

Abstract
The bacterial DNA damage response (the SOS response) is a key pathway involved in antibiotic evasion and a promising target for combating acquired antibiotic resistance. Activation of the SOS response is controlled by two proteins: the repressor LexA and the DNA damage sensor RecA. Following DNA damage, direct interaction between RecA and LexA leads to de-repression of the SOS response. However, the exact molecular details of this interaction remain unknown. Here, we employ the fluorescent unnatural amino acid acridonylalanine (Acd) as a minimally-perturbing probe of the E. coli RecA:LexA complex. Using LexA labeled with Acd, we report the first kinetic model for the reversible binding of LexA to activated RecA. We also characterize the effects that specific amino acid truncations or substitutions in LexA have on RecA:LexA binding strength, and demonstrate that a mobile loop encoding LexA residues 75‒84 comprises a key recognition interface for RecA. Beyond insights into SOS activation, our approach also further establishes Acd as a sensitive fluorescent probe for investigating the dynamics of protein-protein interactions in other complex systems.
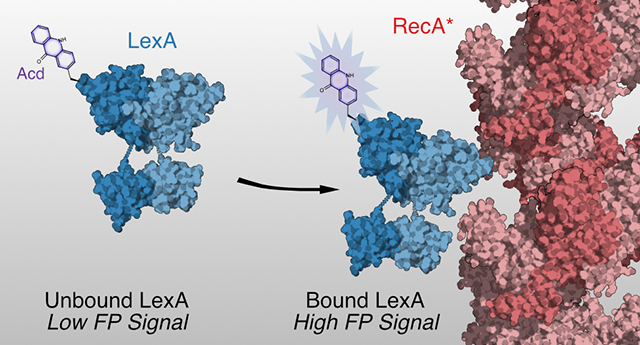
Graphical Abstract
INTRODUCTION
The prokaryotic DNA damage response, known as the SOS response, is precisely regulated given its importance in bacterial survival and adaptation to genotoxic stressors, including antibiotics. The ‘off’ or ‘on’ state of the SOS response is governed by the conserved repressor-protease LexA and its interactions between two different macromolecular partners (Figure 1a).1 One of these partners is genomic DNA. In the absence of DNA damage (the ‘off’ state), LexA repressors bind promoters for SOS regulated genes across the genome and block transcription.2,3 The presence of DNA damage, however, leads to the ATP-dependent polymerization of the sensor protein, RecA, along single stranded DNA (ssDNA), generating “activated” nucleoprotein filaments, termed RecA*.4 RecA* is the second macromolecular partner for LexA. Direct interaction of RecA* with LexA stimulates an intramolecular peptide bond hydrolysis reaction in LexA (auto-proteolysis).5 Cleaved LexA repressor is unable to bind DNA, and expression of SOS genes ensues. The SOS regulon includes a host of genes, ranging from high-fidelity DNA repair proteins to translesion DNA polymerases, which replicate over DNA lesions, but do so with high mutation rates. Correspondingly, genetic inactivation of LexA auto-proteolysis has been shown to decrease bacterial survival and prevent antimicrobial resistance upon treatment with DNA-damaging antibiotics,6–8 making RecA*-mediated LexA auto-proteolysis an appealing target for combating acquired antibiotic resistance.9–11
Figure 1.

Incorporating a minimal fluorescent probe into LexA. a) A schematic of the multiple macromolecular interactions in which the transcriptional repressor LexA participates during repression (left) or activation (right) of the SOS response. b) Modeled incorporation of Acd (blue), a minimally-perturbing fluorescent amino acid, at position 161 in LexA. The active site residues Ser119 and Lys156 are shown in orange. The inset depicts a rotated close-up of the incorporated Acd residue.
E. coli LexA is a dimer in solution,12,13 with each monomer consisting of an N-terminal DNA binding domain (NTD, residues 1–69), a flexible linker (residues 70–74), and a C-terminal protease domain (CTD, residues 75–202) that harbors a Ser119/Lys156 catalytic dyad (Figure 1b).14,15 Within the CTD, a structurally-dynamic peptide loop (residues 79–95) bearing the target scissile bond (Ala84-Gly85) can sample multiple conformations;16 however, crystallographic structural snapshots suggest that only one “cleavable” conformation positions the scissile bond for nucleophilic attack by Ser119. The observation of a conformationally-dynamic loop in LexA has led to debate over the role of RecA* in promoting the rate of LexA auto-proteolysis.13,16
The LexA:RecA* complex has defied numerous efforts aimed at elucidating the strength and nature of its interaction. Past structural studies do not provide sufficient atomic resolution to determine the binding interface for either protein.17,18 Mutagenesis has not identified LexA substitutions that can specifically disrupt RecA*-mediated proteolysis without also slowing the basal LexA auto-proteolysis rate.19–21 Investigations into LexA:RecA* interaction kinetics using surface plasmon resonance (SPR) assays suggest that either the NTD or CTD alone can bind RecA*, but a more precise binding surface is not described.22 In the absence of more compelling evidence, several groups have proposed conflicting models of LexA bound to the high-resolution structure of the activated RecA* filament.22,23 The uncertainty surrounding the size and molecular nature of the interacting regions between LexA and RecA* is undoubtedly reflected in a range of reported estimates for the affinity constant of this interaction.13,19
Given the central role of this interaction in SOS response regulation and the implications of this pathway in acquired antibiotic resistance, we sought to directly investigate the binding of LexA to RecA* through a novel experimental design. Fluorescence spectroscopic methods offer an underutilized approach in the SOS field that offer potentially precise, sensitive, and direct measurements of the protein dynamics inherent to the LexA:RecA* interaction.24 Here, we employ a fluorescent unnatural amino acid as a minimally-perturbing and maximally-sensitive probe that can directly and specifically report on LexA binding to DNA or RecA*.25 By directly measuring binding of LexA to RecA* and localizing key binding determinants, our findings establish a mechanistic foundation for disrupting the RecA*-mediated LexA auto-proteolysis reaction and validate the utility of minimally-perturbing fluorescent probes in the study of protein dynamics.
RESULTS AND DISCUSSION
Design of a Fluorescent Full-Length LexA Binding Reporter
Previously, we identified a number of positions in full-length E. coli LexA that can tolerate the genetic incorporation of acridonylalanine (Acd or δ), a small, intrinsically-fluorescent unnatural amino acid (Figure S1a).25–27 Because the binding region in LexA for RecA* was unknown, we sought to identify a permissive site in LexA for Acd incorporation that would permit RecA* binding. We purified ten different LexA proteins with Acd incorporated at various tolerant positions in a parent construct that was catalytically inactive (LexA-S119A) (Figure S1b), which prevents the auto-proteolytic reaction from complicating analysis. Using fluorescence anisotropy, a sensitive modality that can be used to monitor binding interactions, we screened each Acd-labeled LexA-S119A variant for changes in anisotropy signal when incubated alone, with non-activated RecA, or with specifically activated RecA* (Figure S1c). Equilibrium measurements revealed that most of these labeled LexA proteins demonstrate significant changes in anisotropy upon specific binding to RecA* (Figure S1d). We elected to advance LexA-S119A-Q161δ (LexAδ, hereafter) for further study, in part because Q161δ is removed from both the catalytic dyad and target scissile bond (Figure 1b), two regions in LexA that are suggested to come into close proximity with RecA*.16,28 Following larger scale overexpression and purification, we confirmed successful Acd incorporation at Q161 through SDS-PAGE fluorescence (Figure S2) and mass spectrometry analyses (Figure S3). We further confirmed that the Q161δ substitution does not disrupt native LexA activity by reverting the S119A mutation and determining that the auto-proteolytic activity of this catalytically-active Acd-labeled LexA is similar to that of wild type LexA (Figure S4).
We reasoned that the relatively large fluorescence anisotropy change upon binding of LexAδ to RecA* in our equilibrium measurements could also permit resolution of the kinetics of LexA binding. To test the ability of our Acd-labeled construct to provide time-resolved binding data, we rapidly mixed 250 nM LexAδ with 2 μM activated RecA* in a stopped-flow apparatus and monitored for 60 seconds (Figure 2a). Of note, LexA predominantly adopts its dimeric state at this concentration in solution.12,13 The combination of LexAδ with RecA* revealed a substantial time-dependent change in fluorescence anisotropy (Figure 2b). We attribute the size of this anisotropy change to the unusually long lifetime of Acd (15 ns),29 because longer lifetime fluorophores provide a better dynamic range for the study of macromolecular interactions.30 As expected, this anisotropy change vanishes when either of the ATPγS or ssDNA cofactors, required for RecA activation, are excluded (Figure 2b). The selection of Acd as a fluorescent probe of protein-protein binding thus allowed us to track LexA engagement with RecA* in solution and with high temporal resolution.
Figure 2.

Association kinetics of labeled LexA with RecA. a) Experimental design of association of LexA with RecA. The indicated amount of Acd-labeled LexA-S119A-Q161δ (LexAδ), is rapidly mixed in a stopped-flow apparatus with post-activation reaction mixtures of RecA and fluorescence anisotropy is measured. b) Plots of anisotropy versus time demonstrate the specificity of LexAδ for RecA*. 250 nM of LexAδ were rapidly mixed with 2 μM of RecA reactions containing only RecA (green), RecA + ATPγS (purple), RecA + ssDNA (blue), or RecA + ATPγS + ssDNA (RecA*, red). Data were fit to a smoothing function (solid lines). c) A series of plots of anisotropy versus time shows the dependence of LexA-RecA* association rate on RecA* concentration. Curve labels indicate the concentration of RecA* (μM) that was mixed with 250 nM of LexAδ. The best-fit curves from reaction simulations after globally fitting three independent concentration series experiments are shown as solid black lines for each concentration. Simulation boundaries at a Chi2 ratio of 0.70 are shown as gray ribbons.
Association Rate and Affinity of the LexA:RecA* Binding Step
The specificity of the measured fluorescence anisotropy of LexAδ for the bound versus unbound state positioned us to develop a kinetic model for the natural binding interaction of dimeric LexA with RecA*. As before, we rapidly mixed 250 nM LexAδ with activated RecA* in concentrations spanning a nearly 1000-fold range (0.016–10.24 μM). Examination of time-dependent fluorescence anisotropy changes reveals that the apparent association rate of LexA binding to RecA* increases as a function of RecA* concentration (Figure 2c). Further, at intermediate RecA* concentrations, the LexA binding curves do not approach the same maximum anisotropy values as those at the plateaus for the highest RecA* concentration curves, consistent with a model of reversible binding between LexA and RecA*. Using a reaction simulation and global fitting approach, we confirmed that a one-step, reversible binding model provided a reasonable fit for our association curves (Figure 2c) with all model parameters well-constrained by our data (Figure S5, Table S1). Using our fitted values for the association rate (k1 = 0.061 μM−1 sec−1) and dissociation rate (k−1 = 0.048 sec−1), we calculated an apparent dissociation constant, KD, of 0.79 μM for the interaction of full-length LexA with RecA*.
Our calculated KD value of 0.79 μM for the LexA:RecA* interaction is similar to reported KM values of either 0.5 μM for the RecA*-catalyzed auto-proteolysis of full-length LexA19 or 0.9 μM for the auto-proteolysis of a truncated LexA containing residues 65–202.13 That our measured KD value falls within a range of reported KM values suggests that k2, the step representing the chemistry of LexA auto-proteolysis, may be rate-limiting because it must be slower (i.e. smaller) than the dissociation rate in order for the KD and KM values to be similar. Notably, our results also support an observation that LexA binding to RecA* is demonstrably slower and weaker than reported binding rates and affinities of LexA to operator sites in the E. coli genome.3 This interpretation favors the recently established non-equilibrium view of the SOS response, in which rapid and significant increases in RecA* are required to deplete free LexA and thereby overcome, through mass action, the favored kinetics of LexA binding to operator sites.3
As a means of validating our one-step reversible binding model, we sought to directly isolate and examine the dissociation of LexA from RecA*. Past studies have established that LexA binds rapidly and tightly to SOS operator DNA and that DNA-bound LexA likely does not bind RecA* to a substantial degree.3,31 Therefore, we predicted that the addition of excess operator DNA to a mixture of pre-formed LexA:RecA* complex would result in preferential and rapid sequestration of free LexA by the excess operator DNA (Figure S6a). With the use of the operator DNA trap, the kinetic step of LexA dissociation from RecA* can be isolated in this analysis. Indeed, pre-incubated LexA:RecA* reactions, when mixed with an excess of 44mer double stranded DNA (dsDNA) substrates bearing the consensus SOS operator sequence, produce an observable shift in anisotropy that could be attributed to LexA dissociation from RecA* and subsequent association with dsDNA (Figure S6b). Upon fitting these data, we obtained an estimate of the LexA:RecA* dissociation rate (k−1 = 0.093 sec−1) that was only two-fold faster than the best fit value (k-1 = 0.048 sec−1) from our above binding model.
A Structural Region in LexA Strongly Impacts Binding to RecA*
Efforts to identify a RecA* binding site in LexA have produced conflicting results, from genetic screens that highlight residues 80–84 in the LexA cleavage loop to chemical cross-linking and SPR studies that argue either the NTD (1–69) or CTD (75–202) in isolation can bind to RecA*.19,22 In the past, we and others have shown that certain N-terminal truncations of LexA (65–202 or 75–202) remain competent for RecA*-mediated proteolysis.11,13 Given the reproducibility and specificity of our binding assay, we reasoned we could inspect the effect of alterations to LexA on RecA* binding to narrow down a site in LexA responsible for the RecA* interaction. We devised an experimental approach in which the binding strength (KD) of modified forms of LexA could be assessed. In this design, we allow an unlabeled LexA variant of interest to come to equilibrium with RecA*, so that the availability of free RecA* sites is dependent on the affinity of the unlabeled LexA variant for RecA* (Figure 3a). The sample is next rapidly mixed with fluorescent LexAδ. Because the rate of anisotropy increase is dependent on the concentration of RecA* binding sites, as shown in Figure 2, we can then quantitatively determine the binding affinities of any number of LexA variants using this competitive binding experimental setup. To first validate this approach, we competed unlabeled, catalytically-inactive LexA (LexA-S119A) with its labeled partner, LexAδ. Following rapid mixing, LexAδ demonstrated a concentration-dependent decrease in the observed association rates (Figure 3b). By interpolating the initial association rates at time zero (Figure 3b), we can infer the degree to which the binding of LexAδ is inhibited as a function of the concentration of unlabeled competitor LexA (Figure 3c). The Ki of the unlabeled LexA-S119A is in rough agreement with the directly measured Kd of LexAδ, suggesting that the competition assay is a reliable means to measure affinity of LexA variants for RecA*. Furthermore, the similarity between the measured affinity for the unlabeled LexA-S119A and LexAδ supports the suggestion that Acd is LexAδ is minimally-perturbing with regards to RecA* binding.
Figure 3.

Competitive binding of LexA protein variants for RecA*. a) Experimental setup for measuring the binding affinity of unlabeled LexA variants for RecA*. Top: in a pre-incubation reaction at equilibrium, unlabeled LexA variants at various concentrations are mixed with 2 μM RecA*. Bottom: The observed association of LexAδ depends on the concentration of open LexA binding sites on RecA* in the pre-incubation reaction at equilibrium, which is inversely proportional to the strength of binding for a given unlabeled LexA variant. b) Individual plots of time-dependent anisotropy of LexAδ following pre-incubation or 2 μM RecA* with the indicated concentrations of unlabeled LexA. The data are fit to a smoothing function (solid lines), and the initial association rate at time zero is visualized (dashed lines). c) Plots of percent inhibition of LexAδ binding to 2 μM RecA* as a function of the concentration of various unlabeled LexA fragments: full-length LexA, LexA(1–202), red; N-terminal proteolytic fragment, LexA(1–84), blue; C-terminal proteolytic fragment, LexA(85–202), purple; N-terminal truncation, LexA(75–202), green. Each dataset is fit to a dose response curve (solid line).
Equipped with this experimental system, we sought to examine how LexA binding affinity for RecA* is impacted by various forms of LexA, with a focus on the role for the mobile peptide loop that contains the scissile target bond. We first generated the two protein fragments that naturally result from LexA auto-proteolysis, the N- or C-terminal proteolytic fragment bearing one half of the cleavage loop (1–84 and 85–202) (Figure S7). Notably, in the competition assay, both fragments were dramatically weakened in their ability to inhibit LexAδ binding (Figure 3c and Table S3). These low affinities for the products of auto-proteolysis for RecA* suggest a mechanism for turnover whereby a single RecA* binding interface could bind with intact LexA, promote auto-proteolysis, and then be available for engagement with other intact LexA proteins upon product dissociation.
To further and more precisely probe the molecular interface involved in the RecA* interaction, we next generated a sequence of N-terminal truncations of LexA (Figure S8a and Figure S8b). Remarkably, none of these more N-terminal truncations significantly changed the degree to which LexAδ binding was affected (Figure S8c and Table S4), including truncation of LexA up to residue 75, which marks the start of the structured region of the CTD including the cleavage loop. Our quantitative competitive binding results thus highlight the importance of residues 75–84 in LexA for RecA* binding, given the great degree of difference in the binding curves for 75–202 versus 85–202 (Figure 3c). However, this stretch of residues on its own is not sufficient for binding to RecA* given that the N-terminal proteolytic fragment, LexA(1–84), possesses this region, but is unable to efficiently bind to RecA*. Thus, the combination of residues 75–84, with an intact mobile peptide loop, and the C-terminal catalytic core is required to for molecular recognition and binding to RecA*.
Disruptions to LexA Mobile Loop Impact RecA* Binding
Despite the implication of the intact mobile peptide loop as a critical structural determinant for RecA* binding, the molecular nature of how this loop contributes to binding is not apparent; for example, the interaction could depend on direct contact with residues in the loop or it could require loop flexibility for a recognition interface to form. To further examine possibilities, we selected two amino acid substitutions in the loop itself, G80P and V82M (Figure 4a), that are known to decrease the rates of both basal and RecA*-mediated LexA auto-proteolysis by putatively forcing an unfavorable loop conformation.20 We examined the effect of these substitutions using the LexAδ competition binding assay and found that the apparent affinity of these constructs for RecA* decreased by over ten-fold (Figure 4b and Table S5). Notably, amino acid substitutions at these positions have been shown to dramatically reduce, but not fully eliminate, RecA*-dependent LexA cleavage in vitro and increase susceptibility to DNA damage in vivo.19,20 The effects of these amino acid substitutions in LexA are consistent with the interpretation that a particular cleavage loop conformation, or at the very least conformational flexibility of the loop, is an essential element driving efficient interaction with RecA*.
Figure 4.

Loop variants of LexA impact binding to RecA*. a) Surface representation of the LexA C-terminal domain (PDB 1JHE) with the active site colored orange. The mobile cleavage loop is shown in stick representation (light blue). Sites chosen for auto-proteolysis deficient amino acid substitutions are G80P (green) and V82M (blue). As with both mutant constructs, S119A (red) is the full length 1–202 version. b) Plots of percent inhibition as a function of concentration of unlabeled LexA cleavage loop variants. Data points represent the median value from at least four independent replicates. The solid line represents the best-fit variable-slope sigmoidal dose-response curve for each sample.
A Refined Catalytic Role for RecA* and Therapeutic Implications
RecA* has been previously described as a “co-protease” or catalyst of LexA auto-proteolysis, because it is capable of promoting the auto-proteolysis of multiple LexA “substrates”.13 In the absence of RecA*, however, LexA auto-proteolysis occurs very slowly. At physiologic pH, Lys156 is protonated and likely acts as an energetic barrier to the dynamic peptide loop adopting the cleavable conformation. The observation that exposing LexA to alkaline conditions increases the basal auto-proteolysis rate is consistent with a requirement for deprotonation of Lys156. These structural and biochemical observations have led to the proposal that RecA* accelerates LexA auto-proteolysis by inducing or stabilizing an energetically unfavorable cleavable conformation in LexA.
Despite this model, the role of RecA* as a catalyst has remained poorly defined. Our study sheds light on how binding to RecA* might promote the rate of LexA auto-proteolysis. Our ability to measure the kinetics and affinity of LexA engagement with RecA* narrows the critical binding determinants to the intact mobile cleavage loop in partnership with the C-terminal domain. Poor binding by the native cleavage fragments offers support for the interpretation of RecA* as a catalyst, given that dissociation of products would offer a means for multiple turnover events. Furthermore, our results with LexA variants thought to disfavor the cleavable conformation add support to the model whereby cleavage loop conformation is relevant to RecA* engagement. Our kinetic modeling is most consistent with a single-step reversible binding. While we cannot rule out the possibility that RecA* induces conformational change upon binding, such a conformational change step does not leave any distinct kinetic signature in our rapid mixing experiments. Future studies with LexA variants that incorporate Acd into positions that can be responsive to dynamics within the mobile protein loop may help to select between selective-binding versus induced-fit models for the LexA:RecA* interaction.
Interest in targeting the SOS response for its role in mutagenesis and antibiotic evasion has spurred numerous investigations into the mechanisms underlying its activation. Genetic inactivation of the SOS response regulator, LexA, has been shown to decrease bacterial survival and even reverse resistance to DNA-damaging antibiotics.6–8 The development of small molecule inhibitors to mimic the effects of genetic SOS inactivation has proven promising, but the advancement of inhibitors is in part limited by our insufficient mechanistic insight into SOS activation. The use of Acd-based probes with LexA offer a means to separate the different steps in auto-proteolysis, including the RecA* binding step, which we have probed here, and the subsequent auto-proteolysis of LexA. Given the pressing need for novel approaches to acquired antibiotic resistance, we are hopeful that Acd, as a minimally-perturbing, genetically-incorporated probe, could prove useful to deciphering the mechanism of action of putative inhibitors and to subsequently improving their activity, and that analogous approaches could make use of Acd as a reliable probe of protein dynamics in other complex systems.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Jack Ferrie for assistance with Figure 1b. This work was supported by the National Institutes of Health (NIH, No. R01-GM127593, to R.M.K. and E.J.P.) and the National Science Foundation (NSF, No. CHE-1708759, to E.J.P.). R.M.K. holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. Z.M.H. was supported by the NIH Chemistry Biology Interface Training Program (No. T32-GM071399). Instruments supported by the NSF include the MALDI mass spectrometer (NSF, MRI-0820996) and stopped flow fluorometer (NSF, CHE-1337449).
Footnotes
ASSOCIATED CONTENT
Supporting Information includes experimental methods, supplemental figures and tables, and associated references (PDF).
REFERENCES
- 1.Butala M; Zgur-Bertok D; Busby SJ The bacterial LexA transcriptional repressor. Cell Mol. Life Sci 2009, 66, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brent R; Ptashne M Mechanism of action of the lexA gene product. Proc. Natl. Acad. Sci. U. S. A 1981, 78, 4204–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Culyba MJ; Kubiak JM; Mo CY; Goulian M; Kohli RM Non-equilibrium repressor binding kinetics link DNA damage dose to transcriptional timing within the SOS gene network. PLoS Genet. 2018, 14, e1007405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox MM Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol 2007, 42, 41–63. [DOI] [PubMed] [Google Scholar]
- 5.Little JW Autodigestion of lexA and phage lambda repressors. Proc. Natl. Acad. Sci. U. S. A 1984, 81, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cirz RT; Chin JK; Andes DR; de Crecy-Lagard V; Craig WA; Romesberg FE Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005, 3, e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu TK; Collins JJ Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4629–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mo CY; Manning SA; Roggiani M; Culyba MJ; Samuels AN; Sniegowski PD; Goulian M; Kohli RM Systematically Altering Bacterial SOS Activity under Stress Reveals Therapeutic Strategies for Potentiating Antibiotics. mSphere 2016, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Culyba MJ; Mo CY; Kohli RM Targets for Combating the Evolution of Acquired Antibiotic Resistance. Biochemistry 2015, 54, 3573–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selwood T; Larsen BJ; Mo CY; Culyba MJ; Hostetler ZM; Kohli RM; Reitz AB; Baugh SDP Advancement of the 5-Amino-1-(Carbamoylmethyl)-1H-1,2,3-Triazole-4-Carboxamide Scaffold to Disarm the Bacterial SOS Response. Front. Microbiol 2018, 9, 2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mo CY; Culyba MJ; Selwood T; Kubiak JM; Hostetler ZM; Jurewicz AJ; Keller PM; Pope AJ; Quinn A; Schneck J; Widdowson KL; Kohli RM Inhibitors of LexA Autoproteolysis and the Bacterial SOS Response Discovered by an Academic-Industry Partnership. ACS Infect. Dis 2018, 4, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohana-Borges R; Pacheco AB; Sousa FJ; Foguel D; Almeida DF; Silva JL LexA repressor forms stable dimers in solution. The role of specific dna in tightening protein-protein interactions. J. Biol. Chem 2000, 275, 4708–4712. [DOI] [PubMed] [Google Scholar]
- 13.Giese KC; Michalowski CB; Little JW RecA-dependent cleavage of LexA dimers. J. Mol. Biol 2008, 377, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slilaty SN; Little JW Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang AP; Pigli YZ; Rice PA Structure of the LexA-DNA complex and implications for SOS box measurement. Nature 2010, 466, 883–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Y; Pfuetzner RA; Mosimann S; Paetzel M; Frey EA; Cherney M; Kim B; Little JW; Strynadka NC Crystal structure of LexA: a conformational switch for regulation of self-cleavage. Cell 2001, 106, 585–594. [DOI] [PubMed] [Google Scholar]
- 17.VanLoock MS; Yu X; Yang S; Galkin VE; Huang H; Rajan SS; Anderson WF; Stohl EA; Seifert HS; Egelman EH Complexes of RecA with LexA and RecX differentiate between active and inactive RecA nucleoprotein filaments. J. Mol. Biol 2003, 333, 345–354. [DOI] [PubMed] [Google Scholar]
- 18.Yu X; Egelman EH The LexA repressor binds within the deep helical groove of the activated RecA filament. J. Mol. Biol 1993, 231, 29–40. [DOI] [PubMed] [Google Scholar]
- 19.Lin LL; Little JW Isolation and characterization of noncleavable (Ind-) mutants of the LexA repressor of Escherichia coli K-12. J. Bacteriol 1988, 170, 2163–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mo CY; Birdwell LD; Kohli RM Specificity determinants for autoproteolysis of LexA, a key regulator of bacterial SOS mutagenesis. Biochemistry 2014, 53, 3158–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shepley DP; Little JW Mutant LexA proteins with specific defects in autodigestion. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11528–11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kovacic L; Paulic N; Leonardi A; Hodnik V; Anderluh G; Podlesek Z; Zgur-Bertok D; Krizaj I; Butala M Structural insight into LexA-RecA* interaction. Nucleic Acids Res. 2013, 41, 9901–9910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adikesavan AK; Katsonis P; Marciano DC; Lua R; Herman C; Lichtarge O Separation of recombination and SOS response in Escherichia coli RecA suggests LexA interaction sites. PLoS Genet. 2011, 7, e1002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Speight LC; Samanta M; Petersson EJ Minimalist approaches to protein labelling: getting the most fluorescent bang for your steric buck. Aust. J. Chem 2014, 67, 686–700. [Google Scholar]
- 25.Speight LC; Muthusamy AK; Goldberg JM; Warner JB; Wissner RF; Willi TS; Woodman BF; Mehl RA; Petersson EJ Efficient synthesis and in vivo incorporation of acridon-2-ylalanine, a fluorescent amino acid for lifetime and Forster resonance energy transfer/luminescence resonance energy transfer studies. J. Am. Chem. Soc 2013, 135, 18806–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hostetler ZM; Ferrie JJ; Bornstein MR; Sungwienwong I; Petersson EJ; Kohli RM Systematic evaluation of soluble protein expression using a fluorescent unnatural amino acid reveals no reliable predictors of tolerability. ACS Chem. Bio 2018, 13, 2855–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sungwienwong I; Hostetler ZM; Blizzard RJ; Porter JJ; Driggers CM; Mbengi LZ; Villegas JA; Speight LC; Saven JG; Perona JJ; Kohli RM; Mehl RA; Petersson EJ Improving target amino acid selectivity in a permissive aminoacyl tRNA synthetase through counter-selection. Org. Biomol. Chem 2017, 15, 3603–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin LL; Little JW Autodigestion and RecA-dependent cleavage of Ind- mutant LexA proteins. J. Mol. Biol 1989, 210, 439–452. [DOI] [PubMed] [Google Scholar]
- 29.Hamada H; Kameshima N; Szymańska A; Wegner K; Łankiewicz L; Shinohara H; Taki M; Sisido M Position-specific incorporation of a highly photodurable and blue-laser excitable fluorescent amino acid into proteins for fluorescence sensing. Bioorg. Med. Chem 2005, 13, 3379–3384. [DOI] [PubMed] [Google Scholar]
- 30.Pope AJ; Haupts UM; Moore KJ Homogeneous fluorescence readouts for miniaturized high-throughput screening: theory and practice. Drug Discov. Today 1999, 4, 350–362. [DOI] [PubMed] [Google Scholar]
- 31.Butala M; Klose D; Hodnik V; Rems A; Podlesek Z; Klare JP; Anderluh G; Busby SJ; Steinhoff HJ; Zgur-Bertok D Interconversion between bound and free conformations of LexA orchestrates the bacterial SOS response. Nucleic Acids Res. 2011, 39, 6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.