

Abstract
Telomere fusions inevitably arise as a cell’s last-ditch effort to protect exposed chromosomal ends when telomeres are lost due to aging-associated erosion, breakage, failed replication, or a plethora of other cellular mistakes. Fusion of an exposed chromosomal end to another telomere presumably presents a superficially attractive option to the cell as opposed to the alternative of the impending degradation of the unprotected chromosomal terminus. However, when allowed to progress to mitosis these fusion events subsequently foster non-disjunction or bridge:breakage events — both of which drive highly pathogenic genomic instability and additional chromosomal translocations. Thus, the question becomes how and when telomere fusion events arise and, most importantly, is there a mechanism available to resolve these telomere bridges such that proper repair, and not genomic instability, results? Recent evidence suggests that the formation, and then the resolution of, ultrafine bridges may facilitate this process.
Keywords: Telomeres, Fusions, DNA replication, UFBs, G-quadraplex
Graphical abstract
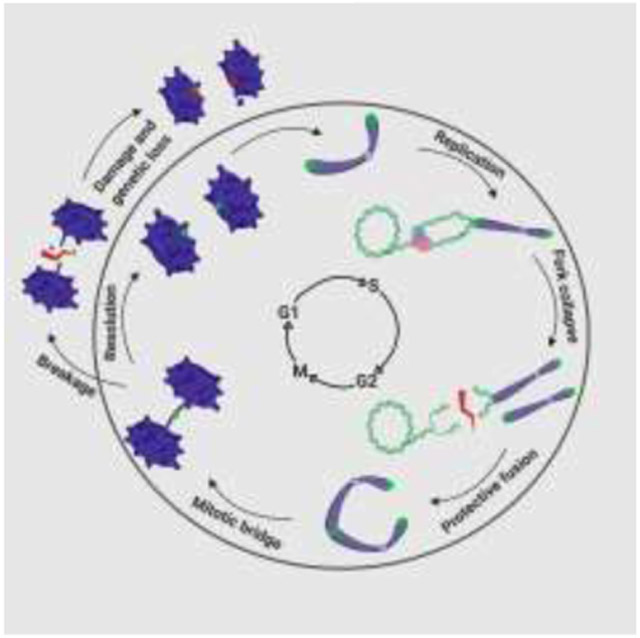
Introduction
Functional telomeres are a critical feature of a stable genome (Figure 1A). Conversely, when telomeres become dysfunctional, telomere fusions and ultimately genomic instability or cellular death ensue. The first line of defense against telomere fusion events is proper telomeric capping, which is provided by a six-subunit telomere protection complex termed Shelterin [1]. The loss of Shelterin subunits via experimental intervention uniformly yields rampant telomere fusions in the absence of telomeric erosion [2,3]. In a complementary fashion, the overexpression of Telomere Recognition Factor 1 (TRF1) and Telomere Recognition Factor 2 (TRF2), which are key Shelterin subunits often frequently overexpressed in cancers, leads to similar telomere fusion outcomes [4,5]. Somewhat confusingly, germline mutations in Shelterin are often more associated with telomere attrition, not telomere fusion [1]. Nonetheless, it is clear that proper (i.e., not too little and not too much) Shelterin expression unequivocally promotes appropriate capping (Figure 1B). Perhaps the least abrupt form of telomere dysfunction induced by Shelterin loss lies in the gradual, global telomere shortening associated with aging [6]. Aged, shortened telomeres result perforce in reduced Shelterin occupancy. Consequently, cells — in the absence of functional checkpoints which should instead trigger senescence — harboring such telomeres have elevated frequencies of telomere sister fusions [7,8] and/or fusions associated with massive fragmentation (aka chromothripsis) [9] (Figure 1C). While it is intuitive that the aberrant conditions of dysfunctional Shelterin expression or advanced aging can cause telomere fusions it needs to be emphasized that these are infrequent conditions/situations. Indeed, Shelterin mutations arise in patient populations only very rarely although when they do occur they are associated with severe pathologies including Dyskeratosis Congenita [10]. Only recently has it also been appreciated that the safeguarding provided by Shelterin is transiently abrogated to permit passage of the replication fork and that this reduction in Shelterin protection probably provides the most frequent window of opportunity for the genesis of telomere catastrophe by way of stalled or failed replication (Figure 1D).
Figure 1. Sources of telomere dysfunction and shortening.
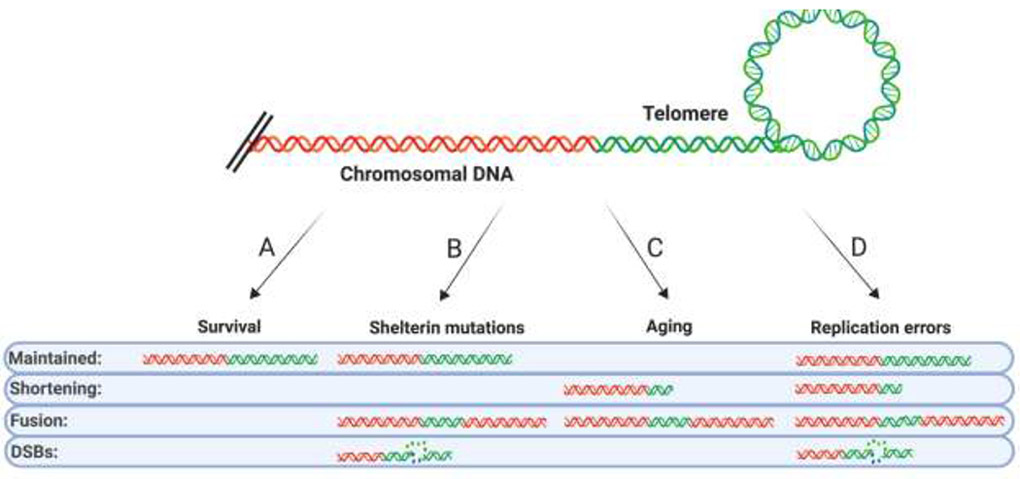
(A) Cell survival depends on faithful maintenance of telomere length. (B) Shelterin mutations induced in a laboratory setting greatly elevate the frequency of telomere fusions. In patients, Shelterin mutations lead to the disease Dykeratosis congenita, which is hallmarked by an increase the rate of telomere shortening and DNA DSBs at the telomere, all of which reduce the number of properly maintained telomeres. (C) Cellular and organismal aging lead to shortened telomeres which are prone to fusion. (D) Telomere replication can accumulate DSBs in the telomere elevating the frequency of fused and shortened telomeres.
Replication-driven fusions
Telomeric DNA is notoriously difficult to replicate due to its repetitive (TTAGGGn) nature, fragile site categorization, and its tendency to form difficult-to-dismantle secondary structures. For example, the transient dissociation of Shelterin binding to telomeric DNA that is required to enable DNA replication also likely permits the formation of G-quadraplex (G4) structures that can stall the replication fork [11,12]. Thus, it becomes incumbent upon cells to be able to limit the frequency of this stalling and/or to efficaciously restart fork progression when the forks do stall. A variety of factors and mechanisms influence these processes (Figure 2).
Figure 2.
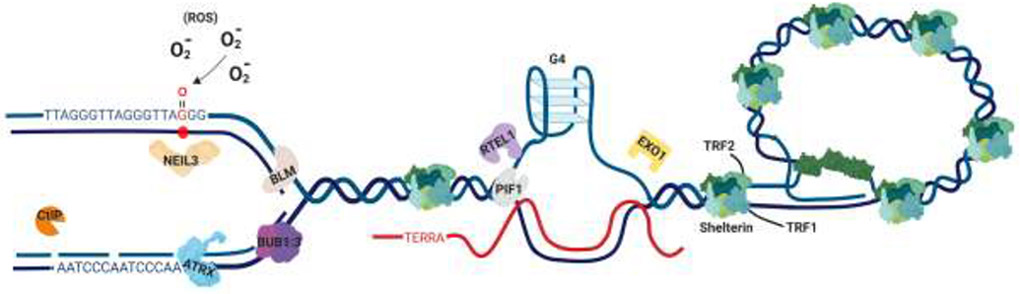
The telomere replication landscape. From left to right is diagrammed on the telomere a replication fork, a G-quadraplex (G4) and a t-loop. A number of proteins relevant to the review are cartooned as well. These include CtIP, NEIL3, ATRX, BLM, BUB1:3, RTEL, PIF1, EXO1, TRF1 and TRF2. In addition, the presence of ROS, the formation of TERRA and the complete Shelterin complex are shown as well. Because of these proteins, unusual structures, modifications and various processes a telomere is a priori difficult to replicate through. Resolution of these replication roadblocks and efficient fork restart are pivotal for telomere integrity.
One of the most extreme ways to induce fork stalling and breakage is to lose the expression of key replication and repair factors. For example, the repair nuclease, C-terminal Interacting Protein (CtIP), was recently shown to be essential for faithful telomere replication by preventing telomere loss and fusion [13]. CtIP is thought to be an “early responder” to stalled and broken forks where it performs the initial nucleolytic processing and recruits other downstream repair factors (Figure 2; orange PacMan™). Another early responder to stressed telomeric forks is the Budding Uninhibited by Benzimidazoles 3 (BUB3):Budding Uninhibited by Benzimidazoles 1 (BUB1) protein complex (Figure 2; magenta:purple symbol). BUB3:BUB1 was found to be recruited to telomeres during S-phase in a TRF2-dependent manner such that it can promote recruitment of subsequent repair factors when forks experience replication stress [14]. To mechanistically complicate this process (akin to a genetic “double whammy”), many telomere replication/repair factors also play a role in Homology-Directed Repair (HDR), thus forcing the cell to utilize the fusion-prone pathway of Non-Homologous End Joining (NHEJ) to repair these lesions in their absence [15]. This holds especially true for Alternative Lengthening of Telomeres (ALT) positive cells, which rely heavily on HDR to maintain their telomeres and which are prone to increased levels of replication stress [16]. For example, multiple groups have implicated the Bloom Syndrome (BLM) helicase, a known HDR factor, as being important for fostering faithful replication under ALT conditions [17,18] (Figure 2, pink symbol). Finally, and consistent with the above observations, the functional loss of the Alpha Thalassemia/Mental Retardation Syndrome X-Linked (ATRX) gene, which is a regulator of telomeric chromatin, drives persistent replication dysfunction at telomeres and is a hallmark of ALT cancers [19] (Figure 2; blue symbol).
Perhaps the major driver of telomeric fork stalling is the secondary structures that telomeric sequence has the propensity to form. In particular, telomeres form G4s at a high frequency due to their G-rich repeats [20] (Figure 2; G4 cartoon). Because these structures are so deleterious for on-going DNA replication they are typically dismantled by specialized helicases. Regulator of Telomere Elongation Helicase 1 (RTEL1), for example, disassembles both G4s as well as terminal loops (T-loops) thereby facilitating replication to the end of the telomere [21] (Figure 2; light purple symbol). However, particularly stable G4s or the failure to recruit the appropriate helicase will still lead to fork stalling. Unsurprisingly then, the loss of RTEL1 yields shortened telomeres and structural variants, which are indicative of breakage-fusion-bridging events [22]. Similarly, the Petite Integration Frequency 1 (PIF1) helicase can unwind lagging strand G4 DNA [23] (Figure 2; light blue symbol). Interestingly, PIF1 can also facilitate resection over G4 DNA [24]. Processing of these G4-proximal regions by Exonuclease 1 (EXO1) has been documented as a backup to helicase unwinding at these sites if fork stalling occurs (Stroik et al., submitted). Not surprisingly then, EXO1 depletion also correlates with shortened telomeres as well as increased sister chromatid fusion events (Figure 2; yellow symbol).
In contrast to the above scenarios, Telomeric Repeat-containing RNA (TERRA) transcription initiated in the sub-telomere prevents replication-induced telomere loss, presumably by positively altering the telomere replication landscape [25]. It’s unclear whether this is due to TERRA promoting a more open chromatin state or perhaps by destabilizing G4 structures; nonetheless it can facilitate successful replication events (Figure 2; red line). However, too many TERRA-induced R-loops has just the opposite effect and essentially promotes telomere loss [26]. Not surprisingly, the precise role of R-loops in facilitating HDR and error-free replication at telomeres versus replication collapse and telomere loss is currently the focus of significant inquiry [27]. Finally, reactive oxygen species (ROS), in some cases caused by mitochondrial dysfunction [28], can give rise to telomeric damage leading to catastrophic telomeric shortening by generating ROS-induced aberrant telomeric DNA structures (Figure 2; ROS). Specifically, ROS has the propensity to modify guanines to produce 8-oxoguanine adducts, which must be repaired either prior to or during replication and when this fails to happen these lesions accumulate leading to increased telomere loss [29]. In further support of the biological relevance of this pathway, the expression of the DNA glycosylase Nei-Like 3 (NEIL3), which can repair a portion of these lesions, prevents telomere fusion and shortening [30] (Figure 2; tangerine symbol).
In toto, a plethora of telomere replication offenses can arise in the form of repair and replication protein loss, RNA:DNA hybrids, free radicals, and aberrant DNA structures — all of which can induce telomeric replication stalling, which, in turn, can generate shortened or fused chromosome ends.
Telomere bridges
Regardless of the precise mechanism by which they occur, the above-listed events share a common feature – telomere shortening and dysfunction with a high propensity for the telomeres to fuse. Telomere sister chromatid fusion largely depends on DNA Ligase 1, while general intra- and interchromsomal translocations are more reliant on DNA Ligases 3 and 4 [8,31]. These aberrant repair events are, in turn, solely dependent on either alternative or classical NHEJ in a manner that is determined primarily by the processing ligase, respectively. Once protective fusion occurs, the connected telomeres/chromosomes inherently represent a ticking time bomb (Figure 3A). The conjoined chromosomes are due to be pulled in opposite directions during anaphase, however, the linkage impedes this process and forms a bridge between the two dividing cells. The majority of telomere bridges are proposed to form due to replication-driven telomere loss followed by fusion of the now radically-shortened chromosome end during S-phase of the cell cycle. Thus, G2-phase and mitosis are the only available opportunities for resolution of the fusion prior to cell division. The mechanism(s), if one exists, to recover telomere fusion events in G2-phase remains largely elusive. On the contrary, several mechanisms have been documented to resolve specific types of telomere fusion intermediates in mitosis when they give rise to a bridge. The best-understood mechanism is simply resolution by physical rupture. Thus, when a telomere sister chromatid or chromosome:chromsome fusion occurs it generates perforce a chromatin bridge, which is a 4’,6-diamidino-2-phenylindole (DAPI)-positive linkage between the two dividing cells (Figure 3A). These — often highly condensed — chromatin bridges can be resolved by simply increasing the physical tension on the bridge until it ruptures. Such resolution may involve breakage at any number of sites throughout the fused chromosomes, but it is paramount to appreciate that whatever resolution events occur, they are always mutagenic (Figure 3E-3G). Indeed, tension-dependent rupture has recently been documented as the dominant resolution pathway for chromatin bridges arising from overexpression of a dominate-negative TRF2 variant (Umbreit et al., bioRxiv; https://doi.org/10.1101/835058). Further, the outcomes of these ruptures encompassed the full of gamut of genomic instability from a single DNA double-stranded break (DSB) to chromothripsis. Physical shearing may not be the only resolution pathway, however. For example, the cytosolic exonuclease TREX1 has also been shown to clip these bridges, facilitating resolution in a more controlled manner [32]. How frequently this later process occurs, however, is currently unclear.
Figure 3. Anaphase bridge sources and outcomes.
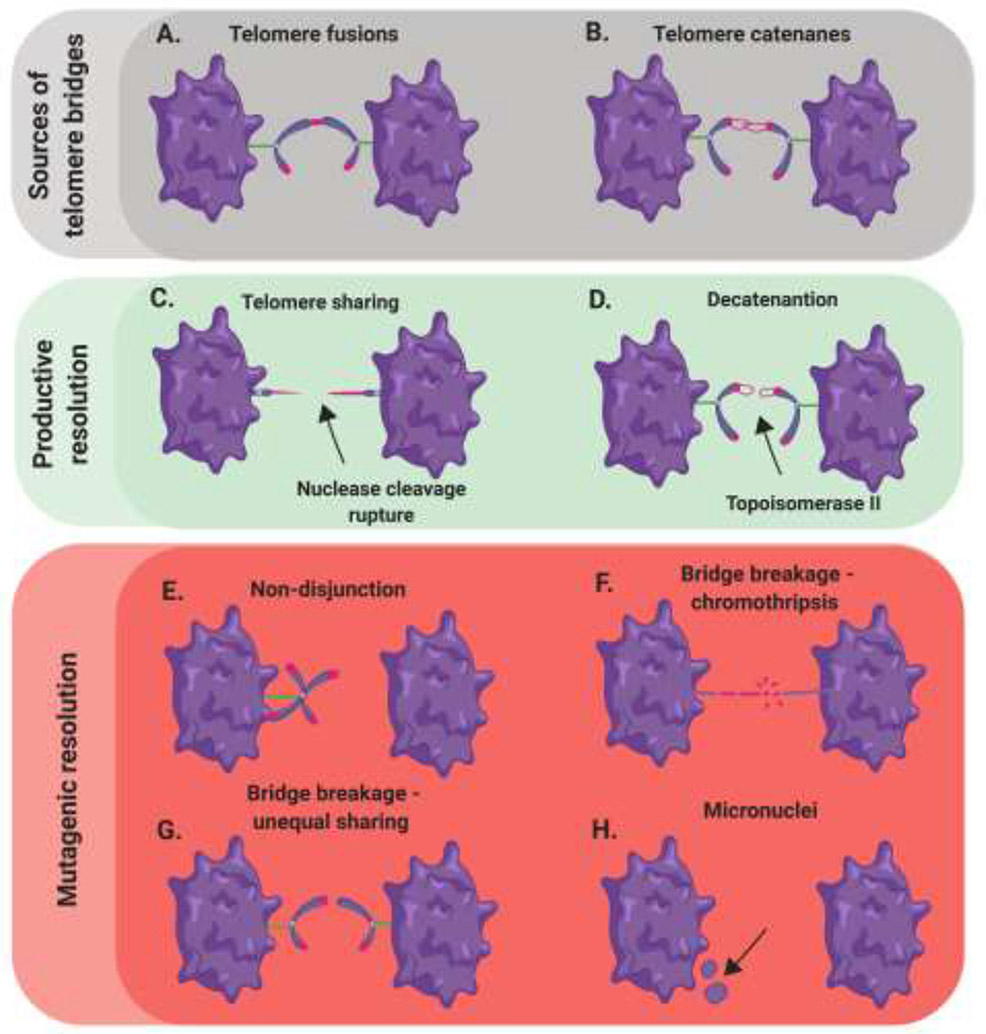
(A,B) End-to-end fusion events and unresolved telomere catenanes generate bridges between dividing cells. (C) Bridge breakage can yield telomere sharing in which both chromosomes separate with telomeric sequence. (D) Catenanes can be resolved by the action of Topoisomerase 2, which untangles this class of DNA bridge. (E) Non-disjunction can occur during anaphase as a result of connected chromosomes. (F) Bridge rupture can induce multiple break points along a bridge resulting in chromothripsis. (G) Singular bridge breakage can occur resulting in all the telomeric sequence covalently linked to one chromosome. (H) Multiple bridge breakage sites can generate micronuclei.
A second, less well-understood mechanism involves the fusion-driven connections giving rise to Ultrafine Bridges (UFBs) – a class of DNA bridges that can’t be visualized by conventional DNA staining methods. These structures are primarily identified by the presence of Replication Protein A (RPA) or Plk-Interacting Checkpoint Helicase (PICH) occupancy depending on their content of single-stranded DNA (ssDNA) or double-stranded DNA (dsDNA), respectively. It is important to note that telomere fusions represent just one of the mechanisms — along with failed decatenation (Figure 3B), incomplete, late replication at common fragile sites, and formation of HDR intermediates — by which UFBs can arise [33]. All of these events represent DNA entanglements (and not necessarily fusions) under tension, which require resolution prior to cellular division to avoid translocations, but for sake of this review we focus primarily on the UFBs caused by telomere fusions. These telomere fusions are typically double-stranded upon entering metaphase and subsequently during anaphase when they give rise to a UFB. Thus, this class of bridges is protectively bound by the PICH protein to stabilize it from the simple tension-dependent rupture described above [34]. The BLM helicase is then recruited to these UFBs in a PICH-dependent manner and is presumed to convert these predominately dsDNA bridges into predominately ssDNA bridges [35]. In support of this model, BLM depletion greatly reduces the formation of RPA-occupied ssDNA bridges in anaphase [33,36]. Prior to BLM unwinding, endonucleolytic and/or exonucleolytic processing to generate an entry site for BLM may also be essential step for repair to proceed (Stroik et al., unpublished). What remains a nearly complete enigma is precisely how single-stranded, telomeric UFBs are processed after this point. Presumably, these bridges undergo additional nucleolytic clipping and are then retracted into their respective cells (Figure 3C). Regardless, if an unprocessed UFB progresses to cytokinesis, breakage can be carried out by dicentric shearing of the offending DNA via standard mechanical rupturing [36]. These bridging:breakage events are in essence telomere translocations, unless of course the break were to occur precisely at the sight of initial joining, which is presumably a very unlikely outcome.
Telomeres connected by chromatin bridges and UFBs are a common class of anaphase abnormalities, especially if TRF2 is overexpressed or a TRF2 dominant-negative mutant protein is expressed [37,38]. TRF2-mediated UFBs are thought to likely be the result of replication fork collapse followed by protective fusion [39]. Similarly, increased anaphase bridging events have been documented with depletion of telomere replication rescue factors such as BLM, CtIP, Fanconi Anaemia Complementation Group M (FANCM) and Werner Syndrome Rec Q-like Helicase (WRN) [40,41] (Stroik et al., unpublished). The failure to decatenate telomeres can also lead to bridging events; however, the structure of a catenane is radically different than a fusion event (Figure 3B). Resolution of the former structure is thought to be largely dependent on topoisomerase II and undergo timely resolution [42] (Figure 3D). Thus, persistent telomeric UFBs are largely thought to result from fusions arising prior in the cell cycle. These fusions require a mitotic UFB processing mechanism that is truly a cell’s “last chance” to fix the aberrant chromosome(s). Importantly, however, unlike the simple rupture mechanism of chromatin bridges, which almost always generates mutagenic outcomes, the formation of and proper resolution of UFBs has at least the potential to yield a cell with a functional genome.
Translocations resulting from broken UFBs
Unfortunately, not all UFBs are processed in a timely fashion and when telomeric UFB entities persist in mitosis, they require a break of some variety to resolve the bridge and yield two unfettered cells. This fusion > bridge > break order is equivalent to (and better known as) a breakage:fusion:bridge (BFB) cycle which may drive any number of telomere translocations in a given cell. Indeed, elevated UFBs derived from sister chromatid bridges have been associated with high levels of genomic hyper-rearrangement [43] (Figure 3E-3G). While these BFB events would at first glance be deleterious, it is not impossible to rule out a positive result from such occurrences. For example, if a telomere-free chromosome end fused protectively with another telomere-capped chromosome, the result of mitotic bridge breakage could be telomeric addition (aka “telomere sharing”) for the once telomere-free chromosome (Figure 3C).
A second source of translocations arise in the form of micronuclei generated during events of improper segregation or multiple BFB shattering events. Depending on the mechanism of breakage during mitosis, the DNA can be separated from their associated nuclei but remain contained within a given cell (Figure 3H). This micronucleus can later be “joined” to another chromosome in the next cell cycle as a mechanism of repair [44,45]. This joining can occur at a chromosome terminus or at a site of a DSB. Importantly, chromothripsis arising from telomere bridge shattering (Figure 3F) can induce multiple translocations apart from the bridging locus translocation itself (Umbreit et al., bioRxiv; https://doi.Org/10.1101/835058). These trickle down “fusions after fusions”, are thought to be predominately NHEJ-mediated but have also been shown to occur in the absence of functional NHEJ machinery [9]. Alternatively, telomere fusions can circumvent a bridging outcome entirely when both chromosomes join one daughter cell instead of going their separate ways (Figure 3E). However, aneuploidies often render one or both of the affected cells inviable and thus this deleterious outcome is rare [46]. Even still, non-disjunctions can yield micronuclei, which could presumably be fused onto other chromosomes in subsequent cell cycles [47]. While these outcomes are drivers of genomic instability and cancer, it should be noted that in exceedingly rare cases they can successfully increase species diversity [48,49]. This later observation, combined with the fact that productive repair, however infrequent, can occur, may be the reason that this system evolved and/or has been evolutionarily retained.
In summary, due to the reoccurring nature of DNA replication we propose that telomere fusions arise from aberrant replication events at a larger frequency than that of dysfunctional Shelterin uncapping or formation of spontaneous DNA DSBs. Further, these fusions likely progress to mitosis generating either a chromatin bridge or a UFB connecting the two dividing cells. Chromatin bridge resolution is predominately dependent on physical rupturing of the bridge whereas UFB resolution likely requires nucleolytic clipping to separate the joined chromosomes. In either event, however, this leads predominately to DNA that is translocated from its original site. Recent work has supported that these mechanisms are likely more sizeable contributors to telomere translocations than previously appreciated.
Acknowledgments
We thank Dr. Duncan Baird for his laboratory’s long-standing contribution to our laboratory’s understanding of telomere dynamics. We thank Dr. Anja K. Bielinsky for her comments on the manuscript. S. S. prepared the original version of this manuscript as well as constructed all of the figures for it. E. A. H. helped with the editing of the manuscript.
Funding
Work in the Hendrickson laboratory was supported in part by grants from the NIH (GM088351) and the NCI (CA190492).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
EAH is a member of the scientific advisory boards for Horizon Discovery and Intellia Therapeutics.
References
- 1.de Lange T: Shelterin-mediated telomere protection. Annu Rev Genet 2018, 52:223–247. [DOI] [PubMed] [Google Scholar]
- 2.Sfeir A, de Lange T: Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336:593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rai R, Chen Y, Lei M, Chang S: TRF2-RAP1 is required to protect telomeres from engaging in homologous recombination-mediated deletions and fusions. Nat Commun 2016, 7:10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisaingo K, Uringa EJ, Lansdorp PM: Resolution of telomere associations by TRF1 cleavage in mouse embryonic stem cells. Mol Biol Cell 2014, 25:1958–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nera B, Huang HS, Hendrickson EA, Xu L: Both the classical and alternative non-homologous end joining pathways contribute to the fusion of drastically shortened telomeres induced by TRF2 overexpression. Cell Cycle 2019, 18:880–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baird DM, Kipling D: The extent and significance of telomere loss with age. Ann N Y Acad Sci 2004, 1019:265–268. [DOI] [PubMed] [Google Scholar]
- 7.Liddiard K, Ruis B, Takasugi T, Harvey A, Ashelford KE, Hendrickson EA, Baird DM: Sister chromatid telomere fusions, but not NHEJ-mediated inter-chromosomal telomere fusions, occur independently of DNA ligases 3 and 4. Genome Res 2016, 26:588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liddiard K, Ruis B, Kan Y, Cleal K, Ashelford KE, Hendrickson EA, Baird DM: DNA Ligase 1 is an essential mediator of sister chromatid telomere fusions in G2 cell cycle phase. Nucleic Acids Res 2019, 47:2402–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cleal K, Jones RE, Grimstead JW, Hendrickson EA, Baird DM: Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin. Genome Res 2019, 29:737–749.(.) 9. Chromothripsis during telomere crisis is independent of NHEJ and consistent with a replicative origin. This study explores telomere-induced chromothriptic rearrangements in the absence of a functional NHEJ pathway. The mutational pattern was found to be consistent with failed replication and subsequent template switching rather than NHEJ-mediated fusions. This data implicates replication as a source of the massive telomere rearrangements and dysfunctions observed in cancer cells.
- 10.Jones M, Bisht K, Savage SA, Nandakumar J, Keegan CE, Maillard I: The shelterin complex and hematopoiesis. J Clin Invest 2016, 126:1621–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valton AL, Prioleau MN: G-quadruplexes in DNA replication: a problem or a necessity? Trends Genet 2016, 32:697–706. [DOI] [PubMed] [Google Scholar]
- 12.Mason-Osann E, Gali H, Flynn RL: Resolving roadblocks to telomere replication. Methods Mol Biol 2019, 1999:31–57. [DOI] [PubMed] [Google Scholar]
- 13.Stroik S, Kurtz K, Hendrickson EA: CtIP is essential for telomere replication. Nucleic Acids Res 2019, 47:8927–8940.(..) 13. CtIP is essential for telomere replication. This work demonstrates that the essential function of human CtIP is likely telomere maintenance. The removal of CtIP results in replication forks that stall at secondary telomeric structures, then break and then result in telomere fusions.
- 14.Li F, Kim H, Ji Z, Zhang T, Chen B, Ge Y, Hu Y, Feng X, Han X, Xu H, et al. : The BUB3-BUB1 complex promotes telomere DNA replication. Mol Cell 2018, 70:395–407 e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones RE, Oh S, Grimstead JW, Zimbric J, Roger L, Heppel NH, Ashelford KE, Liddiard K, Hendrickson EA, Baird DM: Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep 2014, 8:1063–1076. [DOI] [PubMed] [Google Scholar]
- 16.Sobinoff AP, Pickett HA: Alternative lengthening of telomeres: DNA repair pathways converge. Trends Genet 2017, 33:921–932. [DOI] [PubMed] [Google Scholar]
- 17.Sobinoff AP, Allen JA, Neumann AA, Yang SF, Walsh ME, Henson JD, Reddel RR, Pickett HA: BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J 2017, 36:2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, Zhang D: FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A 2017, 114:E5940–E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, Deng Z, Zhang L, Wu C, Jin Y, Hwang I, Vladimirova O, Xu L, Yang L, Lu B, et al. : ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J 2019, 38:e96659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansel-Hertsch R, Beraldi D, Lensing SV, Marsico G, Zyner K, Parry A, Di Antonio M, Pike J, Kimura H, Narita M, et al. : G-quadruplex structures mark human regulatory chromatin. Nat Genet 2016, 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 21.Margalef P, Kotsantis P, Borel V, Bellelli R, Panier S, Boulton SJ: Stabilization of reversed replication forks by telomerase drives telomere catastrophe. Cell 2018, 172:439–453 e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leon-Ortiz AM, Panier S, Sarek G, Vannier JB, Patel H, Campbell PJ, Boulton SJ: A distinct class of genome rearrangements driven by heterologous recombination. Mol Cell 2018, 69:292–305 e296.(..) 22. A distinct class of genome rearrangements driven by heterologous recombination. This work documents chromothripsis and breakage-fusion-bridge events resulting from the loss of RTEL1. These genomic rearrangements are attributed to telomere replication stress inducing misrepair when essential repair and restart factors are missing.
- 23.Dahan D, Tsirkas I, Dovrat D, Sparks MA, Singh SP, Galletto R, Aharoni A: Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res 2018, 46:11847–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jimeno S, Camarillo R, Mejias-Navarro F, Fernandez-Avila MJ, Soria-Bretones I, Prados-Carvajal R, Huertas P: The helicase PIF1 facilitates resection over sequences prone to forming G4 structures. Cell Rep 2018, 24:3262–3273 e3264.(.) 24. The helicase PIF1 facilitates resection over sequences prone to forming G4 structures. PIF1 had been implicated in the dismantling of G4 structures via its helicase activity. Herein, PIF1 was documented to also function as a stimulator of DNA resection proximal to G4s. Further, this stimulation displayed preference for resection over G4s near DNA DSBs, akin to the ones that may form upon replication fork collapse.
- 25.Beishline K, Vladimirova O, Tutton S, Wang Z, Deng Z, Lieberman PM: CTCF driven TERRA transcription facilitates completion of telomere DNA replication. Nat Commun 2017, 8:2114.(..) 25. CTCF driven TERRA transcription facilitates completion of telomere DNA replication. Here, the authors demonstrate an essential function for TERRA transcription in facilitating faithful telomere replication. In the absence of TERRA transcription, telomere sister chromatid loss is prevalent along with UFB and micronuclei formation, likely as a result of replication catastrophe and subsequent protective telomere fusion.
- 26.Lee YW, Arora R, Wischnewski H, Azzalin CM: TRF1 participates in chromosome end protection by averting TRF2-dependent telomeric R loops. Nat Struct Mol Biol 2018, 25:147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toubiana S, Selig S: DNA:RNA hybrids at telomeres - when it is better to be out of the (R) loop. FEBS J 2018, 285:2552–2566. [DOI] [PubMed] [Google Scholar]
- 28.Qian W, Kumar N, Roginskaya V, Fouquerel E, Opresko PL, Shiva S, Watkins SC, Kolodieznyi D, Bruchez MP, Van Houten B: Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc Natl Acad Sci U S A 2019, 116:18435–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fouquerel E, Barnes RP, Uttam S, Watkins SC, Bruchez MP, Opresko PL: Targeted and persistent 8-oxoguanine base damage at telomeres promotes telomere loss and crisis. Mol Cell 2019, 75:117–130 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Chan J, Lambele M, Yusufzai T, Stumpff J, Opresko PL, Thali M, Wallace SS: NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. Cell Rep 2017, 20:2044–2056.(.) 30. NEIL3 repairs telomere damage during S phase to secure chromosome segregation at mitosis. This study documents a role for NEIL3, a DNA glycosylase, in the repair of oxidative lesions during S phase. The loss of this repair pathway greatly elevates telomere dysfunction, which equates to an abundance of anapahse bridges during mitotic division.
- 31.Baird DM, Hendrickson EA: Telomeres and chromosomal translocations : there's a ligase at the end of the translocation. Adv Exp Med Biol 2018, 1044:89–112. [DOI] [PubMed] [Google Scholar]
- 32.Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T: Chromothripsis and kataegis induced by telomere crisis. Cell 2015, 163:1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan YW, Fugger K, West SC: Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat Cell Biol 2018, 20:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez-Casanas M, Chan KL: The unresolved problem of DNA bridging. Genes (Basel) 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarlos K, Biebricher AS, Bizard AH, Bakx JAM, Ferrete-Bonastre AG, Modesti M, Paramasivam M, Yao Q, Peterman EJG, Wuite GJL, et al. : Reconstitution of anaphase DNA bridge recognition and disjunction. Nat Struct Mol Biol 2018, 25:868–876. [DOI] [PubMed] [Google Scholar]
- 36.Guerin TM, Beneut C, Barinova N, Lopez V, Lazar-Stefanita L, Deshayes A, Thierry A, Koszul R, Dubrana K, Marcand S: Condensin-mediated chromosome folding and internal telomeres drive dicentric severing by cytokinesis. Mol Cell 2019, 75:131–144 e133. [DOI] [PubMed] [Google Scholar]
- 37.Bernal A, Molto-Abad M, Dominguez D, Tusell L: Acute telomere deprotection prevents ongoing BFB cycles and rampant instability in p16(INK4a)-deficient epithelial cells. Oncotarget 2018, 9:27151–27170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan YW, West SC: A new class of ultrafine anaphase bridges generated by homologous recombination. Cell Cycle 2018, 17:2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nera B, Huang HS, Lai T, Xu L: Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun 2015, 6:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barefield C, Karlseder J: The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res 2012, 40:7358–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Luis J, Machin F: Fanconi anaemia-like Mph1 helicase backs up Rad54 and Rad5 to circumvent replication stress-driven chromosome bridges. Genes (Basel) 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clarke DJ, Azuma Y: Non-catalytic roles of the topoisomerase IIalpha C-terminal domain. Int J Mol Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiwari A, Addis Jones O, Chan KL: 53BP1 can limit sister-chromatid rupture and rearrangements driven by a distinct ultrafine DNA bridging-breakage process. Nat Commun 2018, 9:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marcozzi A, Pellestor F, Kloosterman WP: The genomic characteristics and origin of chromothripsis. Methods Mol Biol 2018, 1769:3–19. [DOI] [PubMed] [Google Scholar]
- 45.Kato T, Ouchi Y, Inagaki H, Makita Y, Mizuno S, Kajita M, Ikeda T, Takeuchi K, Kurahashi H: Genomic characterization of chromosomal insertions: insights into the mechanisms underlying chromothripsis. Cytogenet Genome Res 2017, 153:1–9. [DOI] [PubMed] [Google Scholar]
- 46.Knouse KA, Wu J, Whittaker CA, Amon A: Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci U S A 2014, 111:13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pampalona J, Roscioli E, Silkworth WT, Bowden B, Genesca A, Tusell L, Cimini D: Chromosome bridges maintain kinetochore-microtubule attachment throughout mitosis and rarely break during anaphase. PLoS One 2016, 11:e0147420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, Denroche RE, Liang SB, Brown AM, Kim JC, et al. : A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo J, Sun X, Cormack BP, Boeke JD: Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast. Nature 2018, 560:392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]