

Abstract
Achieving stable expression of a single transgene in mammalian cells remains challenging; even more challenging is obtaining simultaneous stable expression of multiple transgenes at reproducible, relative expression levels. Previously, we attained copy-number-dependent, chromosome-position-independent expression of reporter minigenes by embedding them within a BAC “scaffold” containing the mouse Msh3-Dhfr locus (DHFR BAC). Here we extend this “BAC TG-EMBED” approach. First, we report a toolkit of endogenous promoters capable of driving transgene expression over a 0.01 to 5-fold expression range relative to the CMV promoter, allowing fine-tuning of relative expression levels of multiple reporter genes. Second, we demonstrate little variation in expression level and long-term expression stability of a reporter gene embedded in BACs containing either transcriptionally active or inactive genomic regions, making choice of BAC scaffolds more flexible. Third, we present a novel BAC assembly scheme, ‘BAC-MAGIC’, for inserting multiple transgenes into BAC scaffolds, which is much more time-efficient than traditional galK-based methods. As a proof-of-principle for our improved BAC TG-EMBED toolkit, we simultaneously fluorescently-labeled three nuclear compartments at reproducible, relative intensity levels in 94% of stable clones after a single transfection using a DHFR BAC scaffold containing 4 transgenes assembled with BAC-MAGIC. Our extended BAC TG-EMBED toolkit and BAC-MAGIC method provide an efficient, versatile platform for stable simultaneous expression of multiple transgenes at reproducible, relative levels.
Keywords: transgene expression, promoter toolkit, copy-number-dependent expression, multi-reporter, gene silencing, BAC recombineering
Graphical Abstract
Transgene expression has been widely used in both basic research and biotechnology. Applications of transgene expression range from the elucidation of gene function by ectopic expression of selected transgenes, to the expression of transgenes for gene therapy, and to the overexpression of genes for production of biopharmaceuticals.1–3 Examples of such applications include the expression of multiple fluorescent proteins for live-cell imaging,4 the expression of the four or more Yamanaka transcription factors for efficient generation of induced pluripotent stem (iPS) cells,5 and the expression of multiple proteins for reconstitution of protein complexes.6,7
Despite the currently widespread use of transgene expression, most transgene expression systems still suffer from serious experimental limitations. Plasmid-, lentivirus- and transposon-based systems, all still show varying degrees of chromosome position effects8 and position effect variegation (PEV).9,10 Moreover, foreign sequences by themselves are targets for epigenetic silencing,11,12 and transgene concatamers can induce the formation of heterochromatin.13,14 Together these transgene silencing mechanisms result in unpredictable transgene expression levels that do not correlate with copy number and are unstable with long-term culture or changes in the cell physiological or differentiated state.15–17
Such limitations are compounded when the simultaneous and reproducible expression of multiple transgenes is required. For example, a common application in the emerging field of synthetic biology is the design of novel gene circuits, involving the expression of multiple proteins, in many cases at precise relative levels.18 While this approach has worked well in prokaryotes and yeast, it has been difficult to implement in mammalian cells due to the lack of suitable multi-transgene expression methods which can overcome chromosome position effects and allow expression of different transgenes at reproducible relative levels. Studies using bicistronic or splicing-based lentiviral systems to express multiple genes in cultured primary cells or established cell-lines have shown variable success.7,19 However, the coexpression of genes through these systems suffer from heterogeneity of cells expressing the transgenes, and by the packaging-size constraints of the lentiviral vectors.20 These factors limit complex metabolic engineering of cells to express three or more transgenes in a predictable manner.
A commonly used approach to countering transgene silencing and variegation has been through the inclusion of cis-elements. These include insulators,21,22 locus control regions (LCRs),23,24 scaffold/matrix attachment regions (S/MARs),25,26 ubiquitous chromatin opening elements (UCOEs)27,28 and anti-repressors;29 some of these regulatory elements have context-dependent and/or vector dependent activity. While these cis-elements improve transgene expression to varying degrees, they are insufficient for chromosome-position independent, copy-number-dependent transgene expression.23,30
Bacterial artificial chromosomes (BACs) carrying ~100–200 kb mammalian genomic DNA inserts harbor most of the cis-regulatory sequences required for expression of the endogenous genes contained within these genomic inserts. Previously we demonstrated how embedding minigene constructs at different locations within the DHFR BAC provided reproducible expression of single or multiple reporter genes independent of the chromosome integration site.31 Similar approaches were used by other labs for high-level recombinant protein production.32,33 Recently, our lab demonstrated stable transgene expression after cell-cycle arrest or after terminal cell differentiation, using the BAC-TG EMBED approach.34 All of these studies tested only BACs containing actively transcribed regions, based on the hypothesis that the expression level of the transgenes inserted into the BACs was determined by the chromatin environments reconstituted by the genomic inserts within the BACs. Indeed, because of this assumption, previous studies have specifically targeted the inserted transgenes to transcription units and even exons.31–33
However, this hypothesis has not been tested. Moreover, overexpression from the genes on the BAC genomic inserts might change the properties of the transfected cells or interfere with other assays of a study. Thus, BACs with no transcription units would be more desirable. Another improvement over our previous BAC TG-EMBED system31,34 would be a toolkit of endogenous promoters capable of driving transgene expression over a wide range of defined, relative expression levels. Viral promoters, including the CMV promoter we used previously, are known to be prone to epigenetic silencing,35,36 while most previously used endogenous and synthetic promoters were selected for their strength.33,37–39 While high-level transgene expression is preferable in applications calling for overexpression, a low or near-physiological expression is important for many other applications, including gene therapy. Additionally, multiple transgenes may need to be expressed simultaneously but at reproducible differential levels.
A potential drawback of using BACs for multi-transgene expression is the extensive time needed for constructing the recombinant BACs containing multiple reporter minigenes. The traditionally used galK positive/negative selection scheme40,41 is especially low efficient for constructing multi-reporter BACs, due to the frequent recombination between the similar sequences on the reporter minigenes and between the repetitive sequences on the BACs, and the slow growth rate of bacteria on minimal plates.
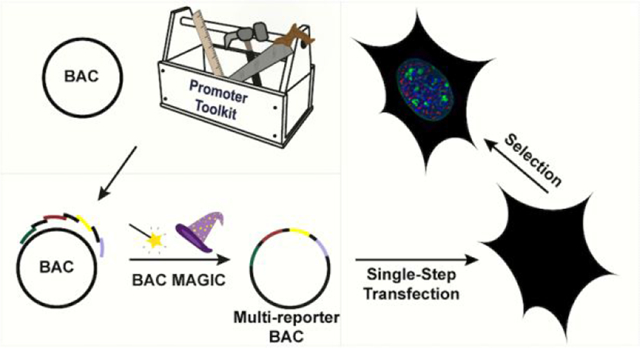
Here we describe further extensions to the BAC TG-EMBED method and a novel BAC assembly scheme, providing a more versatile and efficient platform for a range of future potential applications. First, we describe a toolkit of endogenous promoters, for which we have measured relative promoter strength, that can drive transgene expression at reproducible relative levels over a 500-fold range. Second, we show that multiple BAC scaffolds can be used to drive sustained high-level transgene expression driven by the UBC promoter without selection for up to 12 weeks, including BAC scaffolds containing no active transcription units. Third, we developed a method “BAC-MAGIC” (BAC-Modular Assembly of Genomic loci Interspersed Cassettes) to more rapidly assemble BACs containing multiple transgene expression cassettes. Finally, as a proof-of-principle demonstration of our new, more versatile BAC TG-EMBED toolkit, we demonstrate simultaneous expression of fluorescently tagged proteins labeling three different nuclear compartments, achieving >90% optimally labeled cell clones after a single, stable transfection.
RESULTS AND DISCUSSION
Overview of BAC TG-EMBED toolkit development:
We previously demonstrated the feasibility of the BAC TG-EMBED approach using both the DHFR BAC31 and a BAC containing the human GAPDH gene locus (GAPDH BAC).34 We set out to extend this BAC TG-EMBED methodology in two new directions (Figure 1).
Figure 1. Two-prong experimental approach.
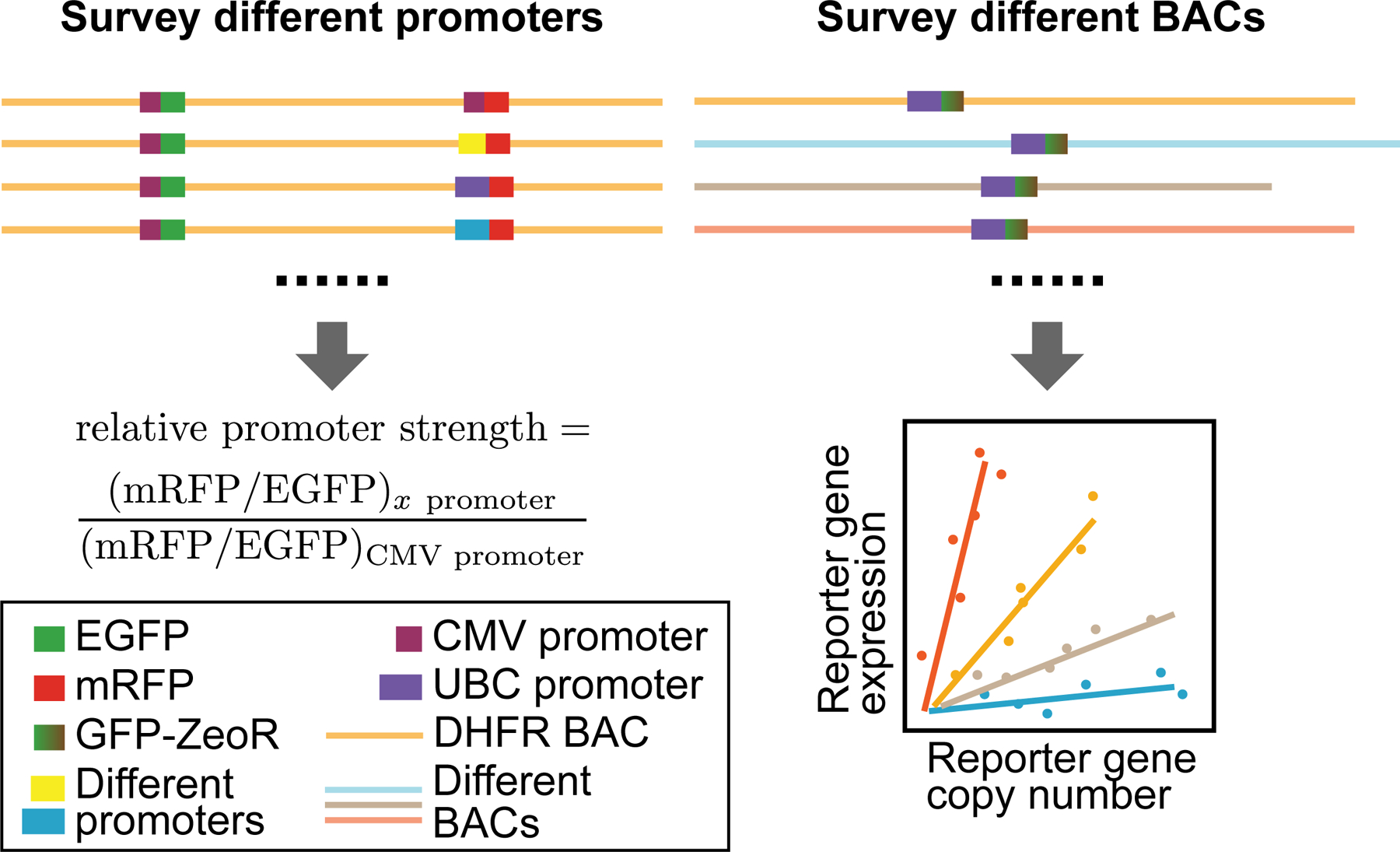
Left: Identification of promoters of different strengths- We measured relative promoter strengths by embedding EGFP and mRFP reporter genes into the DHFR BAC, using the CMV promoter to drive EGFP expression and the test promoter to drive mRFP. The ratio of mRFP and GFP expression, normalized by this same ratio for a CMV test promoter, defines promoter strength relative to CMV. Right: Surveying reporter gene expression in different BAC scaffolds- (Top) The UBC-GFP-ZeoR reporter gene was inserted into BACs carrying DNA from mouse or human genomic regions corresponding to either transcriptionally active or inactive genomic regions. (Bottom) Plotting reporter gene expression (y-axis) versus reporter gene copy number (x-axis) for multiple cell clones stably expressing BAC transgenes: a linear correlation would indicate copy-number dependent, position independent expression, while the slope of this linear correlation would measure reporter gene expression per copy number.
First, to better control transgene expression and to be able to express multiple transgenes at reproducible expression ratios, we explored a set of constitutive promoters with various strengths for transgene expression. A previous similar survey of promoters within BAC scaffolds focused only on strong promoters.33 Moreover this survey compared average expression in pools of cell colonies containing different copy-number BAC insertions.33 Here we used a two-reporter, single-cell ratio assay and also examined promoters with a wide range of promoter strengths. Testing each promoter with each BAC scaffold would have generated too large a number of possible combinations. We therefore decided to test a number of different promoters with the original DHFR BAC.
Second, we used one specific reporter gene construct to survey the effect of different BAC scaffolds on reporter gene expression. Previous similar applications used BAC scaffolds containing multiple endogenous genes which would also be expressed in addition to added transgenes.31–33 Moreover, in a previous, similar application, different strong promoters were tested by insertion into the exon of an active BAC gene.33 Here we compared BAC scaffolds containing expressed genes with BAC scaffolds from gene deserts or regions containing silenced genes. We assayed the level, stability, and reproducibility of the embedded reporter gene expression when inserted into different BAC scaffolds to identify optimal BAC scaffolds for the BAC TG-EMBED system.
A toolset of 7 endogenous promoters for tuning relative transgene expression levels
We selected 7 endogenous promoters to test, either because of their known ability and use to drive transgene expression in a range of cell types (EEF1α, UBC),37,42,43 or because these promoters were derived from housekeeping genes (RPL32, PPIA, B2M, RPS3A, GUSB) expressed uniformly across a wide range of tissue types.44,45 We amplified 1–3 kb of regulatory regions upstream of the transcription start sites of these genes using either human genomic DNA as a template or, for the UBC promoter, using the pUGG plasmid.34
To assay relative promoter strength, we used the two-minigene reporter system developed in our previous study in which we compared expression of CMV-driven EGFP and mRFP minigenes inserted in the same mouse DHFR BAC scaffold.31 We previously showed that the mRFP minigene reporter expression varied less than or equal to 2.4-fold when the mRFP reporter was inserted at 6 different positions ranging 3–80 kb away from the EGFP reporter gene location on the same BAC.31 To compare relative promoter strengths, we fixed the insertion positions of mRFP and EGFP, and measured the relative fluorescence levels of mRFP and EGFP when they were both driven by the CMV promoter versus when the mRFP reporter was driven by an endogenous promoter (Figure 1). Thus, our assay measured the strength of different endogenous promoters relative to the viral CMV promoter, while also measuring the variation in this relative strength in different cells of a mixed clonal population.
For this assay, the EGFP reporter minigene was inserted 26 kb downstream of the Msh3 transcription start site31 (Figure 2a). PCR-amplified promoters from 7 different housekeeping genes were cloned upstream of the mRFP expression cassette (Figure 2b), and then this mRFP expression cassette was introduced 121 kb downstream of the Msh3 transcription start site by BAC recombineering (Figure 2a), generating the dual reporter DHFR BAC. As a control, we used the dual reporter BAC previously constructed31 in which the same mRFP cassette driven by the CMV promoter was inserted at this same location 121 kb downstream of the Msh3 start site.
Figure 2. Dual-reporter assay for promoter strength estimation.
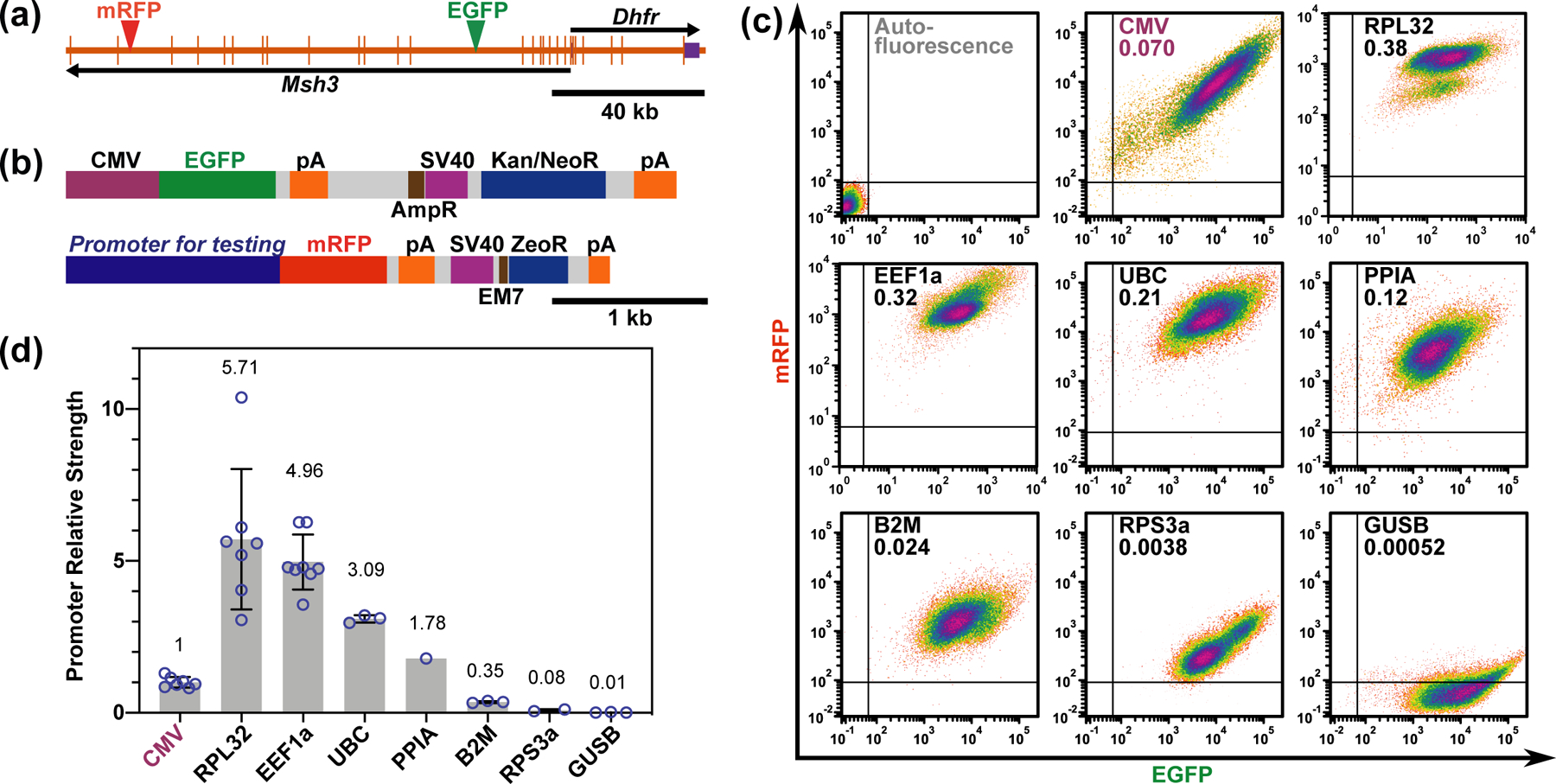
(a) Dual reporter DHFR BAC showing the two genes on the BAC, Dhfr and Msh3, and the insertion sites of the two reporter expression cassettes. Longer vertical bars-exons; shorter vertical bars- UTRs; arrows- direction of transcription; green arrowhead- EGFP expression cassette insertion site; red arrowhead- mRFP expression cassette insertion site. (b) The two-reporter gene/selectable marker cassettes used in the assay. The EGFP cassette (top) contains an EGFP minigene, driven by a CMV promoter, and a Kanamycin/Neomycin resistance gene (Kan/NeoR), driven by a SV40 promoter for expression in mammalian cells, or by an AmpR promoter for expression in bacteria. The mRFP cassette (bottom) contains a mRFP minigene and a Zeocin resistance gene (ZeoR). Different endogenous promoters were inserted immediately upstream of mRFP. ZeoR is driven by a SV40 promoter for expression in mammalian cells, or by an EM7 promoter for expression in bacteria. pA- poly(A) signal. (c) Respective scatter plots showing mRFP fluorescence (y-axis) vs EGFP fluorescence (x-axis) of cells from the mixed clonal populations stably transfected with dual reporter DHFR BACs. Promoters driving the mRFP and the ratio of mRFP/EGFP (promoter strength) are labeled in each plot. (d) Mean promoter strengths relative to CMV. Circles- values from individual experiments; Error bars- standard deviations; Numbers: mean values from individual experiments.
Mixed populations of NIH 3T3 fibroblast stable clones carrying these modified BAC transgenes were analyzed by flow cytometry to measure the relative expression ratio of mRFP and EGFP (Figure 2c). Fluorescent beads were used as an invariant fluorescence standard to calibrate the flow cytometer intensity outputs. The ratio of mRFP to EGFP expression was then normalized by the ratio observed with the original dual-reporter BAC construct in which both reporters were driven by CMV promoter, providing the endogenous promoter strength relative to the CMV promoter.
We observed an overall variation in promoter strength of over 500-folds, ranging from the 4 to 5-folds relative promoter strength of the RPL32 and EEF1α promoters to the 0.01-fold relative promoter strength for the GUSB promoter as compared to the CMV promoter (Figure 2d). This expression ratio appeared to be similar across the cell population, except for the RPL32 which showed a minor subpopulation in all 4 replicates of varying proportion to the main population. This minor subpopulation has a slightly lower RPL32 relative expression, which might be due to cell-cycle dependent expression, for instance in a subpopulation of quiescent cells.
As these promoters are derived from human genes expressed in a wide range of cell lines and tissues,37,42–45 we expect them to support transgene expression in most cell types and independent of cell proliferation or differentiation state. While most of the previous studies on transgene promoters focused on conventional, strong promoters,33,37–39 including a similar approach that expressed minigenes within BAC scaffold,33 we included moderate-strength and weak promoters in our survey. The weak promoters we identified, such as GUSB and RPS3A, could possibly replace the commonly used minimal promoters or inducible promoters where a sustained low-level of transgene expression is needed. Moreover, this wide range of promoter strengths allows reproducible expression of multiple transgenes over a wide range of relative expression levels from a single BAC scaffold, lending itself to such purposes, for example, as the design of synthetic gene circuits, which typically requires expression of different components at reproducible relative expression levels.18
Reporter gene expression as a function of transcriptionally active and inactive BAC scaffolds
To find the best BAC scaffold for the BAC TG-EMBED system, we tested BAC scaffolds from both actively transcribed regions and regions containing silenced genes or no genes. Specifically, we measured the expression as a function of copy number of one specific reporter gene construct inserted into these BAC scaffolds. Previous applications of BAC TG-EMBED showed a linear relationship between copy number and expression level, largely independent of the chromosome integration site, demonstrating copy-number dependent, position independent transgene expression.31,34 For active chromosomal regions, we chose the RP11–138I1 BAC containing the human ubiquitin B gene locus (UBB BAC), the RP23–401D9 BAC containing the “safe-haven” mouse Rosa26 genetrap locus (ROSA BAC),46 and the CITB-057L22 BAC carrying the mouse Dhfr gene locus (DHFR BAC). For inactive chromosomal regions, we chose the CTD-2207K13 BAC (2207K13 BAC) that contains no known gene or regulatory element from a gene-desert region from the human genome, and the CTD-2643I7 (HBB BAC) containing the human HBB gene locus and multiple olfactory genes, all of which are transcriptionally silenced in fibroblasts.47
We selected the UBC promoter for this reporter gene cassette as this promoter had previously been shown to drive high expression across multiple cell types;43 in our dual reporter system the UBC promoter was 3-folds stronger than the CMV promoter (Figure 2d). Moreover, to eliminate any possible transcriptional interference from closely spaced reporter and selectable marker minigenes and to minimize any epigenetic silencing arising from DNA methylation of this reporter gene-selectable marker construct, we used a commercially available GFP-ZeoR fusion protein gene construct in which all CpG dinucleotides had been removed and replaced by synonymous codons (Figure 3a).
Figure 3. Expression of reporter gene embedded in different BAC scaffolds.
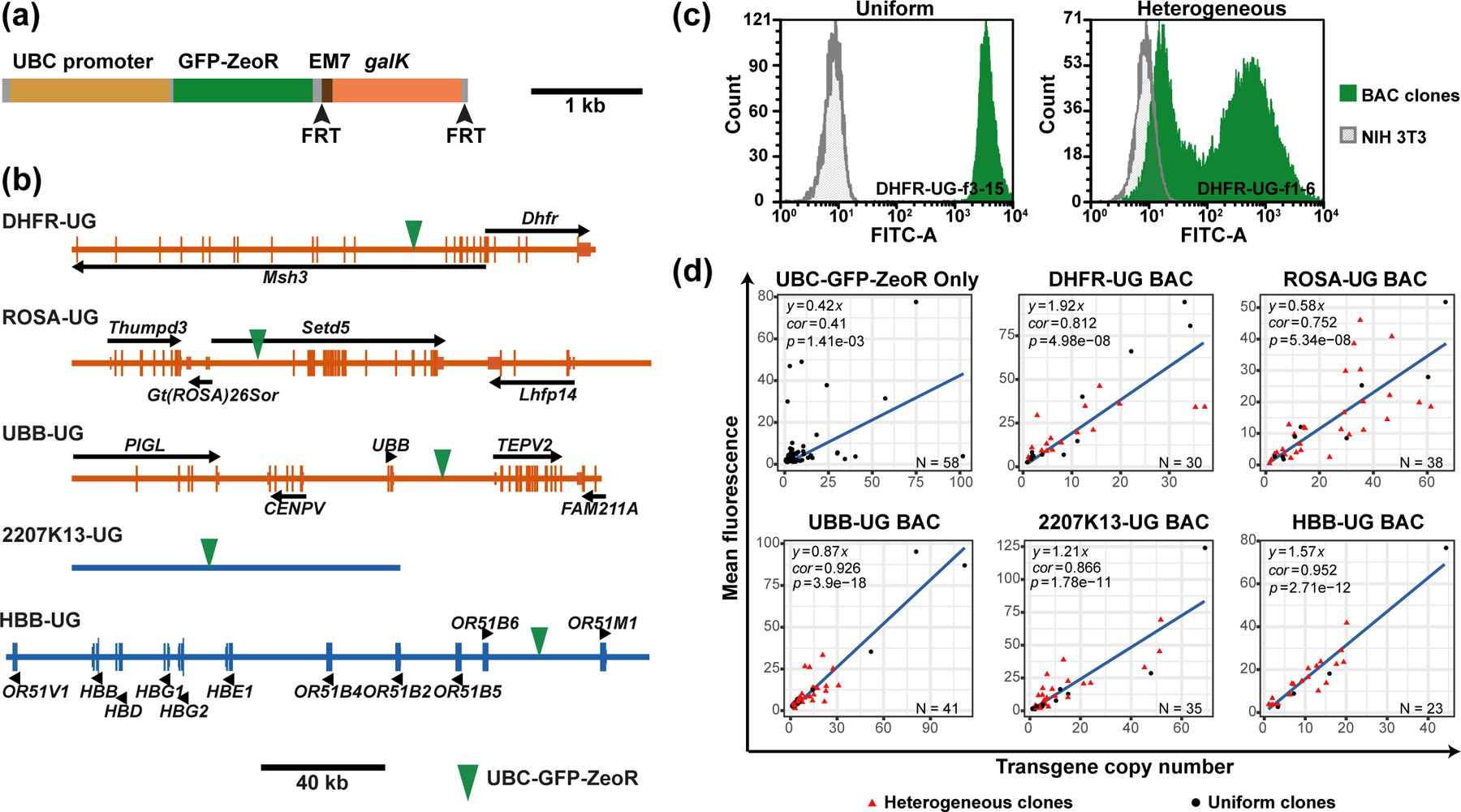
(a) UBC-GFP-ZeoR-FRT-GalK-FRT cassette showing the GFP-ZeoR minigene driven by the UBC promoter and the galK positive/negative selection marker flanked by 34 bp flippase recognition target (FRT) sites (arrowheads). (b) Maps of the BACs used in the study. Longer vertical bars- exons; shorter vertical bars- UTRs; black arrows or arrowheads- direction of transcription; green arrow heads- UBC-GFP-ZeoR insertion site. (c) GFP fluorescence histograms obtained by flow-cytometry for “uniform” (left, green, clone DHFR-UG-f3–15) versus “heterogeneous” (right, green, clone DHFR-UG-f1–6) expressing NIH 3T3 clones carrying the DHFR-UG BAC. x-axis- fluorescence value, y-axis- cell number; gray- autofluorescence of untransfected cells. Fluorescence is measured in arbitrary units. (d) Scatter plots of mean normalized cellular GFP fluorescence (y-axis) vs reporter gene copy number (x-axis) for clonal populations transfected with the UBC-GFP-ZeoR cassette alone or with different BAC scaffolds carrying the UBC-GFP-ZeoR reporter gene. Linear regression fits (black lines, y-intercepts set to 0) are shown with corresponding R-squared values and equations. Red triangles- heterogeneous clones; Black circles- uniform clones; Bottom right of plots: Number of clones analyzed.
We inserted this UBC-GFP-ZeoR reporter gene construct into different BAC scaffolds by BAC recombineering, using galK for positive/negative selection.40,41 To eliminate potential artifacts caused by proximity to active promoters, transcriptional start sites (TSS), or miRNA sequences, we chose insertion sites at least 5 kb away from such sequence elements on either sides (Figure 3b). The UBB, HBB, 2207K13, ROSA, DHFR BACs with the UBC-GFPZeoR reporter gene insertion were named as UBB-UG, HBB-UG, 2207K13-UG, ROSA-UG and DHFR-UG.
After transfection, multiple cell clones (n=20–40) carrying stably integrated BAC arrays were selected for Zeocin resistance and analyzed for reporter gene expression by flow cytometry, using untransfected NIH 3T3 cells to determine background, autofluorescence levels. For each cell clone, we used flow cytometry to measure the mean GFP reporter expression and qPCR to measure reporter gene copy number. These cell clones showed GFP fluorescence mean levels ranging from 10–1000 folds higher than the background autofluorescence.
Our original working hypothesis predicted that the BAC TG-EMBED reporter expression should be uniform in all cells of the same clone. Also, we expected to see a linear relationship between mean reporter gene fluorescence and number of BAC copies, signifying a copy-number-dependent, position independent expression. Furthermore, we expected that the slope of this linear relationship would be higher for BAC scaffolds expected to reconstitute an active chromatin environment permissive for transgene expression as compared to BAC scaffolds expected to reconstitute a more condensed, inactive chromatin environment (Figure 1). In contrast, we expected that the reporter gene cassette transfected without any BAC scaffold would show clonal expression levels that poorly correlated with reporter gene copy number (copy-number-independent expression).
Unexpectedly, the stable cell clones we isolated showed two distinct types of population expression profiles- uniform versus heterogeneous. Uniform clones showed single, relatively narrow expression peaks in the flow cytometry histograms, with more than 90% of the cells showing GFP fluorescence varying only over a 10-fold intensity range (Figure 3c, left). Heterogeneous clones instead showed two peaks with a range of GFP expression varying ~1000-fold, with the lower GFP intensity peak overlapping with the autofluorescence distribution of control cells (Figure 3c, right). We had not previously observed such heterogeneous expression profile using our original DHFR BAC containing the CMV-driven mRFP alone or both the CMV-driven EGFP and CMV-driven mRFP reporter genes.31 However, we had observed ~80% uniform clones for a GAPDH BAC scaffold with the UBC-GFP-ZeoR reporter gene inserted.34 The percentage of clones showing such heterogeneous expression varied from 58% to 83% for the 5 BAC scaffolds surveyed here (Table 1). No similar heterogeneous expression profile was observed when the reporter gene construct was transfected by itself (Table 1).
Table 1.
Percentage of heterogeneously expressing clones transfected with the UBC-GFP-ZeoR cassette alone or with different BAC scaffolds carrying the UBCGFP-ZeoR reporter gene.
| Construct | Heterogeneous clones % | Number of clones |
|---|---|---|
| UBC-GFP-ZeoR | 0 | 58 |
| DHFR-UG | 60% | 30 |
| ROSA-UG | 76% | 38 |
| UBB-UG | 58% | 41 |
| 2207K13-UG | 69% | 35 |
| HBB-UG | 83% | 23 |
As expected, the control transfection of the reporter gene cassette by itself resulted in copy-number-independent expression of the reporter gene (Figure 3d, correlation coefficient = 0.41, p-value = 0.00141), while the reporter gene embedded within the BACs yielded a linear relationship between reporter gene fluorescence and transgene copy number for both uniform (black) and heterogeneous (red) BAC transgene clones (Figure 3d, correlation coefficient = 0.752 to 0.952, p-value = 4.98 × 10−8 to 3.9 × 10−18). Interestingly, uniform clones always had a much better linear correlation (Supporting Information, Figure S1ae, correlation coefficient = 0.909 to 0.994 for uniform clones and 0.545 to 0.884 for heterogeneous clones) and a slightly larger slope (Supporting Information, Figure S1a–e, less than 2-fold difference) than corresponding heterogeneous clones transfected with the same BACs. Moreover, for both uniform and heterogenous clones, residuals increased with transgene copy number (Supporting Information, Figure S2a–f), possibly due to uneven data distribution, as most of the clones analyzed in this study had low transgene copy number.
Surprisingly, we observed no more than a 4-fold variation in expression per copy number among the 5 different BAC scaffolds tested, with no obvious relationship between the observed slope and the type of BAC scaffold (Figure 3d). Although the transcriptionally active DHFR BAC produced the highest slope, the transcriptionally inactive HBB BAC and the 2207K13 BAC containing DNA from a gene desert produced the second and third highest slopes, while the BAC containing DNA from the “safe haven” mouse Rosa26 locus produced the lowest slope.
To determine whether the addition of the highly expressed UBC-GFPZeoR reporter gene disrupted or prevented the formation of condensed chromatin over the HBB and 2207K13 BAC transgene arrays, we visualized BAC transgene arrays using 3D DNA FISH. HBB-UG and 2207K13-UG transgenes formed rounder, and smaller large-scale chromatin domains relative to DHFR-UG arrays of comparable DNA content, which formed more linear structures (Supporting Information, Figure S3a–e). The rounder and more compact shapes seen for the HBB-UG and 2207K13-UG versus DHFR transgene arrays qualitatively were similar to previous results comparing HBB and DHFR BAC transgene arrays in both mouse NIH 3T3 and ESCs using the lac operator/repressor system for visualization.48,49 We conclude, therefore, that the presence of the highly expressed UBC-GFP-ZeoR reporter does not disrupt the more condensed large-scale chromatin structures formed as expected by the HBB-UG and 2207K13-UG BAC transgene arrays.
Interestingly, however, we did observe that the insertion of the UBC-GFPZeoR reporter gene into the HBB BAC lead to these HBB-UG BAC transgene arrays to be repositioned away from nuclear periphery and into the nuclear interior (Supporting Information, Figure S3f–g). We verified that this differed from the peripheral localization of the HBB BAC transgene arrays (HBB lacO clones), which do not contain the UBC-GFP-ZeoR reporter gene, when integrated into NIH 3T3 cells (Supporting Information, Figure S3f–g, HBB-C3 clone).49
Moreover, 2-color DNA FISH revealed that the reporter gene tends to localize at the periphery of the round, compact, HBB BAC transgene array territory, which could explain the high expression of the reporter genes even when present in these condensed arrays (Supporting Information, Figure S4a–c). More specifically, we suggest that this type of positioning activity by the UBC promoter sequences may contribute to its ability to express well despite its placement in condensed, heterochromatic BAC transgene arrays. We previously called this type of looping of DNA within larger, condensed large-scale chromatin structures “dynamic plasticity”.48 In the context of BAC transgene arrays, it creates “subcompartments” of active DNA regions from neighboring BAC copies which cluster together, as well as similar but opposite “subcompartments” of inactive, heterochromatic DNA regions from neighboring BAC copies that position towards the interior of these BAC transgene arrays.48
In contrast, the DHFR BAC transgenes form more linear structures and the reporter gene localizes in foci along their length; the diameter of these linear structures is comparable to the diffraction-limited resolution limit and therefore we cannot tell whether they are inside or outside the transgene arrays.
Overall, these results show that for this UBC-GFP-ZeoR reporter gene, high-level, copy-number-dependent transgene expression using the BAC TG-EMBED method does not require BACs containing active, housekeeping genomic regions, but can also be obtained from a wide range of BAC genomic DNA inserts, including gene-desert regions. UBC may represent a member of a class of active, house-keeping gene promoters that is relatively insensitive to chromosome position effects. This allows choice of a BAC scaffold for the BAC TG-EMBED method that will not co-express any genes other than the introduced transgene cassettes. In contrast, both our previous BAC TG-EMBED studies31,34 and similar work from other laboratories32,33 used only BACs containing highly-transcribed house-keeping genes, due to the assumption that either an active chromatin region or active 5’ cis-regulatory regions would be required for creating a transcriptionally permissive environment for transgene expression. Integration of the UBC-GFP-ZeoR reporter gene into the BAC was required for position-independent, copy-number dependent expression, as its expression was copy-number independent when the same UBC-GFP-ZeoR reporter gene was stably transfected by itself into cells.
Temporal stability of BAC-embedded reporter gene expression in uniform cell clones
We previously showed that the BAC TG-EMBED method provided long-term stability of transgene expression in the presence of continued drug selection.31 However, in the absence of drug selection we observed a 30–80% drop in expression over several months of cell passaging without any apparent drop in the integrated BAC copy number.31
Here we determined the long-term stability of the UBC-GFP-ZeoR reporter gene expression for both uniform and heterogeneous clones for four different BAC scaffolds. Individual clones for each BAC scaffold (3 uniform and 2 heterogeneous for ROSA-UG BAC, 7 uniform and 4 heterogeneous for 2207K13-UG BAC, 8 uniform and 3 heterogeneous for UBB-UG BAC, and 3 uniform and 3 heterogeneous for DHFR-UG BAC) were passaged up to three months in the absence or presence of drug selection and analyzed for reporter gene fluorescence at regular intervals after removal of drug selection.
With the exception of a small number of apparent fluctuations possibly related to transient changes in culture conditions, clones with uniform reporter gene expression showed no significant change either in the mean fluorescence values (Figure 4a) or in the distribution of fluorescence among the same clones (Figure 4b and Supporting Information, Figure S5) over time in the absence of selection for all four BAC scaffolds tested. In the presence of continued selection, uniform clones containing DHFR-UG or ROSA-UG BACs showed no significant reporter gene expression change, while an ~50% or 100% increase was observed for the UBB-UG or 2207K13-UG BAC clones, respectively (Figure 4a). No changes in estimated BAC copy number based on qPCR measurement were observed for any of these clones during this time series. This suggests that epigenetic changes driven by selection pressure may be responsible for these small increases in reporter gene expression.
Figure 4. UBC-GFP-ZeoR reporter gene expression over time.
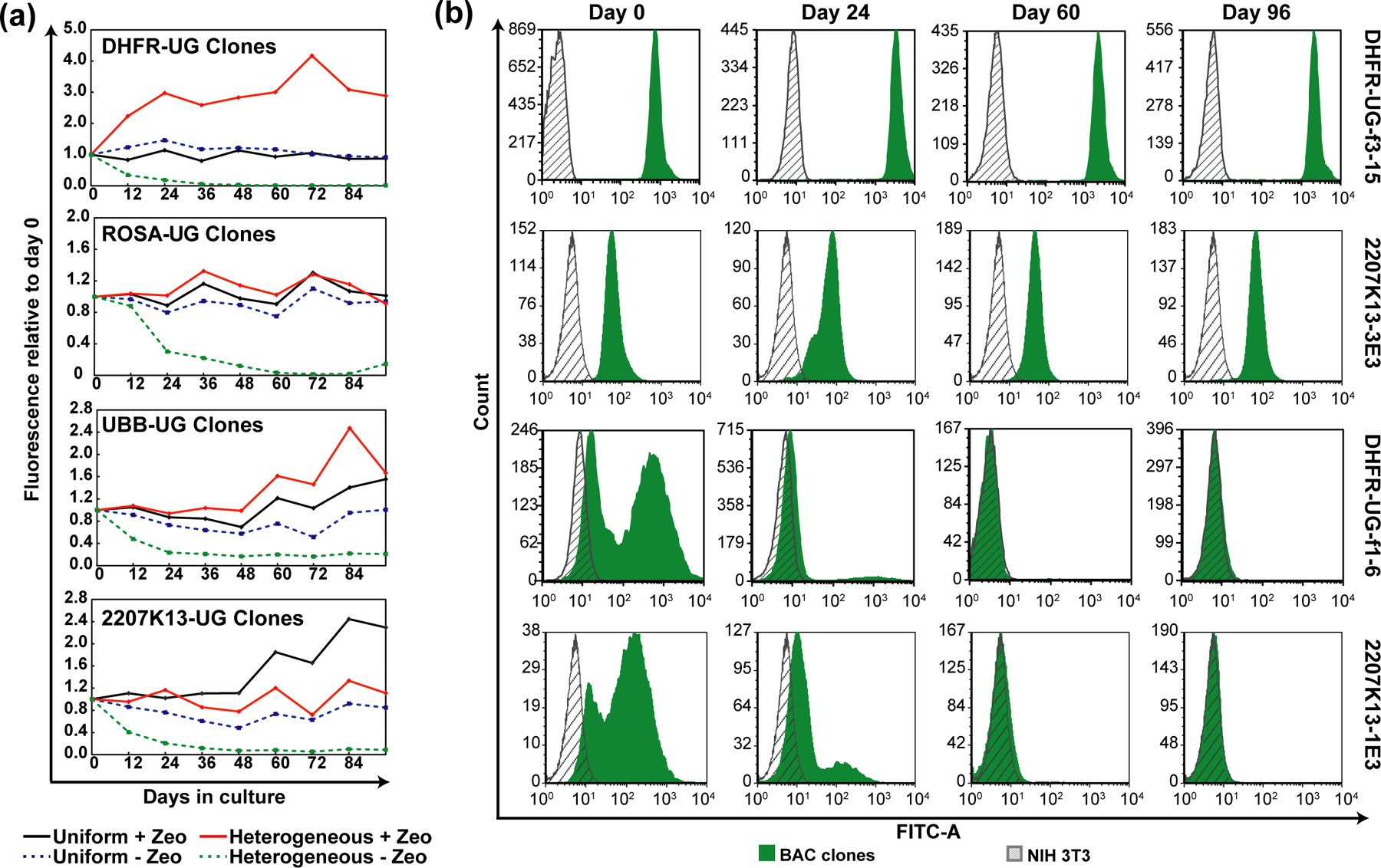
“Uniform” clones show stable expression with or without selection, while “heterogenous” clones show progressive loss of expression without selection. (a) Changes in GFP fluorescence of uniform versus heterogeneous clones, averaged over multiple clones (2–8), carrying indicated BAC transgenes during 96 days of continuous passaging with or without Zeocin selection. x-axis- number of days since removal of Zeocin; y-axis- mean fluorescence values of multiple clones divided by that at day zero; black- “uniform” expressing clones cultured with Zeocin; blue- “uniform” expressing clones cultured without Zeocin; red“heterogeneous” expressing clones cultured with Zeocin; green“heterogeneous” expressing clones cultured without Zeocin; (b) GFP fluorescence histogram of representative “uniform” and “heterogeneous” expressing NIH 3T3 clones at day 0, 24, 60 and 96 without selection. Gray- autofluorescence of untransfected cells; Green- GFP fluorescence of the indicated clones. x-axis- fluorescence; y-axis- cell number.
Notably, in the absence of selection, heterogeneous clones for all tested BAC scaffolds showed a significant and progressive loss of reporter gene expression over time. This led to a significant fraction of cells showing autofluorescence levels of fluorescence by the end of the experiment (Figure 4a). Reporter gene expression-level became progressively more homogenous, but at lower fluorescence levels (Figure 4b and Supporting Information, Figure S5). With selection, UBB-UG and DHFR-UG BAC heterogeneous clones showed a 1.6 to 3-fold increase in reporter gene expression, respectively, while the other BAC scaffold heterogeneous clones showed no significant changes (Figure 4a).
We found the presence of non-integrated BAC transgenes propagating as episomes, and their unequal segregation to be the source of this heterogeneity. The formation and propagation of BAC episomes is beyond the scope of the present study and is reported elsewhere.50 In the absence of continued drug selection, we would expect cells that have lost BAC transgenes will accumulate if there is any selective growth advantage for cells with fewer BAC copies.
We demonstrated stable UBC-GFP-ZeoR reporter gene expression achieved by integrated BAC transgenes in our BAC-TG EMBED system for several months in the absence of drug selection. This is an improvement over the 30–80% drop in expression observed originally with the CMV-mRFP-SV40-ZeoR reporter gene.31 We believe that both the UBC promoter and the CpG free GFP-ZeoR gene body could be contributing to this improvement.
Expression of multiple-reporters by the extended BAC-TG toolkit and BAC-MAGIC
As a proof-of-principle application of our improved toolkit for BAC TG-EMBED, we created a multi-transgene BAC to label simultaneously the nuclear lamina, nucleoli, and nuclear speckles with a single stable transfection. The original DHFR BAC was used for this multi-transgene expression. A SNAP-tagged Lamin B1 reporter minigene was used to label the nuclear lamina, a SNAP-tagged Fibrillarin the nucleoli, and an mCherry-tagged Magoh the nuclear speckles. We used the RPL32 promoter to drive the expression of the SNAP-tagged Lamin B1, and a promoter of intermediate strength, PPIA, for the SNAP-tagged Fibrillarin and the mCherry-tagged Magoh, which are both abundant proteins.
Previously, we used random Tn5 transposition to introduce expression cassettes into BAC scaffolds,51 but this approach is limited in the number of serial insertions that can be made due to the remobilization of existing transposons, its requirement for multiple selectable markers, and the randomness of the insertion sites. Alternatively, BAC recombineering using antibiotic resistance genes as positive selectable markers have been used to insert expression cassettes into precise locations on the BACs. However, like transposition, this method relies on the availability of multiple selectable markers and introduces unwanted selectable markers into the BACs. An alternative BAC recombineering scheme using cycles of galK-based positive selection to insert sequences followed by negative selection to remove galK40,41 have been used to make multiple BAC modifications without addition of unwanted selectable markers. However, the low efficiency of negative selection, due to a high background of competing, spontaneous deletions of mammalian DNA with its high repetitive DNA content, makes this approach quite time and labor intensive. Typically, one month is required for each cycle of insertion of DNA by positive selection, removal of the selectable marker by negative selection, and subsequent screening and testing of DNA from colonies that survive the negative selection to identify the small fraction of colonies containing the desired homology-driven, specific deletion of just the selectable marker.
To accelerate creation of BACs containing multiple transgenes, we created a new BAC assembly approach, BAC MAGIC (BAC-Modular Assembly of Genomic loci Interspersed Cassettes). BAC MAGIC combines the DNA assembler method in yeast52,53 and/or Gibson assembly54 with traditional cloning methods to create a number of BAC recombination modules followed by sequential rounds of BAC recombineering in which one fragment is inserted using one selectable marker followed by addition of a new fragment overlapping the previous fragment using a second positive selectable marker which replaces the first.55 Each round of fragment insertion only requires ~1 week for transformation and screening of clones. In this way, 45 kb of the DHFR BAC was effectively reconstructed such that DHFR sequences remained but 3 fluorescent minigene expression cassettes were added, each spaced by ~10 kb of DHFR sequence (Figure 5a–b, Supporting Information, Figure S6). The large homologous sequences flanking each expression cassette reduces recombination between similar sequences in other expression cassettes already inserted into the BAC, increasing the efficiency of this overall approach.
Figure 5. Construction of the multi-reporter DHFR BAC by BAC-MAGIC.
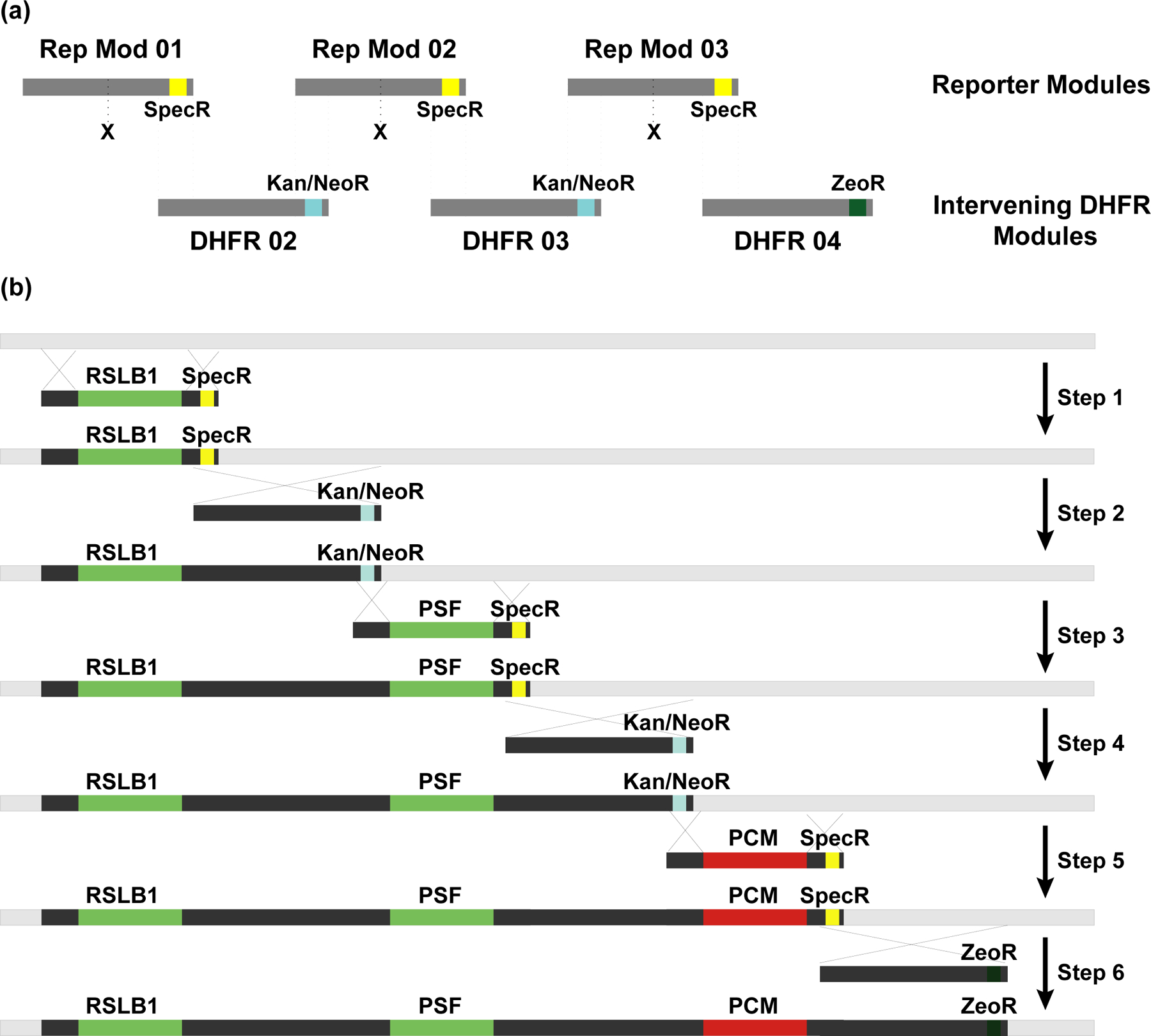
(a) Modular design of BAC-MAGIC. Reporter module 01, 02 and 03 contain reporter gene expression cassettes (X), DHFR BAC homologous sequences (dark gray), and Spectinomycin resistance markers (SpecR, yellow) near the 3’ ends for bacterial selection. Intervening DHFR module 02, 03 and 04 contain DHFR BAC homologous sequences (dark gray), and antibiotic resistance markers near the 3’ ends (Kanamycin/Neomycin resistance marker (Kan/NeoR, blue) in module 02 and 03 for bacterial selection, and Zeocin resistance marker (ZeoR, dark green) in module 04 for dual selection in bacterial or mammalian cells). The dotted lines mark homologous regions between the reporter modules and the intervening DHFR modules. (b) Six sequential steps of BAC recombineering introduce three reporter expression cassettes, RPL32-driven SNAP-tagged Lamin B1 (RSLB1), PPIA-driven SNAP-tagged Fibrillarin (PSF), and PPIA-driven mCherry-Magoh, onto the DHFR BAC (light gray) with ~10 kb of intervening DHFR BAC sequences (dark gray). Homologous regions are indicated by crossed lines.
We began the process using a DHFR BAC. After six rounds of BAC recombineering, we had created a BAC with four expression cassettes (Figure 5b): a SNAP-tagged Lamin B1 minigene, a SNAP-tagged Fibrillarin minigene, a mCherry-Magoh minigene, and a ZeoR selectable marker.
We tested simultaneous expression of the three reporters in 17 independent NIH 3T3 cell clones transfected with the multi-reporter BAC by examining fluorescence in fixed cells under a microscope (SNAP-tagged proteins were labeled with a Fluorescein conjugated SNAP tag substrate before fixation). We observed uniform expression of all the three reporters in 16/17 clones. The loss of SNAP-Lamin B1 expression in one of the clone (Cl#16) may be due to random breakage of the BAC during transfection, as PCR revealed the absence of this minigene from the cell clone. Similarly, 12/14 U2OS human osteosarcoma cell clones showed both SNAP-Lamin B1 and SNAP-Fibrillarin expression after transfection of a BAC containing only these two expression cassettes. Representative images of 4 of the U2OS clones A5, A6, B1 and C4 are shown in Supporting Information, Figure S7.
Within individual cells, a linear correlation was observed between the integrated fluorescence intensity per cell of SNAP-tagged proteins Lamin B1 and Fibrillarin versus mCherry-tagged Magoh in 4/4 representative NIH 3T3 clones (04, 08, 13 and 14, Figure 6a). Moreover, these fluorescently tagged proteins showed uniform rather than variegating expression in different cell nuclei of the same clone observed under the microscope (Figure 6b). A similar correlated expression of SNAP-tagged Lamin B1 and SNAP-tagged Fibrillarin reporters was observed in U2OS cells too (Supporting Information, Figure S7), indicating that the BAC MAGIC approach can be applied to express multiple-reporters in different cell-types.
Figure 6. Simultaneous multi-reporter expression through BAC-MAGIC.
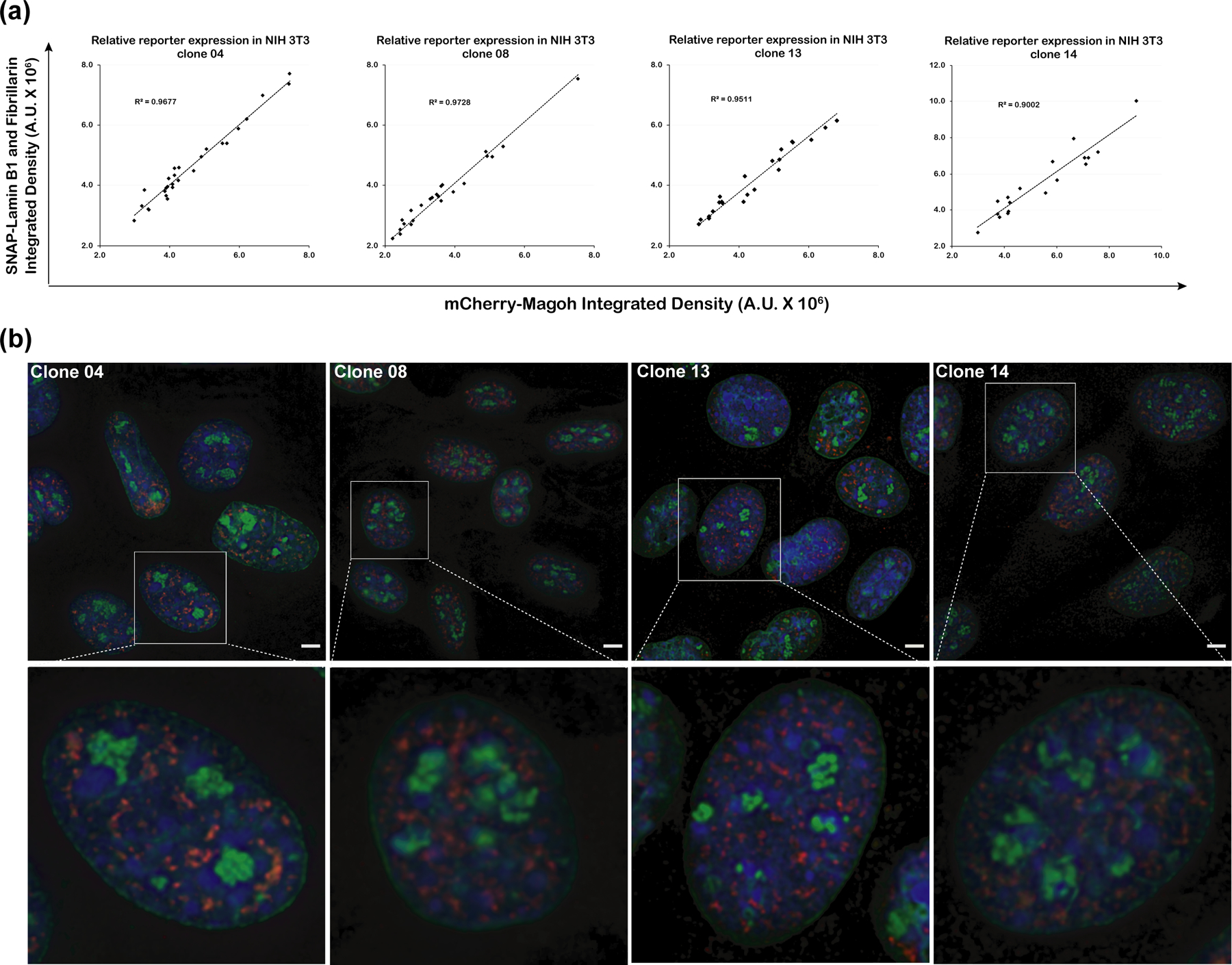
(a) Relative expression of the SNAP-tagged Lamin B1 and Fibrillarin to the mCherry-Magoh reporter in four representative NIH 3T3 cell clones (04, 08, 13 and 14) containing the multi-reporter BAC. Integrated fluorescence intensities per cell of SNAP-fluorescein (y-axis) and mCherry-Magoh (x-axis) are plotted. Linear regression lines (y-intercepts set to 0) are shown with corresponding R-squared values. Number of nuclei of each clone analyzed range from 18 to 27. Red-Clone 04; Blue- Clone 08, Black- Clone 13; Green- Clone 14. (d) Representative images (maximum intensity projections of 2–3 optical sections) from the four cell clones (Clone 04, 08, 13 and 14) showing expression of the three reporter genes. Nuclear lamina is labeled with SNAP-tagged Lamin B1 (green), nucleoli with SNAP-tagged Fibrillarin (green), and speckles with mCherry-Magoh (red). One magnified nucleus from each representative field (top panel) is shown in the bottom panel. Scale bars = 5 μm.
The ability to assemble BACs with multiple minigenes by BAC-MAGIC allows creation of a multi-transgene expressing BAC in several weeks, rather than the 4–5 months which would have been required by multiple rounds of DNA insertion using conventional BAC recombineering or targeted knock-in. Initial attempts to reassemble large, ~50kb regions of DHFR using yeast DNA assembly failed, apparently due to recombination between repetitive elements within the DHFR BAC sequence as well as the expression cassettes. In contrast, assembly of 10–15 kb modules from several DNA fragments using yeast DNA assembly worked with high efficiency. BAC-MAGIC exploits Gibson and yeast DNA assembly to build smaller modules with efficient serial BAC recombineering to reconstruct large BAC constructs containing multiple minigene expression cassettes. More generally, BAC-MAGIC should provide a tool for reconstruction of large eukaryotic DNA sequences containing high numbers of repetitive elements.
Using BAC-MAGIC and the extended BAC-TG EMBED toolkit, we created cell lines expressing three different fluorescently-tagged proteins in a single stable transfection step requiring just several weeks to isolate and expand cell clones. Most cell clones expressed all three tagged proteins at uniform levels and at reproducible relative levels of expression. This contrasts with the 6–12 months we have devoted in previous studies to create similar cell lines expressing multiple tagged proteins56 through a series of individual transfections followed by extensive screening of colonies to identify the small fraction expressing suitable levels of tagged proteins with minimal variegation and/or progressive long-term transgene silencing over time. In addition to the reproducible expression of multiple transgenes, the modular design of BAC-MAGIC is also independent of the cloning capacity limitations of retroviral vectors.
We anticipate that our expanded BAC TG-EMBED toolkit and BAC-MAGIC will facilitate a wide range of applications requiring simultaneous expression of multiple transgenes.
MATERIALS AND METHODS
PCR amplification of endogenous promoters
Primers (Supporting Information, Table S1) were designed using Primer3 program or NCBI primer blast to amplify 1–3 kb promoter regions which included either the entire or part of the 5’ UTRs upstream of the first exons of target genes. We used human genomic DNA extracted from BJ-hTERT cells as the template for PCR. However, the UBC promoter, including a partially synthetic intron, was amplified from plasmid pUGG.34
Construction of dual reporter DHFR BACs
The original dual reporter BAC, DHFR-HB1-GN-HB2-RZ31, was derived from the CITB-057L22 BAC (DHFR BAC) containing mouse chr13:92992156–93161185 (mm9). DHFR-HB1-GN-HB2-RZ has an EGFP expression cassette inserted 26 kb downstream of the Msh3 transcription start site, and a mRFP expression cassette inserted at 121 kb downstream of the Msh3 transcription start site. The EGFP expression cassette contains a CMV promoter-driven EGFP gene and a SV40 promoter-driven Kanamycin/Neomycin resistance gene, while the mRFP expression cassette has a CMV promoter-driven mRFP gene and a SV40-driven Zeocin resistance gene. New dual reporter DHFR BACs were created using a similar strategy to that used to create DHFR-HB1-GN-HB2-RZ, except that new mRFP expression cassettes were used, where the CMV promoter was replaced with alternative, human endogenous promoters. The intermediate DHFR BAC containing only the EGFP expression cassette, DHFR-HB1-GN31, was used to insert these new mRFP expression cassettes using λ Red-mediated homologous recombination.40,41
Plasmid p[MOD-HB2-CRZ]31 contains a CMV driven mRFP and a SV40 driven Zeocin resistance gene, flanked by two ~500 bp regions homologous to the DHFR BAC target site. Plasmid p[MOD-HB2-RCS-Zeo] was created by replacing the CMV-mRFP fragment between NotI and NheI sites of p[MOD-HB2-CRZ] with a synthetic DNA fragment “RCS” containing multiple rare restriction sites (Supporting Information, Table S2). The mRFP fragment generated by digesting p[MOD-HB2-CRZ] with NheI was then inserted into the NheI site of p[MOD-HB2-RCS-Zeo], yielding plasmid p[MOD-HB2-RCS-RZ]. The PCR-amplified endogenous promoters were then inserted into the RCS, generating plasmids p[MOD-HB2-promoter name-RZ]. Promoter functionality was tested by transient transfection of NIH 3T3 cells with these plasmids.
To insert the new mRFP expression cassettes into the DHFR-HB1-GN BAC, one round of λ Red-mediated recombination, using Zeocin resistance as positive selection, was performed according to a published protocol.41 DNA fragments containing the new mRFP expression cassettes with a given promoter with flanking homologous arms were excised from p[MOD-HB2-promoter name-RZ] plasmids by PmeI. SW102, a derivative strain of Escherichia coli (E. coli), was used for recombination. Recombinants were selected on low-salt LB plates containing 25 μg/ml Zeocin and 12.5 μg/ml Kanamycin at 32°C for ~20 hours. Recombinant colonies were screened by PCR amplification of sequences flanking the site of insertion (primers listed in Supporting Information, Table S1). The integrity of BAC constructs was verified by restriction enzyme fingerprinting, where observed band patterns on agarose gels were compared with predicted ones.
Construction of BACs containing the UBC-GFP-ZeoR cassette
Construction of pUGG containing the UBC-GFP-ZeoR-FRT-GalK-FRT cassette was described previously.34 Human BACs RP11–138I1 (UBB BAC), CTD-2643I7 (HBB BAC), CTD-2207K13 (2207K13 BAC) and mouse BAC RP23–401D9 (ROSA BAC) were obtained from Thermo Fisher Scientific. Mouse BAC CITB-057L22 (DHFR BAC) was a gift from Edith Heard (Curie Institute, Paris, France).
The UBC-GFP-ZeoR reporter gene insertion positions (mm9 or hg19) are chr17:16,301,887–16,301,888 in the UBB BAC, chr6:113,043,332–113,043,333 in the ROSA BAC, chr13:93,099,101–93,099,102 in the DHFR BAC, chr1:79,224,725–79,224,726 in the 2207K13 BAC, and chr11:5,390,233–5,390,244 in the HBB BAC.
λ Red-mediated BAC recombineering40,41 using a galK-based dual-selection scheme was used to introduce the UBC-GFP-ZeoR reporter cassette onto the BACs according to published protocols.41 DNA fragments with homology ends for recombineering were prepared by PCR using primers (Supporting Information, Table S1) with 74-bp homology sequences plus 16-bp sequences (forward, 5’- acagcagagatccagt-3’; reverse, 5’-tgttggctagtgcgt-3’) that amplify the UBC-GFP-ZeoR-FRT-GalK-FRT cassette from plasmid pUGG. E. coli strain SW105 was used for BAC recombineering. Recombinants containing the UBC-GFP-ZeoR-FRT-GalK-FRT cassette were selected for galK insertion at 32°C on minimal medium in which D-galactose was supplied as the only carbon source. Recombinant colonies were screened using PCR with BAC specific primers flanking the target regions (Supporting Information, Table S1). Subsequently, FLP recombinase-mediated removal of galK from selected recombinant clones was done by inducing actively growing SW105 cells with 0.1% (w/v) L-arabinose. Negative selection against galK used minimal medium containing 2-deoxy-galactose; deletion of galK in recombinants was again verified using BAC specific primers (Supporting Information, Table S1). The integrity of BAC constructs was verified by restriction enzyme fingerprinting.
Cell culture and establishment of BAC cell lines
Mouse NIH 3T3 fibroblasts (ATCC CRL-1658™) were grown in Dulbecco’s modified Eagle medium (DMEM with 4.5 g/l D-glucose, 4 mM L-glutamine, 1 mM sodium pyruvate and 3.7 g/l NaHCO3) supplemented with 10% HyClone Bovine Growth Serum (GE Healthcare Life Sciences, Cat. # SH30541.03). Human U2OS osteosarcoma cells were grown in DMEM medium supplemented with 10% Hyclone Bovine Growth Serum (GE Healthcare Life Sciences, Cat. # SH30541.03).
BAC DNA for transfection of mammalian cells was prepared with the QIAGEN Large Construct Kit (QIAGEN, Cat. # 12462) as per the manufacturer’s instructions. All BACs except DHFR BAC derived BACs were linearized before transfection: 2207K13-UG BAC with SgrAI (New England Biolabs, Cat. # R0603S), HBB-UG BAC with NotI (New England Biolabs, Cat. # R3189S) and all other BACs with the PI-SceI (New England Biolabs, Cat. # R0696S). Lipofectamine 2000 (Thermo Fisher Scientific, Cat. # 11668019) was used to transfect the cells with the BACs according to the manufacturer’s directions. The dual reporter DHFR BACs and the BACs containing the UBC-GFP-ZeoR reporter gene were transfected into NIH 3T3. Mixed clonal populations of stable transformants were obtained after ~2 weeks of selection (75 μg/ml Zeocin and 500 μg/ml G418 for NIH 3T3 cells transfected with the dual reporter DHFR BACs; 75 μg/ml Zeocin for NIH 3T3 cells transfected with the BACs containing the UBCGFP-ZeoR reporter gene); individual cell clones were obtained by serial dilution or colony picking using filter discs.57 The U2OS osteosarcoma cells transfected with dual reporter DHFR BAC carrying SNAP-tagged lamin B1 and fibrillarin cassettes were selected with 1200 μg/ml G418.
To analyze the stability of reporter gene expression in NIH 3T3 cells, individual cell clones were grown continuously with or without Zeocin (75 μg/ml) selection for 96 days. We used the following clones (Figure 4 and Supporting Information, Figure S5): DHFR-UG BAC- f1-7, f3–13, f3–15 (uniform), f1–6, f2–1, f2–3 (heterogeneous); ROSA-UG BAC- 2D6– 3C11, 3D7 (uniform), 2C12, 3A1 (heterogeneous); UBB-UG BAC- 1C2, 1F1, 1F12, 2F5, 2G4, 4D3, 5C1, 5C7 (uniform), 1A8, 1D5, 6H2 (heterogeneous); 2207K13-UG BAC- 3E3, 5C8, 5E1, 6B9, 6E12, 6F4, 7B2 (uniform), 1E3, 6A2, 6C10, 7B9 (heterogeneous).
Flow cytometry
For analysis of reporter gene expression, cells were grown to ~40%−80% confluence, trypsinized, and resuspended in growth media at ~0.5–1 million cells/ml. For analysis of the expression of mRFP and EGFP, or mRFP alone, cell suspensions were run on a BD FACS AriaII (BD Biosciences) or a BD LSR Fortessa (BD Biosciences), using the PE channel (561 nm laser and 582/15 nm bandpass filter) for mRFP, and the FITC channel (488 nm laser, 505 longpass dichroic mirror and 530/30 nm bandpass filter) for EGFP. For analysis of GFP expression alone, the cell suspensions were run on a BD FACS Canto II Flow Cytometry Analyzer (BD Biosciences), using the FITC/Alexa Fluor-488 channel (488nm laser, 502 longpass dichroic mirror and 530/30 bandpass filter). Rainbow fluorescent beads (Spherotech, Cat. # RFP-30–5A) were used as fluorescence intensity standards. Each sample was run for 1–2 min or until the number of events after gating reached 10–20 thousand.
For cell sorting, cells were resuspended at ~10 million cells/ml in growth media and run on a BD FACS AriaII for up to 30–40 minutes.
Estimation of relative promoter strength
The red and green fluorescence of the mixed-clonal populations stably transfected with the dual-reporter DHFR BACs was measured by flow cytometry. The mean florescence values of all gated cells were divided by the bead intensity values for normalization. The ratio of normalized mRFP to normalized EGFP was calculated as a measure of promoter strength (Equation 1). All promoter strengths were then normalized with the CMV promoter strength (comparing the CMV-driven mRFP to the CMV-driven EGFP expression) to calculate the relative promoter strength (Equation 2) using the CMV promoter as the reference.
| 1 |
| 2 |
Genomic DNA extraction
Genomic DNA was isolated by phenol/chloroform extraction.58 Cultured cells were harvested and washed with 1x Cell Culture Phosphate Buffered Saline (PBS, Corning, Cat. # 21040CV). Sorted cells were pelleted. Up to ~2 million cells were resuspended in 100 μl High-TE buffer (10 mM Tris-Cl, pH 8, 10 mM EDTA, 25–100 μg/ml RNase A (QIAGEN, Cat. # 19101)) and lysed by adding 2.5 μl 20% SDS. After incubation at 37°C for several hours, the lysate was digested by ~0.2 mg/ml Proteinase K (New England Biolabs, Cat. # P8102 or P8107S) at 55°C for ~1 day. 1 M Tris-Cl (pH 8.0), 5 M NaCl and nuclease free water were added to the lysate to bring up the total volume to ~600 μl and final concentrations of Tris-Cl to ~0.1 M and NaCl to ~0.2 M. The lysate was then extracted once with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1 mixture, Fisher Scientific, Cat. # BP1752I-400) and once with an equal volume of chloroform/isoamyl alcohol (24:1 mixture, MilliporeSigma, Cat. # C0549). DNA was precipitated by adding 2.5 volumes of 100% ethanol, washed with 70% ethanol and resuspended in EB (10mM Tris-Cl, pH 8.5).
Estimation of transgene copy number
BAC or plasmid transgene copy number within individual cell clones was measured by real-time quantitative PCR (qPCR), using purified genomic DNA, iTaq universal SYBR Green Supermix (Bio-Rad Laboratories, Cat. # 1725121) and a StepOnePlus (Applied Biosystems). Relative quantitation methods were used for copy number calculation. Primers used for qPCR are listed in Supporting Information, Table S1. Mouse genes Sgk1 and Hprt1 were used as endogenous controls, assuming four copies of each gene per cell in NIH 3T3. A primer pair (Zeo-GFP2for/rev) that binds to the UBC-GFP-ZeoR region was used to estimate transgene copy number, which was calculated by Equation 3 and 4.
| 3 |
| 4 |
Correlation of reporter gene expression and reporter gene copy number
Mean fluorescence intensity (in arbitrary units) of individual clones were measured by flow cytometry and normalized by fluorescent bead intensity to be used as a measure of reporter gene expression. To ensure uniform normalization for all samples, fluorescent beads from the same batch were used for all measurements. Untransfected cells were used to establish background fluorescence levels. Linear correlations of GFP expression level versus transgene copy number for each group of cell clones were calculated using the lm function in R with the y-intercept fixed to 0 (autofluorescence normalized by beads was almost 0). Pearson correlation coefficients and the p-values of the correlation coefficients are calculated by the cor.test function in R.
3D DNA FISH
Biotin or digoxigenin labeled DNA FISH probes were made from pooled PCR products (primers listed in Supporting Information, Table S1) or BAC DNA, using a published protocol59, with the following reagents: AluI, DpnI, HaeIII, MseI, MspI, RsaI (New England Biolabs, Cat. # R0137S, R0176S, R0108S, R0525S, R0106S, R0167S, respectively) and CutSmart Buffer (New England Biolabs); Terminal Deoxynucleotidyl Transferase and reaction buffer (Thermo Fisher Scientific, Cat. # EP0161); dATP (New England Biolabs, Cat. # N0446S) and Biotin-14-dATP (Thermo Fisher Scientific, Cat. # 19524016) for biotin labelling, or dTTP (New England Biolabs, Cat. # N0446S) and Digoxigenin-11-dUTP (MilliporeSigma, Cat. # 11093088910) for digoxigenin labelling.
DNA FISH of interphase nuclei used published protocols60,61 with small modifications. Cells grown on coverslips (12 mm diameter) were fixed with 3–4% paraformaldehyde in Dulbecco’s phosphate buffered saline (DPBS, 8 g/l NaCl, 0.2 g/l KCl, 2.16 g/l Na2HPO4-7H2O, 0.2 g/l KH2PO4) for 10 min, followed by permeabilization with 0.5% Triton X-100 (Thermo Fisher Scientific, Cat. # 28314) in DPBS for 10–15 min. Cells were subjected to six freeze-thaw cycles using liquid nitrogen, immersed in 0.1M HCl for 10–15 min, and then washed 3x with 2x saline-sodium citrate (SSC). Freeze-thaw cycles sometimes were skipped with no noticeable difference in FISH signals. Cells were incubated in 50% deionized formamide (MilliporeSigma, Cat. # S4117)/2x SSC for 30 min at room temperature (RT), and stored for up to 1 month at 4°C. Each coverslip used ~4 μl hybridization mixture, consisted of 5–20 ng/μl probes, 10x of mouse (for NIH 3T3 cells) or human (for HCT116 cells) Cot-1 DNA (Thermo Fisher Scientific Cat. # 18440016 or 15279011,) per ng probe, 50% deionized formamide, 10% dextran sulfate (MilliporeSigma, Cat. # D8906) and 2x SSC. Cells and probes were denatured together on a heat block at ~76°C for 2–3 min and hybridized at 37°C for 16 hrs-3 days. After hybridization, cells were washed 3 × 5 min in 2x SSC at RT, and for 3 × 5 min in 0.1x SSC at 60°C, and then rinsed with SSCT (4x SSC with 0.2% TWEEN 20) at RT. FISH signals were detected by incubation with Alexa Fluor 647 conjugated Streptavidin (1:200; Jackson ImmunoResearch, Cat. # 016-600-084) or Alexa 594 conjugated Streptavidin (1:200; Life Technology, Cat. # S11227) for biotin-labeled probes, or Alexa Fluor 647 conjugated IgG fraction monoclonal mouse anti-digoxin (1:200; Jackson ImmunoResearch, Cat. # 200-602-156) for digoxigenin labeled probes, diluted in SSCT with 1% Bovine Serum Albumin (MilliporeSigma, Cat. # A7906), for 40 min-2 hrs at RT. Coverslips were washed in SSCT for 4 × 5 min, rinsed with 4x SSC and mounted with a Mowiol-DABCO anti-fade medium62 containing ~3 μg/ml DAPI (MilliporeSigma, Cat. # D9542).
Analysis of BAC transgene FISH signals
The area and circularity (4pi(area/perimeter^2)) of z-projected DNA FISH signals were measured using Fiji.63 A macro was developed to semi-automate the analysis process. First, a maximum intensity z-projection of a 3D z-stack image was created using the “Z Project…” function with “projection=[Max Intensity]”. Next, individual nucleus was cropped out from the z-projection image to minimize the detection of background fluorescent aggregates by the computer program. An “Otsu” auto-thresholding function was applied to the DAPI channel of the z-projection to select individual nuclei. The selection was turned into a rectangle and enlarged by 1 μm. Next, a “Maximum Entropy Multi-Threshold” function with “number=3” from the IJ Plugins package (http://ijplugins.sourceforge.net/index.html) was applied to the FISH channel of the cropped nucleus image, generating three thresholds. To prevent endogenous Dhfr-Msh3 loci and background fluorescent aggregates from being identified, the area containing the FISH signal was first identified by applying the “Analyze Particles…” function with “size=0-Infinity display exclude clear add” to the FISH channel masked with the lowest threshold and selecting the largest particle. The FISH signal was then identified by masking the area containing the FISH signal with the middle threshold. The selection of FISH signal was examined manually and any area not corresponding to the BAC transgene arrays were deselected. Finally, the area and circularity of the correctly selected FISH signals were measured by the “Set Measurements…” function with “area perimeter shape redirect=None decimal=3” and the “Measure” function.
The following clones were used for Supporting Information, Figure S3d–e: DHFR-UG-f3–1, -f3–15, -P4–14; HBB-UG-fD2, -H3-50-4, -H4-100-16; 2207K13-UG-K3-50-17, -K4-100-12.
Analysis of BAC transgene nuclear localization
Clone HBB-C3 stably transfected with a HBB BAC containing a Lac operator tandem repeat and no reporter gene is described in a previous study.49 The BAC transgenes were visualized by DNA FISH and images were analyzed using Fiji.63 The orthogonal view of the z-stack images were examined manually and optical sections where the FISH spots were both in focus and were at the middle planes of the nuclei were used for analysis. To detect nuclear periphery localization, the edges of the nuclei were detected by applying a “Gaussian Blur…” function with “sigma=2.50” and subsequently a “Default dark” auto-thresholding function to the DAPI channel. The resulting selection was then shrunk by 0.2 μm. FISH spots overlapping or outside the selection were regarded as localized at the nuclear periphery. FISH spots completely or partially overlapping a chromocenter were regarded as localized at chromocenters.
95% confidence intervals of the proportions were calculated based on binomial distribution, using Equation 5 and 6 (N- total number of nuclei; Nd− number of nuclei with periphery or chromocenter localized BAC transgenes; pU and pL− upper and lower limits of the 95% confidence interval, respectively). Two-sided Fisher’s Exact Test were used to calculate p-values of the proportions of individual clones vs clone HBB-C3.
| 5 |
| 6 |
Microscopy and image analysis
For examining the expression of the three reporter minigenes, SNAP tagged-Lamin B1, SNAP-tagged Fibrillarin and mCherry-tagged Magoh, the cells were first labeled with cell-permeable substrate SNAP-Cell Fluorescein (New England Biolabs, Cat. # S9107S) overnight at 240 nM concentrations. To reduce background of unreacted SNAP-tag substrate, cells were incubated 3× 30 mins with media in the incubator, washed with PBS, and fixed with freshly prepared 4% paraformaldehyde in PBS for 15 min at RT.
3D z-stack images were acquired using a Deltavision wide-field microscope (GE Healthcare), equipped with a Xenon lamp, 60X, 1.4 NA oil immersion objective (Olympus) and CoolSNAP HQ CCD camera (Roper Scientific) or a V4 OMX (GE healthcare) microscope, equipped with a 100X, 1.4 NA oil immersion objective (Olympus) and two Evolve EMCCDs (Photometrics). Images were deconvolved using the deconvolution algorithm62 provided by the softWoRx software (GE Healthcare). Chromatic aberrations were measured using the alignment slide provided by GE Healthcare and the OMX Image registration function in the softWoRx was used to correct the chromatic aberrations in all DNA FISH images according to the manufacturer’s instructions. All image analysis and preparation were done using Fiji.63 Images were assembled using Illustrator (Adobe), Photoshop (Adobe), or GIMP.
Comparison of reporter gene expression levels in for NIH 3T3 cell clones (Figure 6a) was done by projecting deconvolved images stacks and then measuring the integrated intensity within individual nuclei after subtracting background intensity levels measured in the cytoplasm. Regions of interest circumscribing individual nuclei were drawn manually based on the SNAP-lamin B1 signal. Linear correlations of the integrated intensities of the nuclear SNAP-tag and mCherry signals were calculated using Microsoft Excel with the y-intercept fixed to 0.
A non-linear Gamma correction (0.7) to reduce the grey-scale dynamic range followed by a maximum intensity projection of 3–4 z-sections was used to better visualize both lamin and nucleolar staining simultaneously (Figure 6b).
Construction of multi-reporter DHFR BAC by BAC-MAGIC
Overview:
Construction of the 3-reporter BAC was done by serially inserting ~10–15 kb DNA cassettes into the DHFR BAC scaffold by BAC recombineering. These DNA cassettes were constructed from two different DNA plasmid module types: reporter modules and intervening DHFR sequence modules (Supporting Information, Figure S6). DNA cassettes were inserted sequentially into the DHFR BAC using multiple rounds of BAC recombineering and positive selection with one of two different positive selectable markers. After insertion of the first DNA cassette, each subsequent insertion of the next DNA cassette removed the preceding positive selectable marker located at the 3’ end of the preceding cassette while inserting the alternative selectable marker located at the 3’ end of the new cassette. Three reporter gene modules (Rep Mod 01, 02, 03) plus three intervening DHFR sequence modules (DHFR 02, 03, 04) were constructed and then inserted into the DHFR BAC using 6 sequential rounds of BAC recombineering. In this way, 45 kb of the original DHFR BAC effectively was reconstructed such that the original DHFR sequences were retained but the 3 reporter minigenes were inserted into this BAC region with each reporter minigene spaced by ~10 kb of DHFR sequence. We call this overall construction approach BAC-MAGIC (BAC-Modular Assembly of Genomic loci Interspersed Cassettes).
Each DNA cassette was constructed using traditional cloning methods, Gibson assembly,54 and/or DNA Assembler.52,53 Three reporter recipient modules (pRM01-Spec, pRM02-Spec, and pRM03-Spec) were designed to incorporate a rare AgeI restriction site for insertion of reporter expression cassettes of choice, in order to create the final reporter modules for BAC recombineering. Unless mentioned specifically all the enzymes were procured from New England Biolabs. All primers and oligos are listed in Supporting Information, Table S1. Gibson assembly used Gibson assembly cloning kit (New England Biolabs, Cat. # E5510S) as per the manufacturer’s instructions.
DNA Assembler used Saccharomyces cerevisiae (S. cerevisiae) strain VL6–48N (MATα, his3-Δ200, trp1-Δ1, ura3-Δ1, lys2, ade2–101, met14, cir°), transformed with 43 fmol pRS413 vector backbone and 130 fmol of all other fragments using the LiAc/SS carrier DNA/PEG method.64 The S. cerevisiae single-copy shuttle vector pRS413 contains CEN6/ARS autonomously replicating sequence, auxotrophic selection marker HIS3 for propagation in yeast, and pMB1 origin of replication and bla (ApR) marker for selection with ampicillin in E. coli. The 3.8 kb pRS413 vector backbone was PCR amplified from plasmid pRS413 (New England Biolabs) using primer pair RS413-Fw/RS413-Rev for all yeast assembly reactions. The vector backbone and all other fragments made by PCR were digested with DpnI to remove template DNA. Transformants were selected on SC selection media plates lacking histidine [0.17% Bacto-yeast nitrogen base without amino acids (MilliporeSigma, Cat. # Y1251–100G), 0.5% ammonium sulfate, 2% D-glucose, 0.2% Dropout mix (MilliporeSigma, Cat. # Y2001–20G), 2% agar, 80 mg/l uracil, 80 mg/l L-tryptophan, and 240 mg/l L-leucine] at 30°C for 3–4 days. Plasmid DNA were prepared using QIAprep Spin Miniprep Kit (Qiagen, Cat. # 27104) and screened by restriction enzyme fingerprinting. Plasmid DNA from selected yeast colonies was introduced into E. coli strain DH5α and isolated plasmid DNA then further validated by additional restriction enzyme fingerprinting.
Below we describe construction of each reporter and intervening spacer modules and BAC recombineering assembly of these modules to create the 3-reporter BAC. ApE (M. Wayne Davis, University of Utah, http://biologylabs.utah.edu/jorgensen/wayned/ape/) and SnapGene (from GSL Biotech; available at snapgene.com) programs were used to analyze sequence data, design primers, and design cloning strategies.
Construction of plasmid pRM01-RSLB1-Spec (Reporter module 01):
Plasmid pRM01 was made by sequential addition of two DHFR homology regions to plasmid pEGFP-C1 (Clontech). First, the 2.1 kb DHFR homology region (M1F4) was PCR amplified from the DHFR BAC using primer pair M1F4-BamHIfor/M1F4-AgeIrev, double digested with BamHI/AgeI, and ligated with the BamHI/AgeI digested pEGFP-C1 to generate intermediate plasmid pEG-Rep-Module-1a. Next, the 2.0 kb DHFR homology region (M2F12) was PCR amplified from the DHFR BAC using primer pair M2F12-AgeIFor/M2F12-PshRev, double digested with AgeI/PshAI and ligated with the AgeI/SnaBI digested plasmid pEG-Rep-Module-1a to produce plasmid pRM01.
To create plasmid pRM01-Spec (Reporter recipient module 01), a 1.6 kb Spectinomycin resistance gene expression cassette (SpecR), derived from plasmid pYES1L (Thermo Fisher Scientific), was inserted into pRM01, 400 bp upstream of the 3’ end of the M2F12 DHFR homology region by two-fragment Gibson Assembly.54 The two fragments for Gibson assembly were PCR amplified from pRM01 using primer pair GA-RM01-Spec-For/ GA-RM01-Spec-Rev (PCR product size: 7.8 kb), or from pYES1L using primer pair Specfor/SpecRev (PCR product size: 1.6 kb) respectively.
The pRSLB1 (hRPL32-SNAP-Lamin B1) plasmid harboring SNAP-tagged Lamin B1 reporter expression cassette (RSLB1) was constructed by three-fragment Gibson assembly. pEGFP-Lamin B1 plasmid vector backbone 5.3 kb fragment was prepared by AseI/BsrGI double digestion. The hRPL32 promoter (2.2 kb) and SNAP tag (561 bp) fragments were PCR amplified using primer pairs GA-hRPL32-fwd/GA-hRPL32-rev (template plasmid pMOD-HB2-hRPL32-RZ, made in this study), and GA-SNAP-fwd/GA-SNAP-rev (template plasmid pSNAPf, New England Biolabs).
pRM01-Spec was linearized by AgeI and simultaneously dephosphorylated by Shrimp Alkaline Phosphatase (New England Biolabs, Cat. # M0371S). The RSLB1 expression cassette was PCR amplified from plasmid pRSLB1 using primer pair R32CerLBAgeIfor/newPCFAgeIrev (PCR product size: 4.9 kb) and double digested with DpnI/AgeI. The linearized pRM01-Spec and the digested RSLB1 PCR product were ligated to produce plasmid pRM01-RSLB1-Spec, which was digested with AseI to produce the final BAC recombineering 10.3 kb targeting construct.
Construction of plasmid pRM02-PSF-Spec (Reporter module 02):
Plasmid pRM02 was made using similar cloning steps used to produce pRM01 except two different DHFR homology regions were added to pEGFP-C1: 2.0 kb PCR product M2F4 (primer pair M2F4-BamHIfor/M2F4-AgeIrev) replaced M1F4 and 2.0 kb PCR product M3F1 (primer pair M3F1-AgeIFor/M3F1-PshRev) replaced M2F12. Plasmid pRM02-Spec was made the same way as pRM01-Spec except that fragment 1 for Gibson assembly was PCR amplified from plasmid pRM02 using primer pair GA-RM02-Spec-For/GA-RM02-Spec-Rev (PCR product size: 7.8 kb). The final plasmid pRM02-Spec (pRep-module 02-Spec) is Reporter recipient module 02 for the SNAP-tagged Fibrillarin reporter expression cassette (PSF).
To create plasmid pPSF (pPPIA-SNAP-Fibrillarin), the GFP cassette between KpnI/HpaI restriction sites of plasmid GFP-Fibrillarin was replaced with a 730 bp Cerulean cassette PCR amplified from plasmid pCerulean-N1 (New England Biolabs) using primer pair ForCerFib/RevCerFib, resulting in an intermediate plasmid pPCF. Next, the CMV promoter between SnaBI/HindIII sites of pPCF was replaced with the 2.8 kb PPIA promoter PCR amplified from plasmid p[MOD-HB2-PPIA-RZ] (made in this study) using primer pair PPIACerFibFor/ PPIACerFibRev, resulting in plasmid pPPIA-Cer-Fib. Finally, the 720 bp Cerulean cassette between the AgeI/HpaI sites of pPPIA-Cer-Fib was replaced with a 560 bp SNAP tag fragment PCR amplified from plasmid pSNAPf (New England Biolabs) using primer pair Snap-XmaI-For/Snap-HpaI-Fib-Rev and double digested with XmaI/HpaI, producing pPSF.
pRM02-Spec was linearized by AgeI and simultaneously dephosphorylated by Shrimp Alkaline Phosphatase (New England Biolabs, Cat. # M0371S). The 4.6 kb PSF expression cassette was PCR amplified from pPSF using primer pair PSF-AgeI-For/ PSF-AgeI-Rev and double digested with DpnI/AgeI. Their ligation produced plasmid pRM02-PSF-Spec, which provided the 10.4 kb BAC recombineering targeting construct after BamHI/AatII/RsrII triple digestion of pRM02-PSF-Spec.
Construction of plasmid pRM03-PCM-Spec (Reporter module 03):
Plasmid pRM03 was made using similar cloning steps used to produce pRM01 except two different DHFR homology regions were added to pEGFP-C1: 2.1 kb PCR fragment M3F4 (primer pair M3F4-BamHIfor/M3F4-AgeIrev) replaced M1F4 and 2.1 PCR fragment M4F1 (primer pair M4F1-AgeIFor/M4F1-PshRev) replaced M2F12. Plasmid pRM03-Spec was made the same way as pRM01-Spec except that fragment 1 for Gibson assembly was PCR amplified from plasmid pRM03 using using primer pair GA-RM03-Spec-For/ GA-RM03-Spec-Rev (PCR product size: 7.8 kb). The final plasmid pRM03-Spec (pRep-module 03-Spec) is Reporter recipient module 03 for the mCherry-tagged Magoh reporter expression cassette (PCM).
Plasmid pPCM (pPPIA-mCherry-Magoh) was created in two steps. First, the CMV promoter between the NdeI/NheI sites of plasmid pmRFP-Magoh was replaced with the PPIA promoter (2.8 kb), PCR amplified from plasmid pMODHB2-PPIA-RZ using primer pair PPIA-Magohfor/ PPIA-MagohRev and double digested with NdeI/NheI, resulting in intermediate plasmid pPMM. Next, the mRFP tag between the NheI/HindIII sites of pPMM was replaced with a 720 bp mCherry tag PCR amplified from plasmid pQCXIN-TetR-mCherry using primer pair mCherry-NheI-Magoh-For/mCherry-H3-Magoh-Rev, resulting in plasmid pPCM.
To create plasmid pRM03-PCM-Spec (Reporter module 03), plasmid pRM03-Spec was linearized by AgeI and simultaneously dephosphorylated by Shrimp Alkaline Phosphatase (New England Biolabs, Cat. # M0371S). A 4.2 kb PCM expression cassette was PCR amplified form plasmid pPCM using primer pair MMorCF-AgeIfor/newPCFAgeIrev and double digested with DpnI/AgeI. pRM03-Spec and the PCM PCR product were ligated, producing plasmid pRM03-PCM-Spec, which was used as a template for PCR amplification using primer pair M3F4-PCR-Fw/M4F1-PCR-Rev to produce the 9.9 kb BAC recombineering target. After PCR, any remaining template plasmid was digested with DpnI.
Construction of plasmid pRS413-DHFR-Mod-02-Kan (Intervening DHFR module 02):
Plasmid pRS413-DHFR-Mod-02 was made by assembling the vector backbone with four additional fragments using the DNA assembler method.52,53 Fragment 5’-DHM2 (4.3 kb) and fragment 3’-DHM2 (6.3 kb) with an overlap of 659 bp and were both PCR amplified from the DHFR BAC, using primer pair M2F12-AgeIfor/M2F1rev or DHM2-Seq2/M2F4-AgeIrev, respectively. Two bridging oligomers, with a 125 bp homology to the pRS413 vector backbone, and a 125 bp homology to fragment 5’-DHM2 (oligo M2F1-pRS413) or fragment 3’-DHM2 (oligo M2F4-pRS413) were synthesized at Integrated DNA Technologies, Inc. The final Intervening DHFR module 02, plasmid pRS413-DHFR-Mod-02-Kan, was created by ligating a 2.4 kb Kan/NeoR cassette derived from DraI digestion of plasmid pEGFP-C1, with the plasmid pRS413-DHFR-Module-02 linearized by DraIII and blunted by DNA Polymerase I, Large (Klenow) Fragment.
For BAC recombineering an 11.7 kb of targeting construct was amplified from plasmid pRS413-DHFR-Mod-02-Kan using primer pair M2F12-AgeIFor/DH2–4rev and purified by gel extraction after DpnI digestion of the template plasmid.
Construction of plasmid pRS413-DHFR-Mod-03-Kan (Intervening DHFR module 03):
Plasmid pRS413-DHFR-Mod-03 was made by assembling the vector backbone with four additional fragments using the yeast DNA assembler method. Fragment 5’-DHM3 (6.5 kb) and fragment 3’-DHM3 (5.0 kb) with an overlap of 1553 bp were both PCR amplified from the DHFR BAC using primer pair M3F1-AgeIFor/M3F3-BamHIrev or M3-F3For/M3F4-AgeIRev, respectively. Two bridging oligomers, with a 125 bp homology to the pRS413 vector backbone, and a 125 bp homology to fragment 5’-DHM3 (oligo M3F1-pRS413) or to fragment 3’-DHM3 (oligo M3F4-pRS413), respectively, were synthesized at Integrated DNA Technologies, Inc. The final Intervening DHFR module 03, plasmid pRS413-DHFR-Mod-03-Kan, was created by ligating a 2.4 kb Kan/NeoR cassette derived from DraI digestion of plasmid pEGFP-C1, with the plasmid pRS413-DHFR-Mod-03 linearized by SmaI.
For BAC recombineering a 12.2 kb targeting construct was amplified from plasmid pRS413-DHFR-Mod-03-Kan using primer pair DH3–1for/DH3–4rev and purified by gel extraction after DpnI digestion of the template plasmid.
Construction of plasmid pRS413-DHFR-Mod-04-Zeo (Intervening DHFR module 04):
Plasmid pRS413-DHFR-Mod-04 was made by assembling the vector backbone plus 5 additional fragments using the yeast DNA assembler method (4). Fragment 5’-DHM4 (4.9 kb), fragment Mid-DHM4 (5.2 kb) and fragment 3’-DHM4 (5.2 kb) with an overlap of 2663 bp in between 5’-DHM4 and Mid-DHM4, and an overlap of 2542 bp in between Mid-DHM4 and 3’-DHM4, were PCR amplified from the DHFR BAC using primer pair M4F1-AgeIfor/DHM4F2-R, DHM4F2-Fw/DHM4F3-R, or Fw-M4F2-BamHI/RevM4F5-MluI, respectively. Two bridging oligomers, with a 125 bp homology to pRS413 vector backbone, and a 125 bp homology to fragment 5’-DHM4 (oligo M4F1-pRS413), or to fragment 3’-DHM4 (oligo M4F5-pRS413), were synthesized at Integrated DNA Technologies, Inc. The final Intervening DHFR module 04, plasmid pRS413-DHFR-Mod-04-Zeo, was created by ligating a 1.1 kb ZeoR expression cassette PCR amplified from plasmid pSV40/Zeo2 (ThermoFisher Scientific) using 5’ phosphorylated primer pair ZeoMluIFor/ZeoMluIRev, with the plasmid pRS413-DHFR-Module-04 linearized by BmgBI.
For BAC recombineering an 11.6 kb targeting construct was excised out from plasmid pRS413-DHFR-Mod-04-Zeo using KpnI/DrdI restriction enzymes and gel purified.
Assembly of modules to create multi-reporter DHFR BAC:
The six targeting constructs derived from the three reporter modules and the three intervening DHFR modules were incorporated into the DHFR BAC by BAC recombineering, with the following order: Reporter module 01, Intervening DHFR module 02, Reporter module 02, Intervening DHFR module 03, Reporter module 03 and Intervening DHFR module 04. E. coli strain SW102 was used for BAC recombineering. Each round of BAC recombineering used a corresponding antibiotic (50 μg/ml Kanamycin, 50 μg/ml Spectinomycin, or 25 μg/ml Zeocin) as positive selection for incorporation of the current targeting construct as described in section “Construction of dual reporter DHFR BACs”. The colonies were further screened for loss of the antibiotic resistance gene incorporated in the previous round of BAC recombineering by streaking colonies onto a plate containing the corresponding antibiotic. Each round of recombination was validated by restriction enzyme fingerprinting.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institute of General Medical Sciences [GM098319 and, in part, GM58460 to A.S.B.] and by the National Institutes of Health Common Fund 4D Nucleome Program [U54 DK107965 to A.S.B]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
We thank Edith Heard (Curie Institute) for providing DHFR BAC (clone 057L22 from CITB mouse library), Veena K Parnaik (CSIR-CCMB, Hyderabad, India) for GFP-Lamin B1 plasmid, Miroslav Dundr (Rosalind Franklin University of Medicine and Science) for GFP-Fibrillarin plasmid, Huimin Zhao (University of Illinois Urbana-Champaign) for pQCXIN-TetR-mCherry plasmid, Peter Adams (Sanford Burnham Prebys Medical Discovery Institute) for BJ-hTERT cells, and KV Prasanth (University of Illinois Urbana-Champaign) for mRFP-Magoh plasmid. Use of the BD FACS AriaII was assisted by the flow cytometry facility stuff at Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign (UIUC).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available on the ACS Publication website.
Table S1. List of primers used in this study; Table S2. Sequence of “RCS” synthetic DNA fragment; Figure S1: Transgene expression level vs copy number; Figure S2. Linear regression of GFP fluorescence versus transgene copy number; Figure S3. Repressive BAC transgene arrays with UBC-GFP-ZeoR reporter gene form highly condensed chromatin but have altered nuclear localization; Figure S4. Differential localization of the UBC-GFP-ZeoR reporter gene sequence in DHFR vs HBB BAC transgene territories. Figure S5. GFP fluorescence histograms of representative cell clones as a function of time in culture without selection; Figure S6. Maps of Reporter and DHFR modules used for BAC-MAGIC; Figure S7. Expression of dual-reporter BAC in U2OS cells.
REFERENCES
- (1).Walsh G (2018) Biopharmaceutical benchmarks 2018. Nat. Biotechnol 36, 1136–1145. [DOI] [PubMed] [Google Scholar]
- (2).Prelich G (2012) Gene overexpression: uses, mechanisms, and interpretation. Genetics 190, 841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Glover DJ, Lipps HJ, and Jans DA (2005) Towards safe, non-viral therapeutic gene expression in humans. Nat. Rev. Genet 6, 299–310. [DOI] [PubMed] [Google Scholar]
- (4).Rizzo MA, Davidson MW, and Piston DW (2009) Fluorescent Protein Tracking and Detection: Applications Using Fluorescent Proteins in Living Cells. Cold Spring Harb. Protoc 2009, pdb.top64. [DOI] [PubMed] [Google Scholar]
- (5).Takahashi K, and Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- (6).Machida K, Masutani M, Kobayashi T, Mikami S, and Nishino Y (2012) Reconstitution of the human chaperonin CCT by co-expression of the eight distinct subunits in mammalian cells. PROTEIN Expr. Purif 82, 61–69. [DOI] [PubMed] [Google Scholar]
- (7).Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, and Vignali DA (2004) Correction of multi-gene deficiency in vivo using a single “self-cleaving” 2A peptide-based retroviral vector. Nat. Biotechnol 22, 589–594. [DOI] [PubMed] [Google Scholar]
- (8).Akhtar W, de Jong J, Pindyurin AV, Pagie L, Meuleman W, de Ridder J, Berns A, Wessels LF, van Lohuizen M, and van Steensel B (2013) Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927. [DOI] [PubMed] [Google Scholar]
- (9).Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, Dougan G, Dawson MA, Lehner PJ, Göttgens B, Dougan G, Dawson MA, and Lehner PJ (2015) GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Karpen GH (1994) Position-effect variegation and the new biology of heterochromatin. Curr. Opin. Genet. Dev 4, 281–291. [DOI] [PubMed] [Google Scholar]
- (11).Suzuki M, Kasai K, and Saeki Y (2006) Plasmid DNA sequences present in conventional herpes simplex virus amplicon vectors cause rapid transgene silencing by forming inactive chromatin. J. Virol 80, 3293–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Minoguchi S, and Iba H (2008) Instability of retroviral DNA methylation in embryonic stem cells. Stem Cells 26, 1166–1173. [DOI] [PubMed] [Google Scholar]
- (13).Dorer DR, and Henikoff S (1997) Transgene repeat arrays interact with distant heterochromatin and cause silencing in cis and trans. Genetics 147, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Garrick D, Fiering S, Martin DI, and Whitelaw E (1998) Repeat-induced gene silencing in mammals. Nat. Genet 18, 56–59. [DOI] [PubMed] [Google Scholar]
- (15).Laker C, Meyer J, Schopen A, Friel J, Heberlein C, Ostertag W, and Stocking C (1998) Host cis-mediated extinction of a retrovirus permissive for expression in embryonal stem cells during differentiation. J. Virol 72, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Herbst F, Ball CR, Tuorto F, Nowrouzi A, Wang W, Zavidij O, Dieter SM, Fessler S, van der Hoeven F, Kloz U, Lyko F, Schmidt M, von Kalle C, and Glimm H (2012) Extensive methylation of promoter sequences silences lentiviral transgene expression during stem cell differentiation in vivo. Mol. Ther 20, 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hotta A, and Ellis J (2008) Retroviral vector silencing during iPS cell induction: an epigenetic beacon that signals distinct pluripotent states. J. Cell. Biochem 105, 940–948. [DOI] [PubMed] [Google Scholar]
- (18).Brophy JA, and Voigt CA (2014) Principles of genetic circuit design. Nat. Methods 11, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhu Y, Feuer G, Day SL, Wrzesinski S, and Planelles V (2001) Multigene lentiviral vectors based on differential splicing and translational control. Mol. Ther 4, 375–382. [DOI] [PubMed] [Google Scholar]
- (20).Tian J, and Andreadis ST (2009) Independent and high-level dual-gene expression in adult stem-progenitor cells from a single lentiviral vector. Gene Ther. 16, 874–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pikaart MJ, Recillas-Targa F, and Felsenfeld G (1998) Loss of transcriptional activity of a transgene is accompanied by DNA methylation and histone deacetylation and is prevented by insulators. Genes Dev. 12, 2852–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Emery DW, Yannaki E, Tubb J, and Stamatoyannopoulos G (2000) A chromatin insulator protects retrovirus vectors from chromosomal position effects. Proc. Natl. Acad. Sci 97, 9150–9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Guy LG, Kothary R, DeRepentigny Y, Delvoye N, Ellis J, and Wall L (1996) The beta-globin locus control region enhances transcription of but does not confer position-independent expression onto the lacZ gene in transgenic mice. EMBO J. 15, 3713–3721. [PMC free article] [PubMed] [Google Scholar]
- (24).Grosveld F, van Assendelft GB, Greaves DR, and Kollias G (1987) Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell 51, 975–985. [DOI] [PubMed] [Google Scholar]
- (25).Phi-Van L, von Kries JP, Ostertag W, and Strätling WH (1990) The chicken lysozyme 5’ matrix attachment region increases transcription from a heterologous promoter in heterologous cells and dampens position effects on the expression of transfected genes. Mol. Cell. Biol 10, 2302–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kim JM, Kim JS, Park DH, Kang HS, Yoon J, Baek K, and Yoon Y (2004) Improved recombinant gene expression in CHO cells using matrix attachment regions. J. Biotechnol 107, 95–105. [DOI] [PubMed] [Google Scholar]
- (27).Williams S, Mustoe T, Mulcahy T, Griffiths M, Simpson D, Antoniou M, Irvine A, Mountain A, and Crombie R (2005) CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells. BMC Biotechnol. 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Müller-Kuller U, Ackermann M, Kolodziej S, Brendel C, Fritsch J, Lachmann N, Kunkel H, Lausen J, Schambach A, Moritz T, and Grez M (2015) A minimal ubiquitous chromatin opening element (UCOE) effectively prevents silencing of juxtaposed heterologous promoters by epigenetic remodeling in multipotent and pluripotent stem cells. Nucleic Acids Res. 43, 1577–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kwaks TH, Barnett P, Hemrika W, Siersma T, Sewalt RG, Satijn DP, Brons JF, van Blokland R, Kwakman P, Kruckeberg AL, Kelder A, and Otte AP (2003) Identification of anti-repressor elements that confer high and stable protein production in mammalian cells. Nat. Biotechnol 21, 553–558. [DOI] [PubMed] [Google Scholar]
- (30).Truffinet V, Guglielmi L, Cogné M, and Denizot Y (2005) The chicken β-globin HS4 insulator is not a silver bullet to obtain copy-number dependent expression of transgenes in stable B cell transfectants. Immunol. Lett 96, 303–304. [DOI] [PubMed] [Google Scholar]
- (31).Bian Q, and Belmont AS (2010) BAC TG-EMBED: one-step method for high-level, copy-number-dependent, position-independent transgene expression. Nucleic Acids Res. 38, e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Blaas L, Musteanu M, Eferl R, Bauer A, and Casanova E (2009) Bacterial artificial chromosomes improve recombinant protein production in mammalian cells. BMC Biotechnol. 9, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zboray K, Sommeregger W, Bogner E, Gili A, Sterovsky T, Fauland K, Grabner B, Stiedl P, Moll HP, Bauer A, Kunert R, and Casanova E (2015) Heterologous protein production using euchromatin-containing expression vectors in mammalian cells. Nucleic Acids Res. 43, gkv475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chaturvedi P, Zhao B, Zimmerman DL, and Belmont AS (2018) Stable and reproducible transgene expression independent of proliferative or differentiated state using BAC TG-EMBED. Gene Ther. 25, 376–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fitzsimons HL, Bland RJ, and During MJ (2002) Promoters and regulatory elements that improve adeno-associated virus transgene expression in the brain. Methods 28, 227–236. [DOI] [PubMed] [Google Scholar]
- (36).Brooks AR, Harkins RN, Wang P, Qian HS, Liu P, and Rubanyi GM (2004) Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med 6, 395–404. [DOI] [PubMed] [Google Scholar]
- (37).Hong S, Hwang DD, Yoon S, Isacson O, Ramezani A, Hawley RG, and Kim K (2007) Functional analysis of various promoters in lentiviral vectors at different stages of in vitro differentiation of mouse embryonic stem cells. Mol. Ther 15, 1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chen C, Krohn J, Bhattacharya S, and Davies B (2011) A comparison of exogenous promoter activity at the ROSA26 locus using a PhiC31 integrase mediated cassette exchange approach in mouse ES cells. PLoS One 6, e23376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren BZ, and Lahn BT (2010) Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One 5, e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Warming S, Costantino N, Court DL, Jenkins NA, and Copeland NG (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Khanna N, Bian Q, Plutz M, and Belmont AS (2013) BAC manipulations for making BAC transgene arrays Methods Mol. Biol (Shav-Tal Y, Ed.) 1042, 197–210. [DOI] [PubMed] [Google Scholar]
- (42).Mizushima S, and Nagata S (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- (44).She X, Rohl CA, Castle JC, Kulkarni AV, Johnson JM, and Chen R (2009) Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics 10, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, and ter Elst A (2007) Evidence based selection of housekeeping genes. PLoS One 2, e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, and Soriano P (1997) Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc. Natl. Acad. Sci. U. S. A 94, 3789–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bertulat B, de Bonis ML, Della Ragione F, Lehmkuhl A, Milden M, Storm C, Jost KL, Scala S, Hendrich B, D’Esposito M, and Cardoso MC (2012) MeCP2 Dependent Heterochromatin Reorganization during Neural Differentiation of a Novel Mecp2-Deficient Embryonic Stem Cell Reporter Line. PLoS One 7, e47848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sinclair P, Bian Q, Plutz M, Heard E, and Belmont AS (2010) Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J. Cell Biol 190, 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bian Q, Khanna N, Alvikas J, and Belmont AS (2013) β-Globin cis-elements determine differential nuclear targeting through epigenetic modifications. J. Cell Biol 203, 767–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhao B, Chaturvedi P, Zimmerman DL, and Belmont AS (2019) Versatile multi-transgene expression using improved BAC TG- EMBED toolkit, novel BAC episomes, and BAC-MAGIC. bioRxiv 10.1101/708024. [DOI] [Google Scholar]
- (51).Sinclair P, Bian Q, Plutz M, Heard E, and Belmont AS (2010) Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J. Cell Biol 190, 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Shao Z, and Zhao H (2012) DNA assembler: A synthetic biology tool for characterizing and engineering natural product gene clusters Methods Enzymol. 1st ed. Elsevier Inc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Shao Z, Zhao H, and Zhao H (2009) DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 37, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (55).Richardson SM, Mitchell LA, Stracquadanio G, Yang K, Dymond JS, DiCarlo JE, Lee D, Huang CL, Chandrasegaran S, Cai Y, Boeke JD, and Bader JS (2017) Design of a synthetic yeast genome. Science 355, 1040–1044. [DOI] [PubMed] [Google Scholar]
- (56).Khanna N, Hu Y, and Belmont AS (2014) HSP70 transgene directed motion to nuclear speckles facilitates heat shock activation. Curr. Biol 24, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Strukov YG, and Belmont AS (2008) Development of mammalian cell lines with lac operator-tagged chromosomes. Cold Spring Harb. Protoc 2008, pdb.prot4903. [DOI] [PubMed] [Google Scholar]
- (58).Sambrook J, and Russell DW (2006) Purification of nucleic acids by extraction with Phenol:Chloroform. Cold Spring Harb. Protoc 2006, pdb.prot4455. [DOI] [PubMed] [Google Scholar]
- (59).Dernburg AF (2011) Fragmentation and labeling of probe DNA for whole-mount FISH in Drosophila. Cold Spring Harb. Protoc 2011, 1527–1530. [DOI] [PubMed] [Google Scholar]
- (60).Solovei I, and Cremer M (2010) 3D-FISH on cultured cells combined with immunostaining. Methods Mol. Biol 659, 117–126. [DOI] [PubMed] [Google Scholar]
- (61).Cremer M, Grasser F, Lanctôt C, Müller S, Neusser M, Zinner R, Solovei I, and Cremer T (2008) Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol. Biol 463, 205–239. [DOI] [PubMed] [Google Scholar]
- (62).Mowiol mounting medium. Cold Spring Harb. Protoc 2006, pdb.rec10255.
- (63).Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gietz RD, and Schiestl RH (2007) Large-scale high-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 38–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.