

Abstract
Catalytic diastereo- and enantioselective preparation of complex tertiary homoallylic alcohols containing a vicinal quaternary carbon stereogenic center and a versatile alkenylboronic ester is disclosed. Transformations are promoted by 5 mol % of a readily available copper catalyst bearing a bulky monodentate phosphoramidite ligand, which is essential for attaining both high dr and er. Reactions proceed with a wide variety of ketones and allylic 1,1-diboronate reagents that allows for the efficient preparation of diverse array of molecular scaffolds.
Keywords: 1,1-Allyldiboron; Ketones; Quaternary carbon; Enantioselective; Copper
Graphical Abstract
A catalytic diastereo- and enantioselective method for the preparation of complex tertiary homoallylic alcohols containing a vicinal quaternary carbon stereogenic center and a versatile alkenylboronic ester is disclosed. Reactions proceed in the presence of chiral Cu complex with a wide variety of ketones and γ,γ-disubstituted allylic diboronates to afford products in both high dr and er.
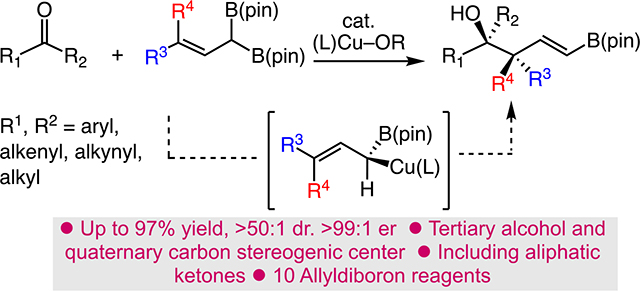
Quaternary carbon stereogenic centers and tertiary alcohols are common motifs found in many bioactive molecules, and their enantioselective preparation, particularly in acyclic systems, are essential transformations in organic synthesis.[1,2] Nonetheless, catalytic enantioselective C–C bond forming reactions that establish both of these critical functional groups remain a significant challenge. To this end, the enantio- and diastereoselective addition of a suitably substituted allyl nucleophile to ketones provides a concise strategy for assembling these key chemical structures.[3] Significant efforts have been directed towards catalytic enantioselective synthesis of tertiary homoallylic alcohols that contain a vicinal tertiary carbon stereocenter.[4] Conversely, the diminished reactivity of ketones and the closer parity in size of the carbonyl substituents serve to hamper development of quaternary carbon equivalents.[5] However, a few examples of enantioselective allylic nucleophile additions to ketones that form quaternary carbon stereocenters have been reported. Enantiospecific reactions of chiral allylboron compounds with ketones proceed efficiently and with excellent levels of diastereoselectivity.[6] In addition, Szabó reported the first catalytic enantio- and diastereoselective synthesis of homoallylic alcohols bearing quaternary carbon stereocenters (Scheme 1A).[7,8] Reactions employ stereodefined γ,γ-disubstituted allylboronic acids and are catalyzed by chiral binaphthyl-based diols. Recent notable advances that circumvent the initial preparation of the organometallic by coupling a diene to a ketone via a tandem Cu-catalyzed borylation/1,2-addition sequence have been disclosed (Scheme 1B).[9] Nevertheless, shortcomings related to the use of unstable boronic acids or the need for sparteine-mediated generation of the allylboronates detract from the implementation of these methods.
Scheme 1.

Catalytic Diastereo- and Enantioselective γ,γ-Disubstituted Allylic Nucleophile Additions to Ketones. B(pin) = (pinacolato)boron.
In light of the aforementioned limitations, we sought to develop a general catalytic allylic nucleophile addition protocol for the stereoselective formation of quaternary carbon centers vicinal to tertiary alcohols (Scheme 1C). Our approach centers on the development of a catalyst controlled 1,2-addition of γ,γ-disubstituted allyldiboronate esters to ketones.[10] 1,1-Diboron reagents represent a nascent class of functional reagents for the enantioselective synthesis of alcohols and amines.[11] In this regard, we envisioned a catalytic reaction that begins with an enantioselective SE2’ transmetalation between in situ-generated (L)-Cu–OMe and a stereodefined allyldiboron (e.g., 2).[12,13] Quaternary allylic Cu A can undergo rapid suprafacial shift to the sterically less congested and energetically more favorable tertiary α-boryl–Cu species B, which can add diastereoselectively to a ketone via C. Notably, with γ,γ-disubstitution, A(1,3)-strain likely limits σ-bond rotation and migration of (L)-Cu, preventing scrambling of alkene geometry.[14] Finally, the sequence (C→D) furnishes tertiary alcohols, following (L)-Cu–OMe regeneration by MeOH, bearing a vicinal quaternary center appended with a versatile alkenylboron. To address the challenge of dr, we postulated that increasing the size of the ligand would lead to allyl-Cu addition that places RL of the ketone in the pseudo-equatorial position in the chair-like transition state of C.[15] Moreover, the reactive allylic Cu species would also counter lower reactivity caused by steric congestion present during C–C bond formation.
Our studies began by examining the ability of in situ-generated phosphoramidite–Cu complexes, which had previously emerged as optimal in promoting reactions with aryl aldimines,[10e] in the reaction of 4 and 5 to afford 6a (Scheme 2). We found that treatment of acetophenone 4 and allyldiboron 5, with 5.0 mol % CuOtBu, 10 mol % H8-binaphthyl-derived L1, and 1.05 equiv MeOH in THF at −40 °C for 16 h furnished 6a as a single regioisomer, favoring generation of the quaternary carbon center (i.e., γ-allylation) in 86% NMR yield, 4:1 dr, and 83:17 er. Switching to ligands L2–L5, which contain increasingly larger aryl groups at the 3,3’-positions, result in an increase in dr (4:1→7:1) and er (83:17→96.5:3.5) at −40 °C. Further improvement in diastereoselectivity to 10:1 dr was observed by the application of phosphoramidite L6 containing 3,5-t-BuC6H3 substituents. Importantly, the slight diminution in enantioselectivity (93.5:6.5 er) could be restored by cooling the reaction of (L6)-Cu to −60 °C, affording 6a in >98% NMR yield, 15:1 dr, and 97:3 er, and represented optimal reaction conditions. In comparison, cooling the reaction with L5 to −60 °C affords 6a in 10:1 dr and 97:3 er, an observation noted with other substrates.
Scheme 2.

Investigation of Chiral Cu Complexes. Reactions performed under N2 atmosphere. Yield and diastereomeric ratios (dr) determined by analysis of 1H NMR spectra of crude reactions with hexamethyldisiloxane as internal standard. Enantiomeric ratios (er) determined by HPLC analysis. See SI for details.
As illustrated in Scheme 3A, a wide variety of aryl-, and heteroaryl-substituted ketones were examined. For example, aryl ketones bearing electron-donating (6b–e) as well as electron-withdrawing (6f–k) groups can be used, including those containing meta- (6l–o) and ortho (6p) substitution. In addition, aryl ketone product 6q is formed in good yield and excellent selectivity, indicating the protocol is tolerant of primary alkyl chlorides. Heteroaryl and polyaromatic ketones are also effective substrates (6r–t); unprotected parent indole was not effective. While the aryl ketone scope is tolerant of substituent electronics, the reaction is sensitive to sterics; most ortho-substituents (such as o-Me) or diaryl ketones such as benzophenone (e.g., 6u) do not participate due to steric congestion. Ketone diversity also extends to enones (7a–b),[16] ynones (7c), and cyclic (7d) and acyclic alkyl ketones (7e–g) (Scheme 3B). The formation of 7d–f demonstrates that transformations in the presence of NBoc amides and silyl ethers proceed in good yield and stereoselectivity. As illustrated by the synthesis of 7h–i, the catalytic protocol extends to symmetrical dialkyl ketones such as acetone (95:5 er) and cyclohexanone (97:3 er) that form enantiomerically enriched tertiary alcohols, without a stereogenic carbinol. Such examples provide support for the highly enantioselective transmetalation that occurs in the initial step (2→A→B, Scheme 1C). Reactions with chiral ketones also proceed with high levels of diastereoselectivity. For example, catalytic addition to (S)-Hajos-Parrish diketone under standard conditions affords 7j as a single diastereomer (>50:1 dr), with exclusive chemoselectivity for the enone over the aliphatic ketone.
Scheme 3.

Diastereo- and Enantioselective Cu-Catalyzed Synthesis of Homoallyl Alcohols and Quaternary Carbon Stereocenters by Allylic Nucleophile Additions to Aryl, Heteroaryl, Alkenyl, Alkynyl, and Alkyl Ketones. Reactions performed under N2 atmosphere on 0.05 mmol scale with 1.0 equiv of ketone, 1.2 equiv. of 5, in 0.350 ml of THF. Yields of purified products after SiO2 chromatography. Experiments were run in duplicate. Diastereomeric ratios (dr) determined by analysis of 1H NMR spectra of purified products. Enantiomeric ratios (er) determined by HPLC or SFC analysis. [a] Reactions run at −50 °C. [b] 2.0 equivalents of CD3OD used in place of CH3OH. See SI for details.
The range of allyldiboron reagents also displays broad scope with regard to varying the substituents of the quaternary carbon stereogenic center (Scheme 4). The requisite stereodefined allyldiboron reagents are readily prepared from the corresponding alkenyl halides (see the SI). The stereospecific nature of the catalytic reaction is exemplified by the enantio- and diastereoselective formation of tertiary alcohols 8a (47:1 dr, 91:9 er) and 8b (32:1 dr, 96.5:3.5 er) bearing Me/Et quaternary carbon stereogenic centers arising for the reaction of both E- and Z-allyldiboron isomers. Additionally, a variety of quaternary carbon stereogenic centers can be prepared, including those containing cyclopropyl (8c), N-heteroaryl (8d), silylether (8e), and homobenzyl (8f) groups. In all cases, yield (>71%), dr (>7:1), and er (>95.5:4.5) remain high. Similar to non-prochiral ketone examples 7h–i (Scheme 3), symmetrically γ,γ-disubstituted allylic diboronates also undergo efficient C–C bond formation to generate chiral tertiary alcohols with a non-stereogenic quaternary carbon center; for example, the tertiary alcohols containing gem-dimethyl (8g), cyclohexane (8h), and cyclobutane (8i) substituents shown in Scheme 4, formed in >75% yield and 88:12–97.5:2.5 er, are representative.
Scheme 4.

Variation of Allyl 1,1-Diboronate Scope. Reactions performed under N2 atmosphere on 0.05 mmol scale with 1.0 equiv of ketone, 1.2 equiv. of 2, in 0.350 ml of THF. Yields of purified products after SiO2 chromatography. Experiments were run in duplicate. Diastereomeric ratios (dr) determined by analysis of 1H NMR spectra of purified products. Enantiomeric ratios (er) determined by HPLC or SFC analysis. See the SI for details.
The relative and absolute stereochemistry of the product, as well as confirmation of the proposed stereochemical model in Scheme 1C, was assigned by single crystal X-ray diffraction of 6q (Scheme 5). As expected, an anti-relationship between the hydroxyl and n-butyl (E-) substituent is observed, corresponding to positioning of the large ketone substituent in the pseudo-equatorial position. In addition, the diastereoselectivity for alkyl ketone-derived 7g was also determined as anti, consistent with the proposed stereochemical model (see SI for details). This is in contrast to the Cu-catalyzed reactions of γ,γ-disubstituted allyldiborons with NPMB-imines, which afford the syn stereoisomer.[10e]
Scheme 5.

X-Ray Structure of 6q. See SI for details.
The variety of functional group rich products synthesized through the catalytic protocol can be transformed in a number of ways to furnish useful enantio- and diastereomerically enriched compounds; five examples are depicted in Scheme 6. Firstly, treatment of 6f and 6g with NaBO3•4H2O results in C–B oxidation and conversion of the vinyl boronic esters to corresponding hemiacetals (9) (Scheme 6A), which can be can be used crude in subsequent reactions. For example, 9 can be deoxygenated with trifluoroacetic acid and triethylsilane to generate tetrahydrofuran 10 in 82% yield,[17] or oxidized with PCC to afford lactone 11 isolated in 51% yield over two steps. Second, both aryl and alkyl ketone-derived alkenylboron products undergo efficient Pd-catalyzed Suzuki-Miyaura cross coupling (Scheme 6B); the conversion of 6c and 7f to N-heteroarene-containing 12 and 13, generated in 55% and 84% yield, are representative. In addition, preparation of the aliphatic aldehyde, without cyclization, can be accomplished by a two-step sequence that first requires protection of the tertiary alcohol (Scheme 6C). In this regard, treatment of 6n with TMSOTf and 2,6-lutidine followed by C–B bond oxidation with NaBO3•4H2O affords β-quaternary carbon aldehyde 14, isolated in 61% yield over two steps.
Scheme 6.

Synthetic Utility of Alkenyl Boronate Products. See SI for details.
In conclusion, we have developed a practical, efficient diastereo- and enantioselective Cu-catalyzed allyl addition to ketones that generates vicinal stereogenic centers – a tertiary alcohol and quaternary carbon. The reaction tolerates a wide variety of aryl, heteroaryl, alkenyl, alkynyl, and alkyl ketones, as well as a diverse array of allyldiboron reagents. In addition, both non-prochiral ketones and symmetrically γ,γ-disubstituted nucleophiles can be employed with high stereoselectivity. The synthetically versatile alkenyl boron products can be elaborated by a number of chemical transformations to useful chiral molecules. Further catalytic stereoselective methods utilizing allylic 1,1-bis-boronates are in progress.
Supplementary Material
Acknowledgements
Financial support was provided by the United States National Institutes of Health, Institute of General Medical Sciences (R01GM116987). Mass spectrometry facilities in the Department of Chemistry at University of North Carolina are supported by the National Science Foundation (CHE1726291). We thank Tia Cervarich of UNC for X-ray structure elucidation of 6q.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].For recent reviews on the enantioselective synthesis of quaternary carbon stereogenic centers, see:Das JP, Marek I, Chem. Commun 2011, 47, 4593–4623.Minko Y, Marek I, Chem. Commun 2014, 50, 12597–12611.Marek I, Minko Y, Pasco M, Mejuch T, Gilboa N, Chechik H, Das JP, J. Am. Chem. Soc 2014, 136, 2682–2694.Quasdorf KW, Overman LE, Nature 2014, 516, 181–191.Liu Y, Han S-J, Liu W-B, Stoltz BM, Acc. Chem. Res 2015, 48, 740–751.Zeng X-P, Cao Z-Y, Wang Y-H, Zhou F, Zhou J, Chem. Rev 2016, 116, 7330–7396.Feng J, Holmes M, Krische MJ, Chem. Rev 2017, 117, 12564–12580.
- [2].For recent reviews on the enantioselective synthesis of tertiary alcohols, see:Riant O, Hannedouche J, Org. Biomol. Chem 2007, 5, 873–888.Hatano M, Ishihara K, Synthesis 2008, 1647–1675.Shibasaki M, Kanai M, Chem. Rev 2008, 108, 2853–2873.(d) Liu Y-L, Lin X-T, Adv. Synth. Catal 2019, 361, 876–918.
- [3].For a recent reviews of catalytic allyl additions to ketones, see:Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794.Shibasaki M, Kanai M, Chem. Rev 2008, 108, 2853–2873.Yus M, Gómez González- J. C., Foubelo F, Chem. Rev 2011, 111, 7774–7854.Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320. See also ref. 1g.
- [4].For representative catalytic enantioselective examples of crotyl additions to ketones, see:Wada R, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2004, 126, 8910–8911.Wadamoto M, Yamamoto H, J. Am. Chem. Soc 2005, 127, 14556–14557.Miller JJ, Sigman MS, J. Am. Chem. Soc 2007, 129, 2752–2753.Kanai M, Wada R, Shibuguchi T, Shibasaki M, Pure Appl. Chem 2008, 80, 1055–1062.Schneider U, Ueno M, Kobayashi S, J. Am. Chem. Soc. 2008, 130, 13824–13825.Rauniyar V, Zhai H, Hall DG, J. Am. Chem. Soc 2008, 130, 8481–8490.Barnett DS, Moquist PN, Schaus SE, Angew. Chem. Int. Ed 2009, 48, 8679–8682; Angew. Chem. 2009, 121, 8835–8838.Wadamoto M, Naodovic M, Yamamoto H, Eur. J. Org. Chem 2009, 5132–5134.Nowrouzi F, Thadani AN, Batey RA, Org. Lett. 2009, 11, 2631–2634.Shi S-L, Xu L-W, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2010, 132, 6638–6639.Yamaguchi M, Morita N, Scheider U, Kobayashi S, Adv. Synth. Catal 2010, 352, 1461–1465.Tsai EY, Liu RY, Yang Y, Buchwald SL, J. Am. Chem. Soc 2018, 140, 2007–2011.Li C, Liu RY, Jesikiewicz LT, Yang Y, Liu P, Buchwald SL, J. Am. Chem. Soc 2019, 141, 5062–5070.
- [5].For examples of enantioselective allyl additions to aldehydes that generate quaternary carbon stereogenic centers, see:Denmark SE, Fu J, J. Am. Chem. Soc 2001, 123, 9488–9489.Denmark SE, Fu J, Org. Lett 2002, 4, 1951–1953.Feng J, Garza VJ, Krische MJ, J. Am. Chem. Soc 2014, 136, 8911–8914.Nguyen KD, Herkommer D, Krische MJ, J. Am. Chem. Soc 2016, 138, 14210–14213.Holmes M, Nguyen KD, Schwartz LA, Luong T, Krische MJ, J. Am. Chem. Soc 2017, 139, 8114–8117.Schwartz LA, Holmes M, Brito GA, Gonçalves TP, Richardson J, Ruble JC, Huang K-W, Krische MJ, J. Am. Chem. Soc 2019, 141, 2087–2096.
- [6].Chen JL-Y, Aggarwal VK, Angew. Chem. Int. Ed 2014, 53, 10992–10996; Angew. Chem. 2014, 126, 11172–11176. [DOI] [PubMed] [Google Scholar]
- [7].Alam R, Vollgraff T, Eriksson L, Szabó KJ, J. Am. Chem. Soc 2015, 137, 11262–11265. [DOI] [PubMed] [Google Scholar]
- [8].For a recent review that includes catalytic enantioselective allyl addition to ketones that avoids the need for generation of pre-formed allylmetal reagents, see:Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron 2015, 71, 2183–2197.
- [9].For a report containing a single example with a diene, see:Feng J-J, Xu Y, Oestreich M, Chem. Sci 2019, 10, 9679–9683. For a related example that involves the dearomative isomerization of alkenylarenes, see:Cheng F, Lu W, Huang W, Wen L, Li M, Meng F, Chem. Sci 2018, 9, 4992–4998.
- [10].(a) Miura T, Nakahashi J, Murakami M, Angew. Chem. Int. Ed 2017, 56, 6989–6993; Angew. Chem. 2017, 129, 7093–7097. [DOI] [PubMed] [Google Scholar]; (b) Miura T, Nakahashi J, Zhou W, Shiratori Y, Stewart SG, Murakami M, J. Am. Chem. Soc 2017, 139, 10903–10908. [DOI] [PubMed] [Google Scholar]; (c) Miura T, Oku N, Murakami M, Angew. Chem. Int. Ed 2019, 58, 14620–14624; Angew. Chem. 2019, 131, 14762–14766. [DOI] [PubMed] [Google Scholar]; (d) Park J, Choi S, Lee Y, Cho SH, Org. Lett 2017, 19, 4054–4057. [DOI] [PubMed] [Google Scholar]; (e) Green JC, Zanghi JM, Meek SJ, J. Am. Chem. Soc 2020, 142, 1704–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For examples of catalytic stereoselective 1,2-additions of diborylalkanes to C=O and C=N bonds, see:Joannou MV, Moyer BS, Meek SJ, J. Am. Chem. Soc 2015, 137, 6176–6179.Joannou MV, Moyer BS, Goldfogel MJ, Meek SJ, Angew. Chem. Int. Ed 2015, 54, 14141–14145; Angew. Chem. 2015, 127, 14347–14351.Murray SA, Green JC, Tailor SB, Meek SJ, Angew. Chem. Int. Ed 2016, 55, 9065–9069; Angew. Chem. 2016, 128, 9211–9215.Park J, Lee Y, Kim J, Cho SH, Org. Lett 2016, 18, 1210–1213.Kim J, Ko K, Cho SH, Angew. Chem. Int. Ed 2017, 56, 11584–11588; Angew. Chem. 2017, 129, 11742–11746.Kim J, Shin M, Cho SH, ACS Catal. 2019, 9, 8503–8508.
- [12]. In principle, direct formation of α-boron-stabilized intermediate B is also possible.
- [13].For examples of site- and stereospecific M–OR SE2’ transmetalation with allylboron reagents, see:Yang Y, Buchwald SL, J. Am. Chem. Soc 2013, 135, 10642–10645.Chausset-Boissarie L, Ghozati K, LaBine E, Chen JL-Y, Aggarwal VK, Crudden CM, Chem. – Eur. J 2013, 19, 17698–17701.Potter B, Edelstein EK, Morken JP, Org. Lett 2016, 18, 3286–3289.
- [14].For recent examples of π-allylmetal E/Z scrambling, see:Tan KL, Jacobsen EN, Angew. Chem. Int. Ed 2007, 46, 1315–1317; Angew. Chem. 2007, 119, 1337–1339.Vieira EM, Snapper ML, Hoveyda AH, J. Am. Chem. Soc 2011, 133, 3332–3335.
- [15].Mejuch T, Gilboa N, Gayon E, Wang H, Houk KN, Marek I, Acc. Chem. Res 2013, 46, 1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16]. Reaction of chalcones suffer from low dr, likely due to epimerization through a benzylic/allylic carbocation.
- [17].Kraus GA, Molina MT, Walling JA, J. Chem. Soc., Chem. Commun 1986, 1568–1569. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.