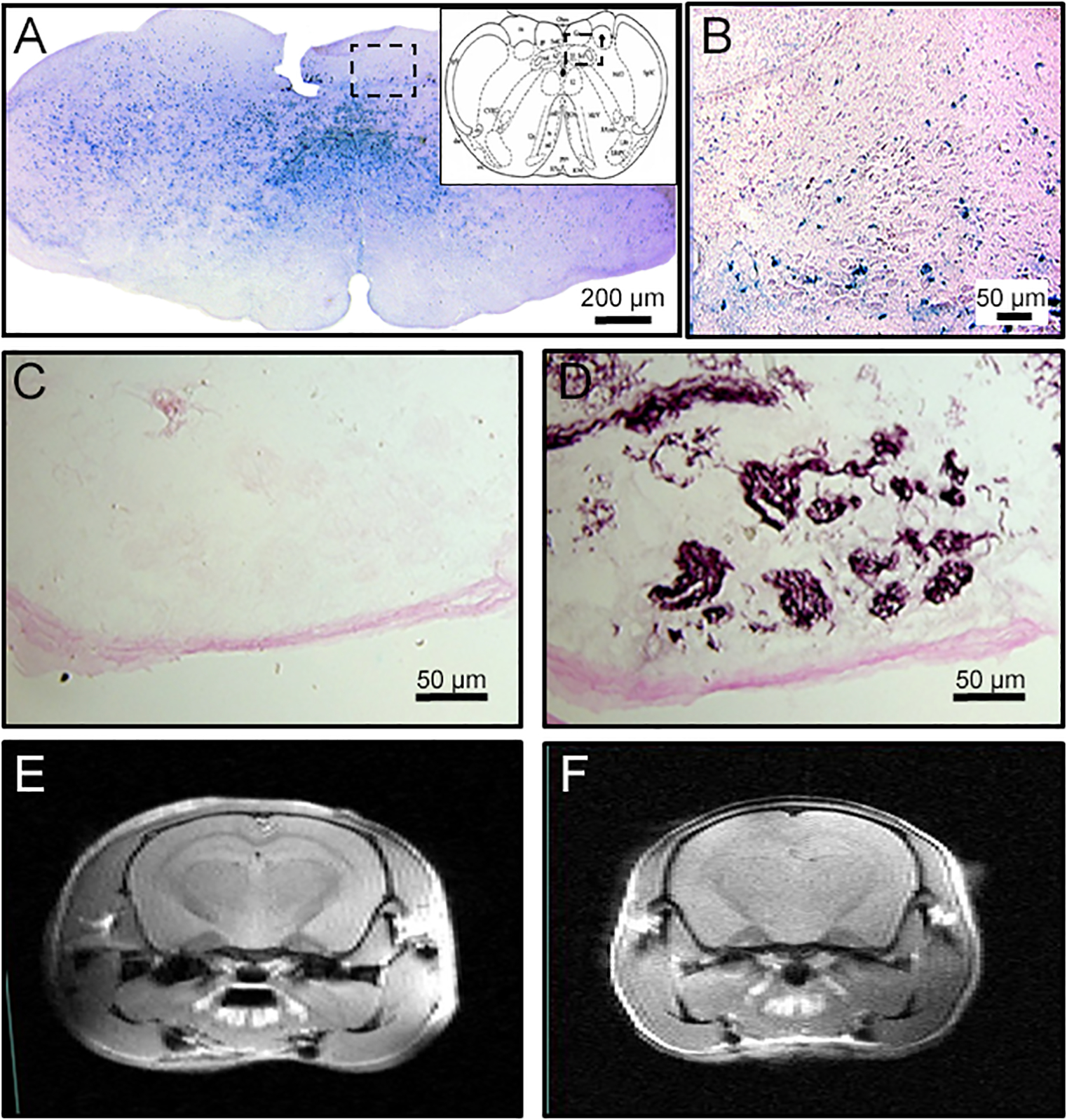
Constitutive deletion of HIF-1α in the central but not peripheral nervous system using the Cre-LoxP strategy. A. Cre is expressed throughout the brainstem as demonstrated by Lac-Z staining (blue) in reporter mice, including the NTS (B, magnified set from A). However, Lac-Z is not observed in in the carotid body (C) which has normal chemosensitive glomus cells as shown with tyrosine hydroxylase staining in a consecutive section (D). MRI images show normal brain structure and ventricles in wild-type (CNS-HIF-1α+/+, E) or CNS-HIF-1α−/− null mice (F).
Furthermore, carotid body chemoreceptor structure appeared normal in CNS-HIF-1α null mice as shown by normal glomus cells expressing TH (Fig. 1D). The overall structure of the brain was normal too, as demonstrated by similar ventricular dimensions in wild type and CNS-HIF-1α null mice (Fig. 1E and F).
To quantify HIF-1α deletion in CNS-HIF-1α null mice, we used RT-PCR to measure DNA amplification in dissections from the dorsal medulla (2−ΔΔCt method, using CNS-HIF-1α−/− as “sample’ and CNS-HIF-1α+/+ as “control”, 7 mice per group) and found 91 ± 13% (SD) deletion of the HIF-1α gene relative to c-jun.
2. HIF-1α deletion in neurons of adult mice tends to decrease the HVR after CH but does not produce a significant change in ventilation
We measured the HVR to acute hypoxia (10% O2) in CNS-HIF-1α−/− and CNS-HIF-1α+/+ mice under control conditions and after 7 days of CH. All groups demonstrated an HVR with increased V̇I breathing at 10% compared to 21% O2, and there were no differences between genotypes in normoxic control conditions (Fig. 2A). There was a significant interaction between acute and chronic O2 levels (p ≤ 0.03), although the 3-way interaction with genotype was not significant (p > 0.16). However, there was a trend for decreased ventilatory O2-sensitivity with HIF-1α deletion in the CNS after CH. Post hoc testing showed V̇I breathing 10% O2 increased significantly with CH in wild-type but not CNS-HIF-1α−/− (Fig. 2A, p ≤ 0.05, Fisher’s test after multivariate ANOVA). Ventilation in normoxia increased after CH in both genotypes, although the post hoc analysis did not show significant differences. However, small increases in V̇I coupled with small decreases in metabolic rate in normoxia after CH (Table 1), can result in the significant hyperventilation and hypocapnia in normoxia which is a hallmark of acclimatization.
Figure 2.
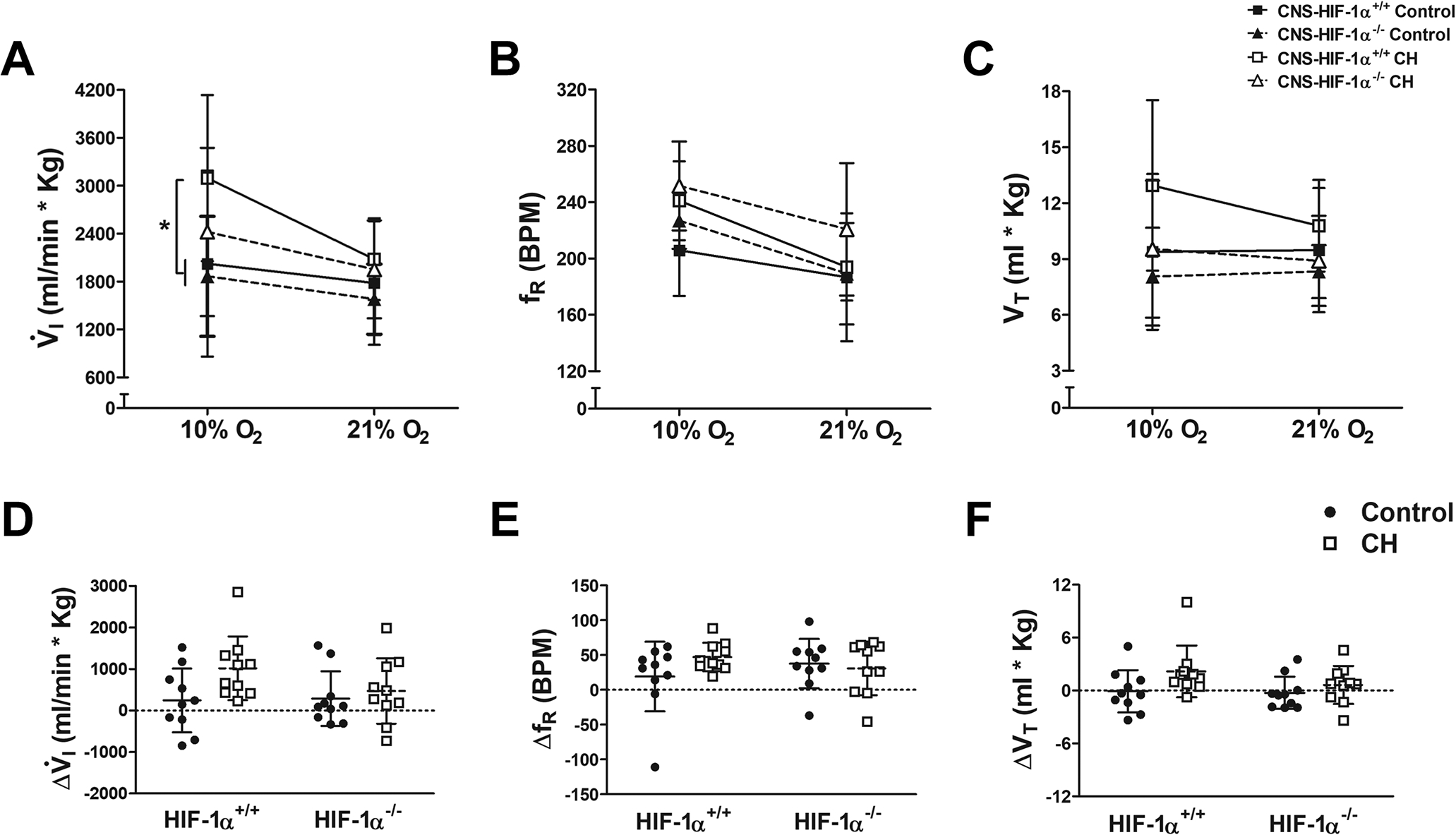
Ventilatory responses to hypoxia in wild type (CNS-HIF-1α+/+, squares and solid lines) and CNS-HIF-1α−/− null mice (triangles and dashed lines) during normoxic control conditions (filled symbols) and after 7 days of chronic hypoxia (CH, open symbols). A. For V̇I, the only significant interaction with ANOVA was acute vs. chronic hypoxia level (p=0.03). Post hoc testing showed V̇I in hypoxia after chronic hypoxia was significantly greater than in chronically normoxic mice in wild-type but not CNS-HIF−/− (* p ≤ 0.05, Fisher’s test after multivariate ANOVA, n=10 mice per group). This was due to similar effects on VT (C) with no significant effects on fR (B). The magnitude of the HVR (D) increased with CH more in wild-type than in CNS-HIF null mice but this was not significant (E and F).
Table 1.
decreases with acute hypoxia and was not different between normoxic control and CH mice, or CNS-HIF-1α+/+ and CNS-HIF-1α−/− mice.
| (mL/(min*Kg)) | CNS-HIF-1α+/+ | CNS-HIF-1α−/− | ||
|---|---|---|---|---|
| 21% O2 | 10% O2 | 21% O2 | 10% O2 | |
| Control | 79 (10) | 44 (6)* | 73 (20) | 39 (12)* |
| CH | 76 (21) | 52 (7)* | 64 (22) | 46 (7)* |
Values in table show mean and also standard deviation in parenthesis.
p ≤ 0.05, 10% O2 vs. 21% O2, Fisher’s test after Multivariate ANOVA, n = 6 mice per group.
The magnitude of the HVR, although it tended to increase with CH more in wild-type than in CNS-HIF-1α−/−, did not show significant differences when we measure the effect of CH and HIF status (Fig. 2D, p > 0.05, Two-way ANOVA).
Changes in the pattern of breathing (Fig. 2B and C, fR and VT) were similar to those in V̇I. The trend for a decrease in the hypoxic response of fR and VT with CH in CNS-HIF-1α−/− null mice was similar to that for V̇I (Fig. 2D–F) but was not significant either (p > 0.15 for fR and p > 0.37 for VT).
O2 consumption decreased in acute hypoxia (p < 0.05) and was not different between genotypes or in CH versus normoxic controls (Table 1).
3. Deletion of HIF1α in the NTS significantly blunts the HVR in mice exposed to CH
These experiments were designed to determine the effects of specific spatial and temporal deletion of HIF-1α on ventilatory acclimatization to chronic hypoxia. As such, HIF-1α deletion in these experiments was limited to glutamatergic neurons in the caudal and medial NTS (as detailed later in Figs. 5 and 7). With the inducible expression of the Cre recombinase due to AAV injection, the mice had normal HIF-1α levels in their CNS during development, from in utero to adulthood. Figure 3 illustrates the effects of HIF-1α deletion only in the NTS and only during exposure to CH on the HVR.
Figure 5.
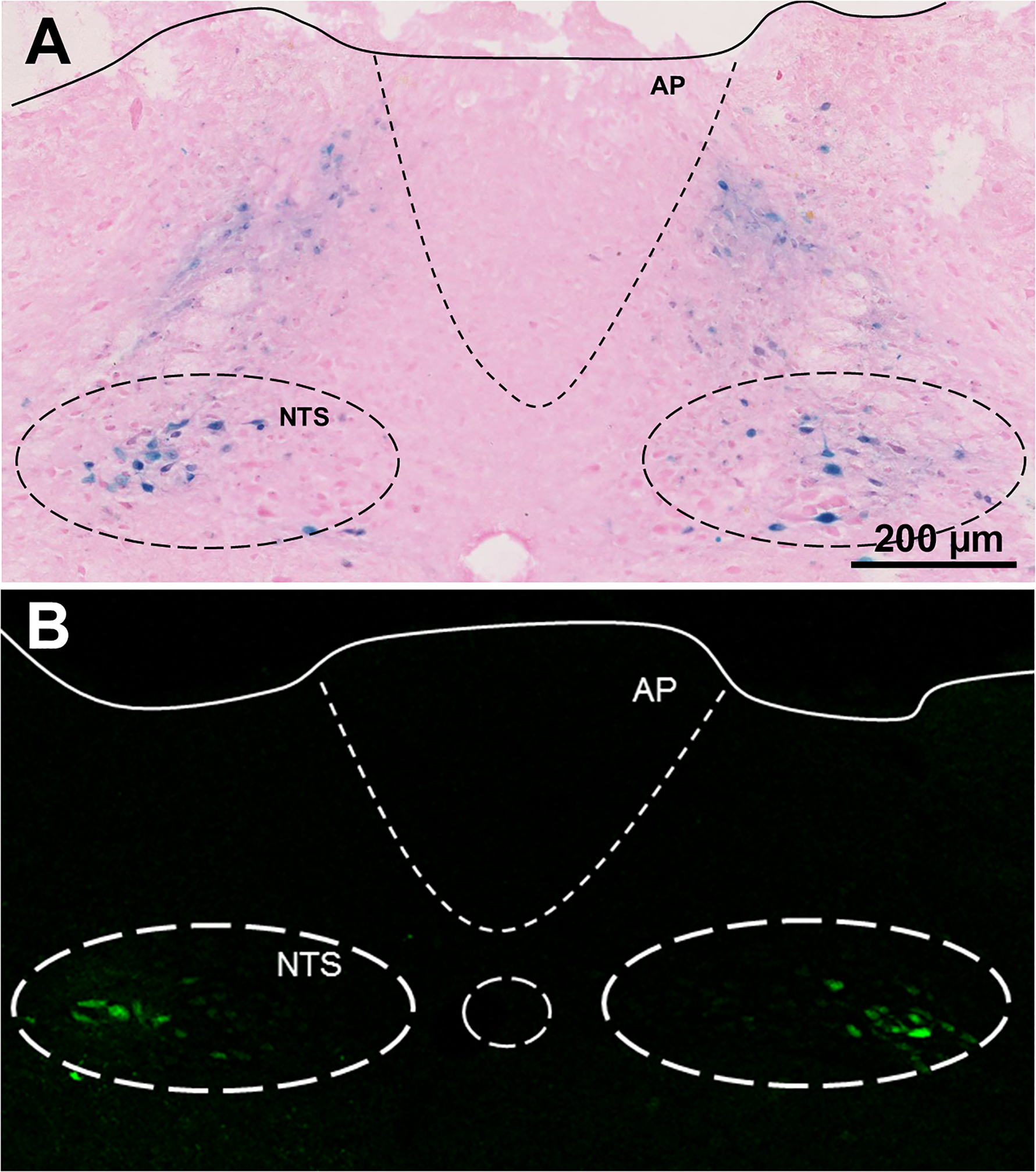
Expression of Adeno-associated virus (AAV) after microinjection in the NTS. A. AAV encoding β-galactosidase produces a blue precipitate localized in the area of bilateral microinjections in LoxP-HIF1-α mice. B. Similar results for microinjecting AAV encoding GFP-Cre. AP: Area postrema; NTS Nucleus tractus solitarius.
Figure 7.
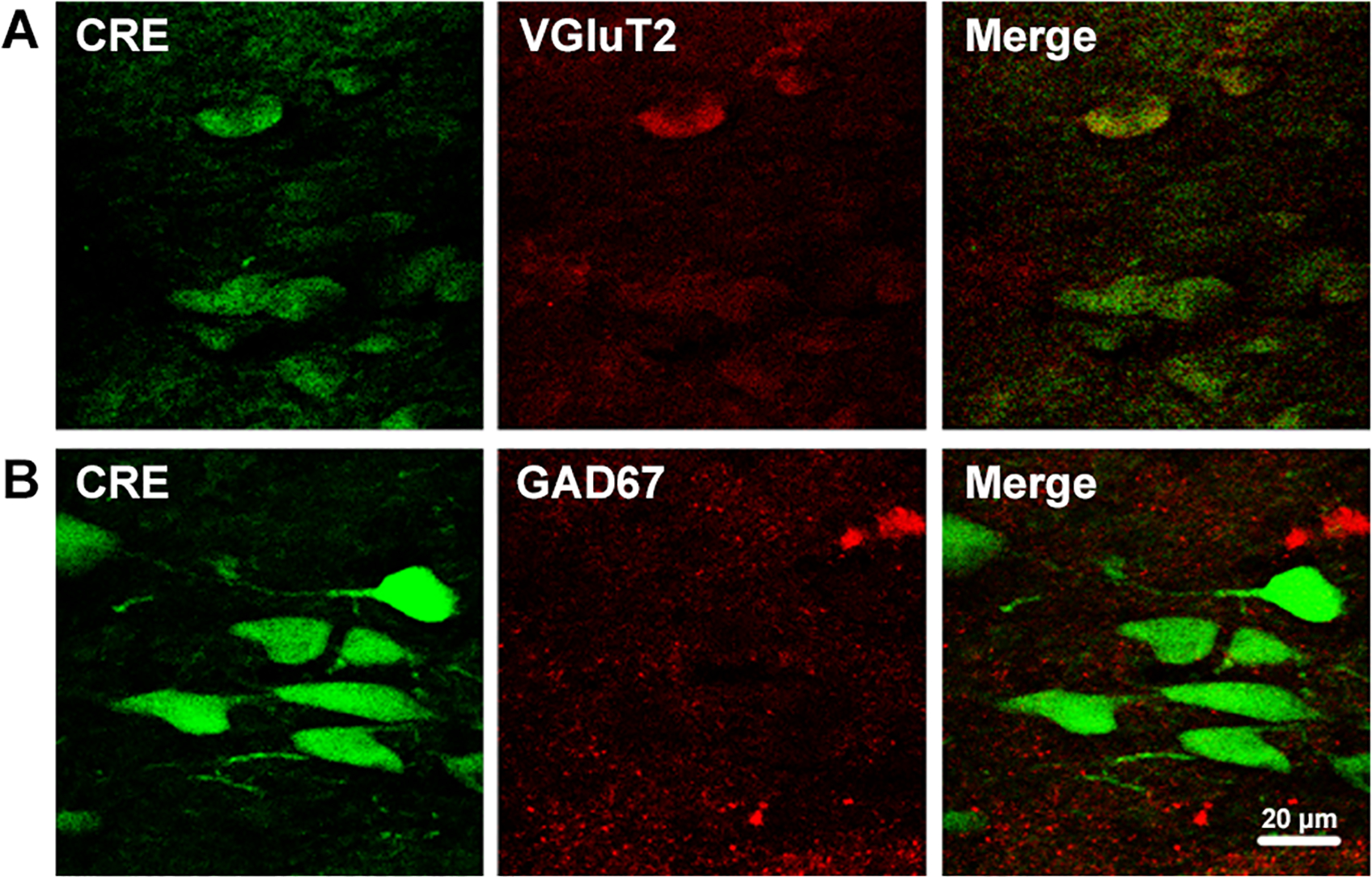
Representative photographs showing the NTS of a mouse exposed to CH, and the neurons infected with AAV containing GFP-Cre recombinase (Cre, green) that co-localizing (yellow) with red signals for VGluT2 (A) but not GAD67 (B).
Figure 3.
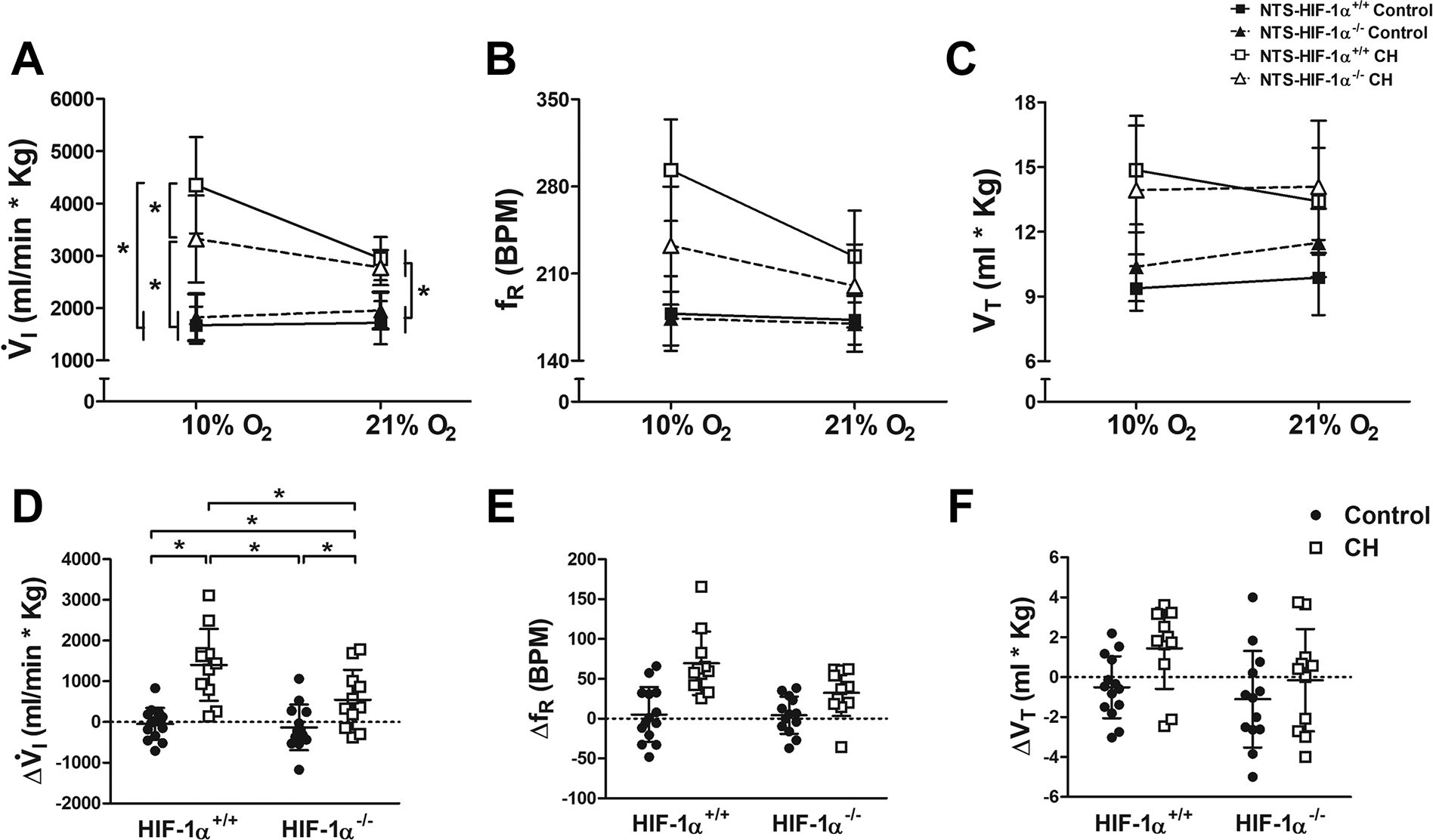
Ventilatory responses to hypoxia in wild type (NTS-HIF-1α+/+, squares and solid lines) and NTS-HIF-1α−/− null mice (triangles and dashed lines) during normoxic control conditions (filled symbols) and after 7 days of chronic hypoxia (CH, open symbols). A. For V̇I, the 3-way interaction between acute O2 level, chronic O2 level and genotype by ANOVA was significant (p=0.044) and all of the 2-way interactions were significant (p≤0.0025). In acute normoxia (21% O2), V̇I was significantly greater after CH regardless of genotype (p ≤ 0.0003, Fisher’s test after multivariate ANOVA, n=11 to 14 for the 4 groups of mice). In acute hypoxia (10% O2), however, the increase in V̇I with CH was significantly greater in wild-type (NTS-HIF-1α+/+) than in NTS-HIF1-α−/− mice (*p ≤ 0.05, Fisher’s test after multivariate ANOVA, n=11 to 14 for the 4 groups of mice). This was mainly due to similar effects on fR (B) while VT increased similarly with CH independent of genotype (C). D. The magnitude of the HVR increased significantly more with CH in wild-type versus NTS-HIF1-α−/− null mice (2-way interaction p=0.044 with multivariate ANOVA, *p ≤ 0.05 Fisher’s post-hoc test, n=11 to 14 for the 4 groups of mice). The effects of CH on fR (E) and VT (F) responses to hypoxia showed similar trends with genotype but were not significant (p=0.056 and 0.429, respectively, for 2-way interaction with ANOVA).
In normoxic control conditions, there was no difference in the HVR or pattern of breathing (fR and VT) between genotypes (Fig. 3 A–C, black symbols). Neither genotype showed an absolute increase in ventilation with acute hypoxia in normoxic control conditions (Fig. 3A and D, black symbols), which is often observed in rodents during poikilocapnic hypoxia protocols such as that used here (Soliz et al., 2005; Vincent et al., 2007; Hodges & Richerson, 2008; Pamenter & Powell, 2016). V̇I decreased but metabolic rate decreased more in acute hypoxia (Table 2), so the mice were actually hyperventilating. This is illustrated by the increase in ventilatory ratio for with acute hypoxia for all genotypes under all conditions (Fig. 4, black symbols).
Table 2.
and decrease with acute hypoxia (p<0.0001) and are lower in CH than normoxic control conditions (p<0.0001) but there are no significant differences between wild type (NTS-HIF1-α+/+) and NTS-HIF1-α−/− mice (p=0.52 and 0.77 for and , respectively).
| (mL/(min*Kg)) | NTS-HIF-1α+/+ | NTS-HIF1-α−/− | ||
|---|---|---|---|---|
| 21% O2 | 10% O2 | 21% O2 | 10% O2 | |
| Control | 45 (7) | 26 (7)* | 53 (15) | 28 (6)* |
| CH | 59 (15)# | 45 (8)*# | 59 (19) | 46 (18)*# |
| (mL/(min*Kg)) | NTS-HIF-1α+/+ | NTS-HIF1-α−/− | ||
| 21% O2 | 10% O2 | 21% O2 | 10% O2 | |
| Control | 40 (6) | 25 (5)* | 47 (13) | 26 (6)* |
| CH | 50 (14)# | 40 (7)*# | 47 (12) | 40 (13)# |
Values in table show mean and also standard deviation in parenthesis.
p ≤ 0.05, 10% O2 vs. 21% O2;
p ≤ 0.05 CH vs Control, Fisher’s test after multivariate ANOVA, n=10–13 for different groups of mice.
Figure 4.
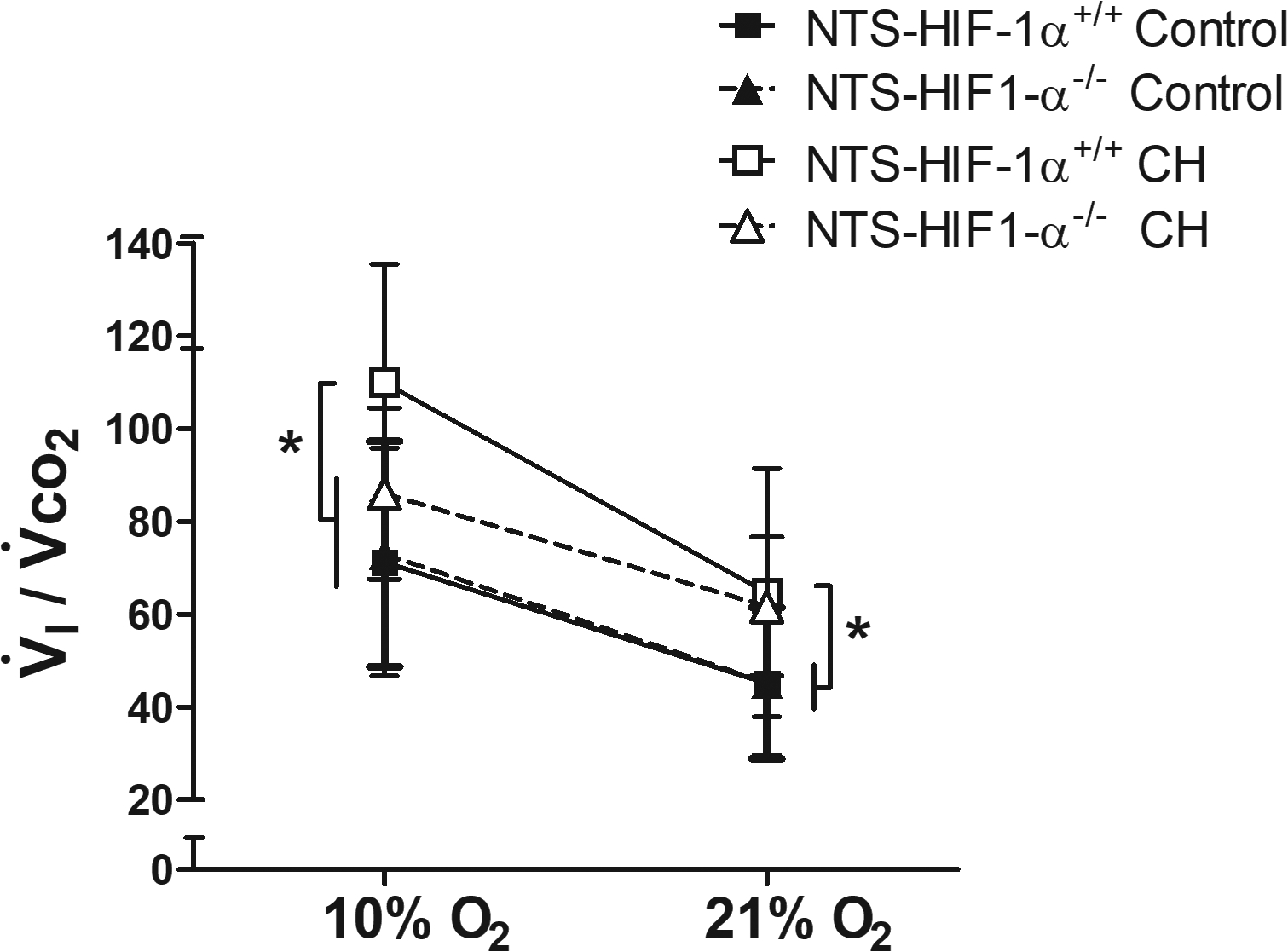
Correcting the hypoxic ventilatory response for changes in metabolic rate with acute hypoxia shows that differences after CH with HIF-1α deletion in the NTS cannot be explained by differences in arterial blood gases. Changes in (which is inversely proportional to arterial ) are qualitatively similar to those in V̇I with CH and genotype (Fig. 3), i.e. significantly less in acute hypoxia in NTS-HIF1-α−/− compared to wild-type (NTS-HIF-1α+/+) mice after CH. This means arterial would be greater in NTS-HIF1-α−/− null mice and should stimulate V̇I more so their ventilatory response must be depressed compared to wild-type mice. * p ≤ 0.05, Fisher’s test after 3-way ANOVA interaction p=0.0002, n=10–13 for the 4 groups of mice.
Chronic hypoxia (CH) significantly increased V̇I in both genotypes breathing 10% or 21% O2, but it increased V̇I in wild-type mice significantly more than in NTS-HIF-1α−/− mice breathing 10% O2 (Fig. 3A, p ≤ 0.05, Fisher’s test after multivariate ANOVA). ANOVA showed a significant 3-way interaction between acute and chronic O2 and genotype (p < 0.05) as well as significant 2-way interactions between all of these variables (p < 0.03). This effect on V̇I was primarily from differences in the effect of CH on fR between genotypes (Fig. 3B), while VT increased similarly with CH in both genotypes (Fig. 3C). The magnitude of the HVR increased significantly more with CH in wild-type than in NTS-HIF-1α−/− mice (Fig. 3D, p ≤ 0.05 Fisher’s post-hoc test), and again this is mainly from an effect on fR versus VT (Fig. 3E–F).
Table 2 shows that acute hypoxia significantly decreased metabolic rate in both genotypes before and after CH. To control for any differences in arterial blood gases that might stimulate ventilation differently in these cases, we normalized ventilation to CO2 production. Figure 4 show , which is inversely proportional to arterial . The ratio was similar for both genotypes in normoxic control conditions breathing 10 or 21% O2. Exposure to CH increased in both genotypes breathing 21% O2. Hence, normoxic ventilatory drive increased in both wild-type and NTS-HIF-1α−/− mice, and arterial was regulated at the same new and lower value in both genotypes. In contrast, CH only increased during 10% O2 breathing significantly in wild-type and not in NTS-HIF-1α−/− mice. Hence, this measure of ventilatory acclimatization also supports a role for HIF-1α in the NTS.
4. AAV injection confined to neurons in the NTS
Microinjection of AAV-Cre in the NTS resulted in focal infection and expression of Cre as illustrated with a LacZ reported gene (Fig. 5A). Earlier attempts using a different AAV vector (serotype 9 compared to 2) and more superficial microinjections showed a diffuse pattern of infection, including areas where the AAV appeared to follow nerve tracks to other regions such as the ventral medulla (data not shown). Animals used for physiology experiments or immunohistochemistry had injections in the intermediate and caudal NTS, and AAV infections were restricted to this area as confirmed by GFP expression (Fig. 5B).
To determine if AAV was infecting a specific type of neurons in the NTS, we used immunohistochemistry. First we co-localized DAPI (indicating nuclei) and GFP (indicating AAV infection and Cre-expression) in medullary sections from AAV microinjected mice and found that not all of the neurons in the NTS expressed CRE (Fig. 6A). We also co-localized GFP with cFOS (indicating early activation in neurons) in AAV microinjected mice exposed to acute and chronic hypoxia ( Torr for 3 hour and 7 days, respectively). cFOS was not observed in normoxic controls (Fig. 6A), but it increased in the cytoplasm with 3 hours of hypoxia (Fig. 6B) and in the nuclei with chronic hypoxia (7 days, Fig. 6C). We used 3 hours of hypoxia as a relatively acute exposure, i.e. long enough to provide a stimulus for inducing detectable cFOS as a marker of increased neuronal activity in response to carotid body chemoreceptor input but much shorter than the 5 days needed to show measurable differences in plasticity in the CNS for ventilatory acclimatization (cf. (Dwinell & Powell, 1999). GFP co-localized with neurons expressing cFOS in both cases, indicating that AAV, and therefore Cre expression and HIF-1α deletion, occurred in neurons in the NTS that were activated by hypoxia. We also used immunohistochemistry to characterize the neurochemical phenotype of NTS neurons infected by AAV (Fig. 7). Cre-recombinase co-localized with VGluT2 (Fig. 7A) but not GAD67 (Fig. 7B), indicating we deleted HIF-1α in glutamatergic NTS neurons but not GABAergic neurons in the NTS.
Figure 6.
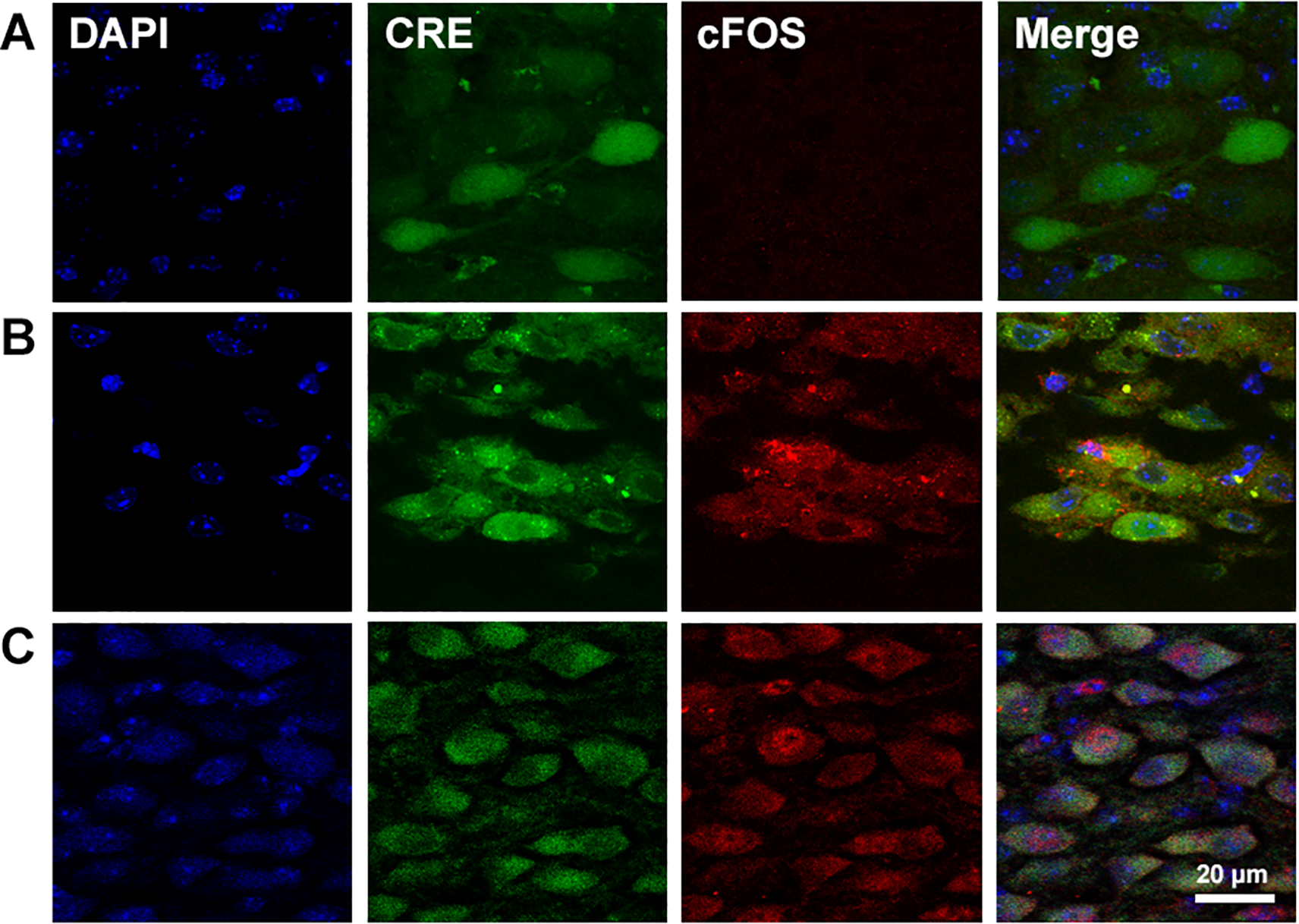
Micro-photographs showing neurons in the NTS infected with AAV in a mouse exposed to control normoxic conditions (A), to 3 hours of hypoxia (B) or 7 days of CH (C). Cellular nucleus were stained with DAPI (blue). Expression of GFP-Cre recombinase (Cre, green) allows to observe AAV-infected neurons. Expression of cFOS (red) was observed in cytoplasm of AAV-infected neurons after 3 hours of hypoxia (B) and in the cell nucleus in the CH mouse (C), but not in the control mouse (A). The overlapped image (merge) shows that AAV is infecting neurons that are activated by 3 hours of hypoxia.
IV. Discussion
This study examined the role of HIF-1α in the CNS for ventilatory acclimatization to hypoxia. Ventilation in CNS-HIF-1α−/− mice showed that CNS-constitutive deletion of HIF-1α tended to decrease the ventilatory response to 10% O2 compared to CNS-HIF-1α+/+ mice after CH. However, NTS-HIF-1α deletion significantly decreased ventilation during acute hypoxia in mice after CH, indicating a critical role of HIF-1α in the NTS for normal ventilatory acclimatization to hypoxia. As discussed below, this cannot be explained by differences in metabolic or CO2-sensitive ventilatory drives and HIF-1α expression in glutamatergic neurons in the NTS appears to be critical (although a role for HIF-1α in other types of neurons cannot be excluded). Changes in normoxic ventilatory drive after acclimatization to CH were not affected, supporting the idea that there are different mechanisms for different aspects of VAH as discussed below also.
The results obtained using CNS-HIF-1α−/− mice partially agreed with those found in heterozygous HIF-1α mice in the whole body (Kline et al., 2002). Kline et al. found that heterozygous HIF-1α mice have similar responses to normoxic and acute hypoxic challenge compared to homozygous HIF-1α mice in control conditions. Nevertheless, after 72 hours of CH, homozygous, but not heterozygous HIF-1α mice, develop increases responses during normoxia and acute hypoxia. The authors attribute this difference to the decreased carotid body activity, since the frequency of discharge in isolated preparation of carotid bodies extracted from heterozygous HIF-1α mice is lower, and the responses to the Dejour’s test are diminished in the same mice. On the other hand, Hodson et al. (2016) concluded that HIF-2α was more important than HIF-1α for ventilatory acclimatization to 7 days of hypoxia. They found an increase in the HVR of normoxic control or CH mice with inducible deletion of the gene of the prolyl hydroxylase domain 2 (PHD2), and deletion of HIF-2α but not HIF1α prevented this increase in HVR of PHD2 null mice (Hodson et al., 2016). These authors attributed changes in ventilation to cellular proliferative responses to hypoxia in the carotid bodies and the effects of HIF-2α throughout the body may explain why HIF-1α deletion in the NTS did not completely eliminate VAH in our experiments. However, no one else has tested the independent contribution of HIF-1α expression during CH in central respiratory centers before to directly compare with our results.
Our results support the idea that HIF-1α causes changes in central integrative mechanisms of the ventilatory chemoreflex pathway from arterial chemoreceptors to alter the HVR with chronic hypoxia. We established that CNS-HIF-1α−/− mice have decreased VAH, emphasizing the contribution of the CNS to acclimatization to hypoxia. Constitutive deletion of HIF-1α did not affect the acute response to hypoxia in control normoxic mice. In contrast, after exposure to CH, the CNS-HIF-1α+/+ mice had a significant increase in the V̇I and the VT responses to acute 10% O2 that was not observed in CNS-HIF-1α−/− animals (Fig. 2A). The change in V̇I between 21% and 10% O2 were not significantly different between CNS-HIF-1α+/+ and CNS-HIF-1α−/−, mice after CH (Fig. 2D) but there was a trend for decreased changes in both frequency and tidal volume responses to hypoxia (Fig. 2E–F). Since the CNS-HIF-1α deletion in these mice is distributed in the neurons in other parts of the CNS, compensatory effects from other respiratory areas, or at different times during development, may explain why the differences were not significant in these experiments while they were with HIF-1α in the NTS.
To determine the contribution of neurons specifically in the NTS to ventilatory acclimatization to hypoxia, we performed localized deletion of HIF-1α in the NTS by inducing Cre-recombinase expression through AAV microinjections. Similar to the results obtained from the CNS-HIF-1α-deletion model, this did not change the ventilatory responses to acute hypoxia observed in normoxic control mice (Fig. 3 and 4). After the exposure to CH, both NTS-HIF-1α+/+ and NTS-HIF-1α−/− had increased V̇I, fR and VT compared to normoxic control mice during 21 or 10% O2. Ventilation in 21% O2 after CH was the same in NTS-HIF-1α+/+ and NTS-HIF-1α−/− mice. But V̇I breathing 10% O2 in NTS-HIF-1α−/− mice was significantly less than in NTS-HIF-1α+/+ mice (Fig. 3A). The same was true for changes in with HIF-1α deletion in the NTS (Fig. 4). Hence, HIF-1α in the NTS, in addition to HIF-1α in the carotid bodies (cf. Kline et al., 2002; Hodson et al., 2016), is a necessary signal for normal VAH in mice.
Previous experiments performed in our laboratory demonstrated that glutamatergic neurons in the NTS contributes to the VAH. Microinjectiong the N-methyl-D-aspartate (NMDA) receptor antagonist MK801 into the NTS of rats exposed to CH significantly decreases ventilation in 10% but not 21% O2; MK801 microinjections in the NTS did not have an effect on the animals that acclimatized to normoxic conditions (Pamenter et al., 2014a). These results are similar to those obtained with the NTS-HIF-1α−/− mice here (Fig. 3 and 4). Therefore, it is possible that HIF-1α in the glutamatergic neurons of the NTS may regulate the NMDA-mediated response to 10% O2 after CH exposure. However, α-amino-3-hydroxy-5-methyl-4-isoxazolepropinoic (AMPA) receptor blockade and combined AMPA + NMDA receptor blockade in the NTS produce different effects (Pamenter et al., 2014b) so the interactions between different aspects of glutamatergic neurotransmission in the NTS and VAH remain to be determined.
The exact mechanisms of action of HIF-1α on ventilatory acclimatization remain to be determined but we can hypothesize multiple pathways based on HIF-1α gene targets. For example, HIF-1α is important for, at least, erythropoietin, inflammatory signaling, cathecholamines, nitric oxide and heme oxygenase, all of which have been shown to be involved in plasticity of the HVR (reviewed by Pamenter and Powell, 2016). Considering this diversity, a transcriptomic approach may be the next best step in understanding this system and identifying candidate pathways that could interact, in addition to individual genes and gene products, for experimental manipulation to test mechanisms.
Finally, the fact that NTS-HIF-1α−/− mice changed ventilation in 10% O2, but not in 21% O2, indicates that neuronal HIF-1α present in the NTS contributes to specific aspects of plasticity in VAH. Apparently other areas or other signals in the CNS are involved in the plasticity that increases normoxic ventilatory drive in VAH. These new results also support other evidence for distinct mechanisms contributing to different aspects of VAH observed in acute normoxia versus hypoxia (reviewed by Pamenter and Powell, 2016). This is important because the ability to manipulate individual components of ventilatory acclimatization could be useful in developing new therapeutic approaches for patients with chronic hypoxemia.
Critique of Methods.
The differences in ventilation with genotype and acclimatization cannot be explained by differences in metabolism and CO2 on ventilatory drive. Exposure to CH increased the and , but the changes were similar in NTS-HIF-1α+/+ and NTS-HIF-1α−/− mice exposed to CH (Table 2). Normalizing ventilation to CO2 production, as , reveals a similar pattern of response to that observed for V̇I Indeed, only increased significantly more with CH in NTS-HIF-1α+/+ compared to NTS-HIF-1α−/− mice (Fig. 4). The ratio is inversely proportional to arterial so arterial would be greater in NTS-HIF-1α−/− mice versus wild type breathing 10% O2 after CH. This should increase ventilatory drive so decreased ventilation in hypoxia after acclimatization when HIF-1α is deleted in the NTS is not explained by differences in CO2. Summarizing, Fig. 4 shows that deleting HIF-1α in the NTS significantly blunts the increase in hypoxic V̇I with acclimatization independently of metabolism or arterial .
The inducible model of HIF-1α deletion developed by Ryan et al. (2000) and used here provides site-specific deletion of HIF-1α. Our CNS-HIF-1α−/− shows normal CNS development and brain morphology (Fig. 1A and 1E–F). These results contrast with those reported by Tomita et al. (2003), which observed defective CNS development in the hippocampus with abnormal cerebral ventricles and severe hydrocephalus in mice with deletion of HIF-1α in the CNS. The authors used a nestin-promoter driven Cre as Nestin is expressed in the ventricular zone of the developing neurons system such as the telencephalon and spinal cord (Tomita et al., 2003). We used CaM kinase IIα as promotor, as it is expressed throughout the adult brain with high levels in forebrain and moderate levels in cerebellum and brainstem. In addition, Tomita et al. used endogenous HIF-1α locus with a loxP-flanked locus between the E13, E14 and E15 allele sites, which deletes the carboxy end of the protein, decreasing the ability of HIF-1α to activate transcription (Jiang et al., 1996). Our mice were floxed at the E2 allele site, which deletes the helix-loop-helix motif essential for HIF-1α dimerization with HIF-1β avoiding the transcriptional activation (Jiang et al., 1997).
The injection of the AAV in the NTS produced a circumscribed deletion of HIF-1α confined specifically to the area of injection (Fig. 5). We used the AAV serotype 2, which demonstrates a restricted area of action (Aschauer et al., 2013; Watakabe et al., 2015) and is targeted to infect mainly neurons (Davidson & Breakefield, 2003; Watakabe et al., 2015) and not other CNS cells such as astrocytes (Merienne et al., 2013). This contrasts with other AAV serotypes, such as serotype 9, which showed disperse expression in nerve tracks far from the site of injection (data not shown).
Our results also showed that the AAV serotype 2 did not infect all types of neurons equally since the AAV coupled GFP co-localized with the Vglut2 but not with GAD67, supporting the idea that AAV serotype 2 injected in the NTS infects glutamatergic but not GABAergic neurons (Fig. 7). The AAV cell preference has been shown for several AAV serotypes (Davidson & Breakefield, 2003). For example AAV2 used in the neural retina is more effective in rod photoreceptors cells tan in cones; and AAV5 used in the cerebellum is transduced with higher efficiency in Purkinje cells bot not in granule cells (Davidson & Breakefield, 2003). Therefore, it is plausible that in our injected mice, there is preference for a specific type of neurons, but the mechanisms of selective glutamatergic-neuron infection require further study. Also, it is important to note that although we only observed AAV expression in glutamatergic neurons in the NTS, and not in GABAergic neurons, we cannot exclude any specific neuronal phenotype from a role in acclimatization from these experiments. Our conclusion is that deletion of HIF-1α in glutamatergic neurons in the NTS can block normal ventilatory acclimatization to hypoxia in mice.
4. Conclusions
In this study we examined for the first time the effects of a constitutive CNS and inducible NTS-specific deletion of HIF-1α on ventilatory acclimatization to chronic sustained hypoxia. HIF-1α in glutamatergic neurons in the NTS is necessary for the normal increase in ventilation during acute hypoxia after acclimatization. However, HIF-1α in the NTS does not affect the HVR in normoxic mice or the metabolic responses to acute and chronic hypoxia. Further, HIF-1α in the CNS does not appear to affect changes in normoxic ventilatory drive with chronic hypoxia indicating differential effects of HIF-1α on the different aspects of ventilatory acclimatization to hypoxia. The genes that are activated by HIF-1α in central respiratory centers in chronic hypoxia remain to be defined and could advance our understanding of changes in the control of breathing in patients with chronic hypoxemia and healthy subjects acclimatizing to high altitude.
Supplementary Material
Key Points.
We hypothesized that hypoxia inducible factor 1α (HIF-1α) in central nervous system (CNS) respiratory centers is necessary for ventilatory acclimatization to hypoxia (VAH); VAH is a time-dependent increase in baseline ventilation and the hypoxic ventilatory response (HVR) occurring over days to weeks of chronic sustained hypoxia (CH).
Constitutive deletion of HIF-1α in CNS neurons in transgenic mice tended to blunt the increase in HVR that occurs in wild-type mice with CH.
Conditional deletion of HIF-1α in glutamatergic neurons of the NTS during CH significantly decreased ventilation in acute hypoxia but not normoxia in CH mice.
These effects are not explained by changes in metabolic rate nor CO2 and there were no changes in the HVR in normoxic mice.
HIF-1α mediated changes in gene expression in CNS respiratory centers are necessary in addition to plasticity of arterial chemoreceptors for normal VAH.
IX. Acknowledgements
The authors are grateful to Dr. Miriam Scadeng for the MR images and for experimental support from Glenn Adams, Steve Berruecos, Dr. Ellen Breen, Trevor Cooper, Wayne McNulty and Dr. John Scheel (all at UCSD at the time of the experiments).
Funding
Supported by HHS | NIH | National Heart, Lung, and Blood Institute (NHBLI), RO1 HL-081823 (F.L.P.) and 1RO1HL145470 T.S.S.
Footnotes
Competing Interests
The authors have nothing to disclose.
V. Bibliography
- Aschauer DF, Kreuz S & Rumpel S (2013). Analysis of Transduction Efficiency, Tropism and Axonal Transport of AAV Serotypes 1, 2, 5, 6, 8 and 9 in the Mouse Brain. PLoS One 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigham AW & Lee FS (2014). Human high-altitude adaptation: Forward genetics meets the HIF pathway. Genes Dev 28, 2189–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop T & Ratcliffe PJ (2015). HIF Hydroxylase Pathways in Cardiovascular Physiology and Medicine. Circ Res 117, 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson BL & Breakefield XO (2003). Viral vectors for gene delivery to the nervous system. Nat Rev Neurosci 4, 353–364. [DOI] [PubMed] [Google Scholar]
- Dragatsis I & Zeitlin S (2000). CaMKII??-cre transgene expression and recombination patterns in the mouse brain. Genesis 26, 133–135. [DOI] [PubMed] [Google Scholar]
- Dwinell MR & Powell FL (1999). Chronic hypoxia enhances the phrenic nerve response to arterial chemoreceptor stimulation in anesthetized rats. J Appl Physiol 87, 817–823. [DOI] [PubMed] [Google Scholar]
- Finley JCW & Katz DM (1992). The central organization of carotid-body afferents-projections to the brainstem of the rat. Brain Res 572, 108–116. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR & Richerson GB (2008). Interaction between defects in ventilatory and thermoregulatory control in mice lacking 5-HT neurons. Respir Physiol Neurobiol 164, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson EJ, Nicholls LG, Turner PJ, Llyr R, Fielding JW, Douglas G, Ratnayaka I, Robbins PA, Pugh CW, Buckler KJ, Ratcliffe PJ & Bishop T (2016). Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J Physiol 594, 1179–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey KA, Low MJ, Kelly MA, Juarez R, Szewczak JM & Powell FL (2000). Ventilatory responses to acute and chronic hypoxia in mice : effects of dopamine D2 receptors. J A 89, 1142–1150. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Rue E, Wang GL, Roe R & Semenza GL (1996). Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271, 17771–17778. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Zheng JZ, Leung SW, Roe R & Semenza GL (1997). Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem 272, 19253–19260. [DOI] [PubMed] [Google Scholar]
- Kline DD, Peng Y-J, Manalo DJ, Semenza GL & Prabhakar NR (2002). Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1a. Proc Natl Acad Sci 99, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merienne N, Douce J Le, Faivre E, Déglon N & Bonvento G (2013). Efficient gene delivery and selective transduction of astrocytes in the mammalian brain using viral vectors. Front Cell Neurosci 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Goldrath AW, Nizet V & Johnson RS (2014). HIF Transcription Factors, Inflammation, and Immunity. Immunity 41, 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamenter ME, Carr JA, Go A, Fu Z, Reid SG & Powell FL (2014a). Glutamate receptors in the nucleus tractus solitarius contribute to ventilatory acclimatization to hypoxia in rat. J Physiol 592, 1839–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamenter ME, Nguyen J, Carr JA & Powell FL (2014b). The effect of combined glutamate receptor blockade in the NTS on the hypoxic ventilatory response in awake rats differs from the effect of individual glutamate receptor blockade. Physiol Rep; DOI: 10.14814/phy2.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamenter ME & Powell FL (2016). Time Domains of the Hypoxic Ventilatory Response and Their Molecular Basis. Compr Physiol 6, 1345–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell FL, Huey KA & Dwinell MR (2000). Central nervous system mechanisms of ventilatory acclimatization to hypoxia. Respir Physiol 121, 223–236. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, Mcnulty W, Elson D, Gassmann M, Arbeit JM & Johnson RS (2000). Hypoxia-inducible Factor-1 α Is a Positive Factor in Solid Tumor Growth Advances in Brief Hypoxia-inducible Factor-1 ␣ Is a Positive Factor in Solid Tumor Growth. Cancer 4010–4015. [PubMed] [Google Scholar]
- Semenza GL (1998). Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr Opin Genet Dev 8, 588–594. [DOI] [PubMed] [Google Scholar]
- Semenza GL (1999). Regulation of Mammalian O2 Homeostasis by Hypoxia-Inducible Factor 1. Annu REv Dev Biol 15, 551–578. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2003). Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3, 721–732. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2011). Oxygen Sensing, Homeostasis, and Disease. 537–547. [DOI] [PubMed]
- Semenza GL (2013). Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu Rev Pathol Mech Dis 9, 47–71. [DOI] [PubMed] [Google Scholar]
- Shimoda LA & Semenza GL (2011). HIF and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 183, 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson TS (2015). Altitude Adaptation: A Glimpse Through Various Lenses. High Alt Med Biol 16, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliz J, Joseph V, Soulage C, Becskei C, Vogel J, Pequignot JM, Ogunshola O & Gassmann M (2005). Erythropoietin regulates hypoxic ventilation in mice by interacting with brainstem and carotid bodies. J Physiol 568, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ & Dahan A (2010). The Ventilatory Response to Hypoxia in Mammals: Mechanisms, Measurement, and Analysis. Physiol Rev 90, 675–754. [DOI] [PubMed] [Google Scholar]
- Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N, Sakamoto H, Gassmann M, Kageyama R, Ueda N, Gonzalez FJ & Takahama Y (2003). Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol Cell Biol 23, 6739–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SG, Waddell AE, Caron MG, Walker JKL & Fisher JT (2007). A murine model of hyperdopaminergic state displays altered respiratory control. FASEB J 21, 1463–1471. [DOI] [PubMed] [Google Scholar]
- Vohwinkel CU, Hoegl S & Eltzschig HK (2015). Hypoxia signaling during acute lung injury. J Appl Physiol 119, 1157–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watakabe A, Ohtsuka M, Kinoshita M, Takaji M, Isa K, Mizukami H, Ozawa K, Isa T & Yamamori T (2015). Comparative analyses of adeno-associated viral vector serotypes 1, 2, 5, 8 and 9 in marmoset, mouse and macaque cerebral cortex. Neurosci Res 93, 144–157. [DOI] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL & Prabhakar NR (2011). Hypoxia-Inducible Factor 1 Mediates Increased Expression of NADPH Oxidase-2 in Response to Intermittent Hypoxia. 2925–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.