

Abstract
The primary cilium is an organelle which nearly all cells within the body contain. Its function is to sense the extracellular environment through its abundance of receptors and linked signaling pathways, working as an antenna. Ciliary defects lead to different pathologies. In particular, many tumors lose primary cilia, and this is linked with negative implications for the cell such as an increase in malignancy. In this work we will go through the knowledge of the role of primary cilia in normal conditions, how it is involved in diverse signaling pathways, and in disease, particularly in cancer, highlighting its tumor suppressor properties.
Keywords: primary cilia, cancer, HDAC6
Graphical Abstract

1. Primary cilia, its function and architecture
Primary cilia were first reported in a variety of vertebrate cells by Kowalevsky in 1867 and described protruding into the lumen from rabbit epithelial cells along the length of the nephron in 1898 by the Swiss anatomist KW Zimmerman, and named as we currently know it by Sergei Sorokin in 1968 [1, 2]. Primary cilia, from the Latin word ‘eyelashes’, are cellular organelles that are present on almost all type of cells of the body, including stem, epithelial, endothelial, connective tissue, muscle cells, and neurons, while absent on differentiated cells of myeloid or lymphoid origin, hepatocytes, mature adipocytes and skeletal muscle [3, 4]. The cilium contains a microtubule scaffold which protrudes from the cell, and is ideally positioned to sense and interact with the cellular environment, comparable to an ‘antenna’. The axoneme is the main protruding part of primary cilia. Contrary to motile cilia that have a 9+2 microtubule arrangement, the axoneme of primary cilia contains a 9+0 microtubule arrangement and lack dynein, a motor protein needed for motility (Figure 1A). The microtubules are composed of polymerized alpha- and beta-tubulin dimers stabilized by acetylation, glycylation and glutamylation. Its length can vary between 5–10 μm and around 300 nm diameter. Inside of the cell, the nine doublet microtubules of the axoneme anchor on the triplet microtubules of the mother centriole in the basal body and the two regions are connected by a transition zone [5]. A centriole is mandatory for cilia formation. The primary cilia membrane is continuous with the plasma membrane, but it is partitioned from the rest of the plasma membrane by a transition zone and show enrichment of lipids and proteins. Septins on the transition zone impede ciliary membrane proteins to diffuse to rest of the plasma membrane.
Figure 1. Primary cilium, receptors and signaling pathways.

A. Illustration of transversal plane of primary and motile cilia. Primary cilia have a 9+0 microtubule arrangement while motile cilia contain a 9+2 arrangement and have dynein. B. The primary cilia sense the extracellular environment through its multiple ciliary receptors that results in activation or inhibition of different signaling pathways. LKB1: liver kinase B1; P2YR: Purinergic receptors; PC-1: polycystin-1; PC-2: polycystin-2; PDGFRα: Platelet-derived growth factor receptor α; PTCH: patched receptor; SMO: Smoothened receptor; TGR5: G-protein-coupled bile acid receptor 1; TRPV4: Transient Receptor Potential Vanilloid 4.
Among its numerous functions, the cilium senses the extracellular environment through chemoreception in olfactory neurons, photoreception in photoreceptor cells and mechanosensing of fluid flow in kidney epithelial cells for example. They also can sense the extracellular environment through osmotic-, thermo-, and gravity- receptors that are then transduced into different intracellular pathways that generate cellular responses such as Sonic hedgehog, PDGF, TGFβ, Wnt, Hippo, NOTCH signaling pathways. For example, in kidney tubule cells, primary cilia can activate calcium flux as a response of liquid flowing through its mechanoreceptors. Specialized non-motile cilia in photoreceptors and olfactory receptors use G protein-coupled receptors to respond to sensory stimuli [6]. In cholangiocytes, the epithelial cells which surround the hepatic lumen of bile ducts, the primary cilium functions as a sensor of diverse bile stimuli with proteins such as polycystin-1 (a cell surface receptor) and polycystin-2 (a calcium channel), that works as a mechanosensory complex on the cilia surface after the bending of primary cilia by luminal flow; TRPV4, that works as an osmosensory channel activated by extracellular hypotonicity; P2Y12, which is activated by nucleotides, producing changes in intracellular cAMP levels, and TGR5 that functions as a chemoreceptor of biliary acids [7–10] (Figure 1B).
2. Mechanisms of ciliary assembly and disassembly
Ciliary assembly and disassembly are tightly linked with the cell cycle. Indeed, the cilium and mitotic spindle assembles in a mutually exclusive manner. To understand ciliary loss in cancer, it is necessary to comprehend the mechanisms of ciliary dynamics. Primary cilia assemble when the cell exits mitosis or differentiates in the post-mitotic G0/G1 phases of the cell cycle. This dependence to the cell cycle resides on the fact that the primary cilia anchors on the basal body. The basal body is derived from the two centrioles of the centrosome [11]. During the first stage, the basal body associates with a Golgi derived vesicle and migrates to the cell surface. Specific motifs on vesicle membrane proteins lead them from Golgi to the cilia [12]. Additionally, the lateral transport between the plasma membrane and the cilia is controlled by a specialized membrane at the ciliary base regulated by the filament-forming small G-protein Septin2 and by interaction with the ezrin/radixin/moesin (ERM)-actin network [13–15]. Arl and Rab GTPases of the Ras superfamily are involved in cilium assembly [16, 17]. CP110 and Cep97 are interacting centrosome proteins which loss is linked with ciliary protrusion [18, 19]. CP110 caps the distal end of the centriole to prevent ciliary assembly. CEP290 is needed for ciliogenesis and controls vesicular trafficking into the centrosome through Rab8 [20]. Other proteins such as ninein, CEP170, and ODF2 are needed for anchoring the cilium to the basal body. CEP164 works as a connector between the basal body and plasma membrane. Once the primary cilium begins to extend, further growth is dependent on the bidirectional movement of multiprotein complexes on the axoneme known as Intraflagellar Transport (IFT), which allows the transportation of ciliary cargo through the microtubules. In this regard some of these components are essential for ciliary assembly, expression and maintenance. In the case of IFT88, downregulated expression leads to ciliary loss [21]. Cargo proteins are trafficked along the microtubule from the cytoplasm to the tip of cilium through kinesins (KIF3A, KIF3B, KAP in mammals) associated with IFT complex B and back down via dynein (DHC2 in mammals) associated with IFT complex A. Tubulin subunits and other components are incorporated on the distal tip of the axoneme, and once assembled, the cilium maintains dynamic instability with new tubulin continually incorporated into the tip [13]. The rate of this transport defines the length of the cilia. GSK3 and von Hippel-Linday are associated with ciliary maintenance [18].
The basal body works as the initiation nucleation site for the growth of the axoneme microtubules within differentiated or quiescent cells in G1/G0. The basal body is composed of the centrioles, therefore many proteins involved in cell division are involved with primary cilia assembly and disassembly such as PLK1 which is a mitotic kinase involved in cell cycle progression through phosphorylation; Aurora A Kinase, that is a centrosome mitotic kinase involved in the entry to the S phase; and NEK2 involved on the disassembly of axoneme microtubules. For example, the ciliary protein IFT88 regulates G1-S transition in non-ciliated HeLa cells as a centrosome protein [18, 22]. Fa2p deletion is involved with delay at the G2-M transition in Chlamydomonas [18, 23]. Cnk2p assess the cell size prior to mitosis in Chlamydomonas[18, 24]. CP110 is regulated during the cell cycle by CDK2 [20]. The centrioles are needed for chromosome segregation in cell division. Consequently, cilia are disassembled and absent during the mitotic process, while the basal body converts into the centrioles that organize the mitotic spindles and centrosomes. The cilium must be disassembled prior mitosis. Primary cilia disassembly usually follows at the G2-M transition.
Aurora kinase A is a centrosome associated kinase that activates at mitotic entry and is involved in the mechanism of cilia disassembly on mammalian cells [25]. The adaptor protein HEF1/NEDD9 and calmodulin in presence of calcium are responsible of activating Aurora A Kinase that phosphorylates histone deacetylase 6 (HDAC6). HDAC6 is a deacetylase that, contrary to many of HDACs, has cytoplasmic functions. This protein deacetylates the ciliary axoneme, destabilizing the microtubules, leading to ciliary disassembly [26]. Normal cholangiocyte cells that overexpress HDAC6 reduce ciliary frequency and acquire a malignant phenotype involving an increase in proliferation rate and anchorage independent growth [26]. Upstream of HEF1 proteins which may be important for ciliary disassembly are the ligands of integrin receptor, GPCR, Ca2+ and TGFβ [18]. The mitotic kinase Plk1 [27] activates HEF1, and Nek2 [28] phosphorylates Kif24, promoting microtubule disassembly through microtubule depolymerization, and are also involved in tumorigenesis (Table I).
Table I.
Key Players in primary cilia assembly and disassembly
| Protein | Role | Reference |
|---|---|---|
| Ciliary Assembly | ||
| Septin2 | Lateral transport between the plasma membrane and the cilia | [13–15] |
| Arl / Rab GTPases | Targeted transport of membrane proteins to the cilia | [16, 17] |
| CP110 | Caps the distal end of the centriole | [18, 19] |
| Cep97 | Required for recruitment of C110 to the centrosome | [18, 19] |
| Cep290 | Control vesicular trafficking through Rab8 | [20] |
| Ninein, CEP170, OFD2 | Anchors the cilium to the basal body | [18] |
| IFT88 | Intraflagellar transport | [21,22] |
| KIF3A, KIF3B, KAB | Kinesins | [13] |
| DHC2 | Dyneins | [13] |
| GSK3, von Hippel-Linday | Components of a pathway that maintains the cilia | [18] |
| Ciliary Disassembly | ||
| PLK1 | Mitotic kinase involved in cell cycle progression through phosphorylation | [27] |
| Aurora A Kinase | Centrosome mitotic kinase involved in the entry to the S phase. Activates HDAC6 | [25] |
| NEK2 | Disassembles axoneme microtubules | [28] |
| HEF1/NEDD9 | Activates Aurora A Kinase | [25] |
| HDAC6 | Destabilizes microtubules by deacetylation leading to ciliary disassembly | [26] |
Serum starvation in cultured cells is known to enhance ciliogenesis and autophagy. Autophagy is important for ciliogenesis. It is known that the degradation of OFD1 by autophagy at the centriole satellites is needed for ciliogenesis [29]. But autophagy also has a role in the degradation of ciliary proteins. The inhibition of autophagy restores ciliogenesis in the Hürthle cell line XTC.UC1 and promoted the accumulation of IFT88 and ARL13B [30]. In KECs cells the autophagy machinery colocalizes at the cilia and the cells that have an impairment of autophagy form cilia longer and faster than normal KECs cells [31]. Additionally, the ubiquitin-proteasome system has a role in ciliogenesis. The removal of trichoplein by the ubiquitin-proteasome machinery causes Aurora-A inactivation and leads to ciliogenesis in RPE1 cells [32]. Similar is the case of NDE1, a negative regulator of ciliary growth that is degraded through the ubiquitin proteasome system increasing ciliary length [33]. Usp14 is a protein of the ubiquitin-proteasome system involved in ciliogenesis, cilia growth and Hh signaling [34].
3. Signaling pathways regulated by primary cilia
Many signaling pathways that are related to cancer are linked to the primary cilia. In this regard, Hippo, mTOR, NOTCH, Sonic hedgehog, PDGF, TGFβ and Wnt signaling pathways have been shown to be regulated through ciliary dependent mechanisms with an outcome on cell apoptosis, autophagy, differentiation, proliferation, size and tumorigenesis [35, 36].
Sonic hedgehog pathway proteins are enriched in the cilia. This pathway is involved in development and adult tissue homeostasis and shown to be involved in cancers [37]. In the Sonic hedgehog (Shh) pathway, the Patched (PTCH) receptor located in the ciliary membrane binds the Shh ligand. The seven transmembrane protein Smoothened (SMO) is outside of the cilia in vesicles close to the cell membrane. Inactive PTCH1 inhibits its entry. The zinc finger transcription factors Gli1, Gli2 and Gli3 binds to the inhibitor SuFu during the inactivated state, facilitating their proteasomal degradation and the generation of the Gli3 repressor through partial proteolysis of Gli3. Kif7 acts also as a negative regulator of this pathway having a role in controlling localization of hedgehog signaling molecules through organizing the cilium tip compartment by the regulation of the dynamics of microtubules plus ends. When Shh binds to PTCH, this receptor activates and leaves the cilia, allowing the entrance of SMO and consequently, the Gli factors detach form SuFu allowing migration and action into the nucleus [37]. When activated, the Gli proteins trigger the expression of genes involved in proliferation, survival and epithelial-to-mesenchymal transition [38] (Figure 2A). Cilia may regulate tumorigenesis through the function of Hedgehog pathway in different cell types [18, 39]. Cilia are involved with the canonical Hedgehog pathway and the ciliary loss may stimulate tumor development in some kinds of cancer [40, 41]. In fact, it stimulates tumor development on meduloblastoma and basal cell carcinoma. Another example is that GLI3 repressor is loss when cells are deciliated in chondrocytes, activating this pathway (Chang, 2012). The protein ARL13B is responsible of the transportation of Hedgehog components across the cilia [42].
Figure 2. Signaling pathways.

A. Hedgehog signaling. In absence of Hg ligands PTCH accumulates in the cilia and inhibits the entrance of SMO. SuFu binds to Gli proteins inhibiting its transcription factor function. The partial proteolysis of Gli3 produces a repressor form that inhibit the expression of Hg targets. In presence of Hg ligands (i.e. Shh) occurs the activation of PTCH inducing its degradation. Without PTCH, SMO can accumulate in the cilia preventing the degradation of Gli proteins. The Gli proteins can translocate to the nucleus and activate target gene expression. B. Wnt signaling. In the canonical pathway, β-catenin is degraded by a protein complex that includes Apc and Axin. With the activation of the Frizzled receptor the destruction complex is inhibited by the disheveled protein resulting in the stabilization of β-catenin, its translocation to the nucleus and the activation of the expression of target genes. The non-canonical way is involved on the establishment of cell polarity through small GTPases activation. Inversin has a switch role between the canonical and non-canonical pathway. C. Tyrosine kinases signaling. PDGFRα colocalizes in the cilia. The activation transduces in the activation of MEK1/2, AKT (trough the activation of PI3K) and ERK1/2 resulting in cell cycle entry.
The Wnt signaling pathway involves the Wnt ligands interacting with the Frizzled family of receptors. The canonical Wnt pathway includes the stabilization of β-catenin and the translocation into the nucleus where it interacts with transcription factors involved in cell proliferation, differentiation and metabolism. The protein Disheveled (Dvl) is activated by this pathway and inhibits the beta-catenin degrading protein complex composed of Apc and Axin. The non-canonical pathway is independent of β-catenin and is involved in establishment of cell polarity on the plane of the epithelium through activation of the small GTPases Rho and Rac (Figure 2B). Many factors of this pathway are present on the basal body: vangl-2, GSK3 beta, Apc and Inversin [37]. It has been stated that the progression of tumors associated with defects in cilia are linked to a constitutive activation of the Wnt/β-catenin pathway [43]. The Inversin protein works as a molecular switch between the canonical and non-canonical Wnt pathway. Specifically, Inversin inhibits Dvl protein of the canonical pathway by degradation and its required for the non-canonical Wnt pathway for polarization. The downregulation of the ciliary protein Kif3a, caused constitutive phosphorylation of Dvl [44]. Additionally, the activation of the non-canonical pathway induces the formation of a Dvl2-Plk1 complex that stabilizes HEF1 and activation of Aurora A, leading to ciliary disassembly.
The NOTCH signaling pathway includes Delta-like and Jagged protein ligands and four NOTCH receptors. The activation of the Notch receptor generates a proteolytic cleavage that result in an intracellular fragment that translocate into the nucleus for activation of genes related with proliferation. NOTCH1 and NOTCH3 have ciliary localization [45]. The induced ciliary loss of basal corneal epithelial cells decreases this signaling pathway and is linked with hyperproliferation [46].
Primary cilia have been associated with Tyrosine kinase receptors. The best described is PDGFα. The PDGF pathway has two receptors PDGFRα and PDGFRβ and four ligands. The union of the ligand produces dimerization of the receptors and autophosphorylation of thyroxines. This activates downstream molecules such as ERK1/2, MEK1/2 and AKT kinases resulting in cell cycle entry (Figure 2C). The receptor PDGFRα colocalizes with cilia [36, 47]. The platelet-derived growth factor (PDGF) pathway is dysregulated in different cancerous tissues and studies revealed that the primary cilium is essential for its ligand-induced activity [48]. TGFβ has a dual role on tumorigenesis, having an inhibitor role at the beginning of tumor growth and a tumor growth enhancer at later stages. TGFβ-RI and TGFβ-RII are located at the base of the primary cilia [36, 49]. The effectors ERK1/2 and SMAD3 are present at the base of the cilia after TGFβ induced activation [49].
mTOR is a serine/threonine kinase that participates in different cellular processes such as cell size regulation, metabolism, growth, proliferation and survival. Primary cilia regulate mTOR through activation of the ciliary protein LKB1 in MDCK cells [50]. Additionally, the activation of P2Y11 receptors by nucleotides and the subsequent activation of LKB1, leads to inhibition of migration and invasion in normal ciliated cholangiocytes [51].
The Hippo pathway is a regulator of cellular proliferation, survival, differentiation and homeostasis. Its dysregulation is related to cancer development. The cascade regulates the function of transcriptional activators YAP/TAZ inhibiting their proliferative and anti-apoptotic nuclear activities. This pathway regulates and is controlled by the cilia [52]. NPHP proteins can induce TAZ activity [53]. Additionally, the upstream Hippo proteins MST and SAV1 inhibit ciliary disassembly, phosphorylating Aurora A [54].
The factors that are needed to repair damaged DNA in cancer are also linked to primary cilia. Specifically, to the centrosome that is localized in the basal body at least during parts of the cell cycle. These factors include the DNA repair proteins BRCA1, BRCA2, PARP1, and NBS1. Proteins that sense DNA damage and initiate repairing responses such as ATM, ATR, CHK1 and CHK2 and the cell cycle checkpoint and transcriptional regulator p53 also colocalizes with the basal body [35]. p53, BRCA1 and PARP1 are linked to centrosome physiology since p53 and PARP1 deficiencies, and BRCA1 mutations leads to centrosome amplification that may also lead to aberrant number of primary cilia [55, 56]. Additionally, CEP164 interacts with ATM, being phosphorylated as a response to DNA damage and its downregulation decrease phosphorylation of other proteins of the DNA damage response such as CHK2 and CHK1 [57]. Centrosomes are affected by distortion of the pericentriolar material and duplication by factors that damage DNA [58].
4. Ciliopathies
The importance of cilia becomes evident when there are defects in ciliary structures. These ciliary defects can lead to diseases that have been denominated ciliopathies. Ciliopathies comprise a group of disorders which result in abnormal formation or function of cilia. Developmental and single gene disorders are responsible for different kind of ciliopathies such as polycystic kidney disease, nephronophthisis, retinitis pigmentosa, Bardet–Biedl syndrome, Joubert syndrome, and Meckel syndrome.
Polycystic Kidney disease (PKD) is a disorder characterized by the onset of kidney cysts which also present increased proliferation. It is caused by mutations in PKD1 involved in encoding the protein PC-1, a likely membrane mechano-sensor ciliary receptor, and PKD2, involved in encoding the ciliary protein PC-2, a non-selective cationic channel permeable to calcium. The recessive form of this ciliopathy is caused by the mutation of PKHD1 that encodes the integral transmembrane protein fibrocystin that is found on primary cilia [59]. Half of the neonates carrying this mutation die due to pulmonary hypoplasia. In adult survivors, complications are associated with renal insufficiency, renal diseases and hepatic fibrosis [60]. Polycystic liver disease (PLD) is a manifestation of PKD in the liver that can also exist alone as a result of mutations of the genes PRKCSH, SEC63, LRP5, ALG8, SEC61B, GANAB and PKHD1, PKD1 and PKD2 which products are ciliary proteins. The patients develop cysts, though are usually asymptomatic, yet severe hepatomegaly may cause serious complications [60].
Nephronophthisis (NPH), is a genetic disorder of the kidneys which affects mainly children. The abnormalities lead to increased urine production (polyuria), excessive thirst (polydipsia), general weakness, and extreme tiredness (fatigue) [61]. NPH is an inherited disease which occurs in an autosomal recessive manner. There are three clinical subtypes that are recognized based on age of onset of the disease. Nephrocystin-3 protein encoded by NPHP3 gene, is required for normal ciliary development and function and it’s involved in the adolescent form of nephronophthisis [62]. This protein inhibits disheveled-1-induced canonical Wnt-signaling activity and control the non-canonical Wnt signaling which regulates planar cell polarity [63].
Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision. Due to this disease, the retina is unable to respond to light, deteriorating night and peripheral vision. RP starts with night blindness, inability to discriminate colors from one another, leading ultimately to a progressive tunnel vision. This is the most common manifestation of inherited retinal diseases with a high degree of genetic, allelic, and phenotypic heterogeneity. About 20–30% of RP cases are associated with non-ocular clinical phenotypes [64]. RP can be inherited in an autosomal dominant, autosomal recessive and X-linked manners. McCray et al., in 2019 showed disrupted ciliary orientation in patients with X-linked retinitis pigmentosa [65]. Ciliary genes and cilia have an important role in retinal structure and function. Cilia- and flagella-associated protein 410 (CFAP410) plays a role in cilia formation and/or maintenance and also regulates cell morphology and cytoskeletal organization [66]. This CFAP410 protein is causative for axial spondylometaphyseal dysplasia (axial SMD), which presents with retinitis pigmentosa (RP) [67]. In 2019, Kurashige et al. described that RP is associated with CFAP410 mutation [68]. Centrosome-associated protein CEP250 is an important active component of the centrosome. It also plays a vital role in centrosome cohesion, centriole duplication and biogenesis, cell cycle progression control, and is also associated with RP [68]. A nonsense mutation was reported in CEP250 in a consanguineous family with nonsyndromic RP [69].
Bardet–Biedl syndrome (BBS) is a ciliopathic human genetic disorder. BBS presents as a rare disease found in approximately 1 in 250,000 people worldwide. Many patients of this disease have a decreased ability to sense smells due to a change in the size of the “olfactory bulb” in the brain. Symptoms also include obesity, particularly with fat deposition along the abdomen. BBS proteins are necessary to maintain primary cilia structure and function [70]. It is an inherited autosomal recessive disorder caused by mutations in more than 20 different genes which includes, BBS1, BBS2, ARL6 (BBS3), BBS4, BBS5, MKKS (BBS6), BBS7, TTC8 (BBS8), BBS9, BBS10, TRIM32 (BBS11), BBS12, MKS1 (BBS13), CEP290 (BBS14), WDPCP (BBS15), SDCCAG8 (BBS16), LZTFL1 (BBS17), BBIP1 (BBS18), IFT27 (BBS19), IFT72 (BBS20), and C8ORF37 (BBS21). Moreover, there is no clear link between the mutations identified and disease severity.
Joubert syndrome is a brain development disorder due to absence or underdevelopment of the cerebellar vermis. The signs and symptoms of this condition vary among affected individuals. Most common symptoms are lack of muscle control (ataxia), abnormal breathing patterns (hyperpnea), sleep apnea and abnormal eyes movement. Joubert syndrome is known as a rare disease and it’s an autosomal recessive inheritance disorder. The prevalence of Joubert syndrome is 1/80,000 [71]. The genetic bases of Joubert syndrome are extremely complex. More than 30 gene mutation has been identified, among them NPHP, AHI1, CEP290, TMEM67, CC2D2A, and C5orf42 and X-linked genes, such as OFD1. ADP-ribosylation factor-like protein 3 (ARL3) is a small GTP-binding protein which cycles between an inactive GDP-bound and an active GTP-bound form [72]. ARL3 play a crucial role for ciliogenesis and formation of the axoneme. It was reported that the ARL3 mutation is implicated in Joubert Syndrome by ciliary disruption [73]. ARL13B is a cilium-specific protein required to control the microtubule-based, ciliary axoneme structure. ARL13B maintain uniform distribution of proteins along the ciliary membrane by binding with tubulin. Mutations of this gene are also responsible for Joubert Syndrome [74].
Meckel syndrome is a rare inherited autosomal recessive disorder with 100% mortality rate. Symptoms includes oral clefts genital anomalies, central nervous system (CNS) malformations and liver fibrosis. It was first reported by Johann Friedrich Meckel in 1822. The genes involved includes MKS1, MKS2, MKS3, MKS4, MKS5, and MKS6 [75]. A study also reported that mutation in B9D1 and B9D2 genes are responsible for Meckel syndrome. The transition zone (TZ) is a cilium domain which maintain the ciliary compartment separated from the rest of the cell for its sensory and signaling activity. Mutations in TZ proteins like MKS6 results in cilia dysfunction [76]
Interestingly, there are points of contact between ciliopathies and tumor cells, such as the loss of response to extracellular signals, increased cell proliferation, cell polarity alteration, and abnormal extracellular matrix control leading to fibrosis. It has been observed that there is a decrease of cilia expression on tumors [40].
5. Cilium and Cancer
It has been proposed that the primary cilium may work as a tumor suppressor organelle. This hypothesis is supported by the fact that primary cilia contains signaling pathways linked with cancer and additionally many different types of tumors lose primary cilia [40]. On the other hand, primary cilia has been reported to give resistance to kinase inhibitors, depending on the driver oncogenic lesion and aberrant cilia lengthening, such as with Kif7 downregulation [77].
In the case of astrocytoma/glioblastoma only 8 to 25% of the cells in vitro and in biopsies shows ciliary expression [78]. In medulloblastoma primary cilia seems to have a dual function. The knockout of ciliary proteins such as Kif3a, blocked tumor formation on a medulloblastoma mouse model that has only one Ptch allele [79]. But mouse that has expression of an active form of Gli2 only showed tumor formation when cilia was depleted [80]. In Glioblastoma it has been observed that ciliogenesis is perturbed in cell lines and in patient glioblastoma multiforme tumor samples [81].
Basal cell carcinoma, that is the most common skin cancer, usually presents a dysregulation of the Sonic hedgehog (Shh) pathway. Deciliation in keratinocytes that have an active form of SMO, abolished tumor development, while deciliation that have an active form of Gli2 induced tumorigenesis [39]. In Melanoma patients, loss of primary cilia have been seen in advanced stages [82]. EZH2, an oncogene involved in metastatic BRAF and NRAS melanoma, may be responsible for the silencing of ciliary genes in melanoma development, and the loss of cilia enhances pro-tumorigenic WNT/β-catenin signaling [83].
The cilia in colonic epithelial cells can be reduced by downregulation of TTLL3, a tubulin glycine ligase. This ciliary reduction leads to an increase of proliferation and increased tumor development in mice [84]. The decreased primary cilia expression promotes Wnt signaling during colon carcinogenesis.
Ovarian cancer present a decreased number of cilia and this decrease is associated with an increased expression of Aurora A kinase in the basal body, and correlated with decreased Hedgehog signaling and PDGRFα expression [85]. In breast cancer there is a decreased presence of cilia and is lost at a very early stage [86]. In prostate cancer, a decrease of ciliary expression at different stages during its formation and increased levels of Wnt signaling have been observed [87].
Renal cell carcinoma shows loss of cilia and this is related with a downregulation of the von Hippel-Linday (VHL) tumor suppressor gene that participates on controlling oxygen levels and microtubule stabilization through the activation of HIFα and Aurora A kinase, that leads to the activation of HDAC6 [88]. The VHL downregulation regulates beta-catenin, increasing the levels of Aurora A kinase. Other report shows an increase on NEK8 with the VHL downregulation that also leads to ciliary loss [89]. Mutations of the tumor suppressor gene FLCN, that its protein has shown to be localized in the primary cilia and basal body, are linked with Birt-Hogg-Dubé syndrome that manifest with renal cysts and predisposes to an increased risk for kidney tumor development [90]. Tuberous sclerosis complex is caused by the mutation of the tumor suppressor genes TSC1 or TSC2, and it can lead to renal manifestations such as renal cell carcinoma and renal cystic disease. TSC1 localizes in the base of primary cilia and its downregulation produces increased ciliary length [91].
In pancreatic adenocarcinoma the absence of cilia is independent of ongoing proliferation, and the ciliary loss can be reversed by inhibition of KRAS pathways [92]. The absence of cilia in early stage patient samples have been described [92]. In this particular cancer it has been demonstrated that HDAC2 is responsible for promotion of ciliary loss, and the inhibition of HDAC2 decreases the expression of Aurora A that is needed for ciliary disassembly, and thus promotes ciliary restoration [93]. On the other hand a study shows that the presence of primary cilia correlates with poor prognosis and lymph node metastasis in pancreatic ductal adenocarcinoma [94].
Mice that lack primary cilia on the thyroids due to a thyroid follicular epithelial cell-specific deletion of IFT88 showed follicular cells with malignant phenotype and developed papillary solid proliferative thyroid follicles with malignant features [95]. Additionally a decrease of primary cilia expression on rhabdomyosarcoma [96] and chondrosarcomas [97] have been reported.
On cholangiocarcinoma there is a reduction of cilia in vivo and in vitro in human patient samples and cells respectively [26]. The deciliation of normal cholangiocyte cells using drugs such as chloral hydrate or using gene-silencing techniques on ciliary proteins such as IFT88 induces proliferation, anchorage-independent growth, and invasion.
Furthermore, it also induces activation of Hedgehog and MAPK pathways that are normally negatively regulated by the cilia, both involved in the malignant cholangiocarcinoma phenotypes. The mechanisms that may explain the deciliation of cholangiocytes in cholangiocarcinoma involve the overexpression of HDAC6 [26]. Our laboratory demonstrated the dysregulation of microRNA (miRNAs) in CCA are responsible for the overexpression of HDAC6 leading to ciliary disassembly [98].
6. Ciliotherapies: Restoration of ciliary structure/function as a therapeutic approach
Cilia is considered as a tumor suppressor organelle. Many cancer cells have developed mechanisms to inhibit ciliogenesis and stimulate ciliary disassembly. The loss of primary cilia is linked with increased proliferation. Therefore, efforts to stimulate restoration of cilia on tumor cells may work as a potential therapeutic approach. HDAC6 inhibition with pharmacological or genetic approaches induces partial cilia restoration and reverses the malignant phenotype in cholangiocarcinoma. HDAC6 is overexpressed in cholangiocarcinoma, and its targeting restores ciliary expression, decrease proliferation and anchorage independent growth, and down regulates Hedgehog and MAPK pathways. This is a ciliary dependent mechanism, as cells that were transfected with shRNA targeting the ciliary protein IFT88, interrupted ciliogenesis, and the malignant phenotype couldn’t be reverted by tubastatin-A, an HDAC6 inhibitor. In vivo experiments in a model of cholangiocarcinoma showed that the inhibition of HDAC6 reduces tumor growth and induces partial restoration of cilia [26] (Figure 3). Additionally, HDAC6 inhibition also decreases proliferation and invasion capacity of chondrosarcoma tumor cells and restores the expression of primary cilia [99]. The use of pan-HDACs inhibitors is controversial because the potential adverse effects and broad epigenetics changes. In contrast specific HDAC6 inhibition is promising since viable, fertile, and normally developing, HDAC6 knock out mice model are available. The immune response is slightly affected and the mice present a small increase in cancellous bone mineral density but show not detrimental development [100]. Other HDACs may also be related with the ciliary loss. It is reported that the silencing of HDAC2 restores primary cilia in pancreatic ductal adenocarcinoma cells [93]
Figure 3. Working model.
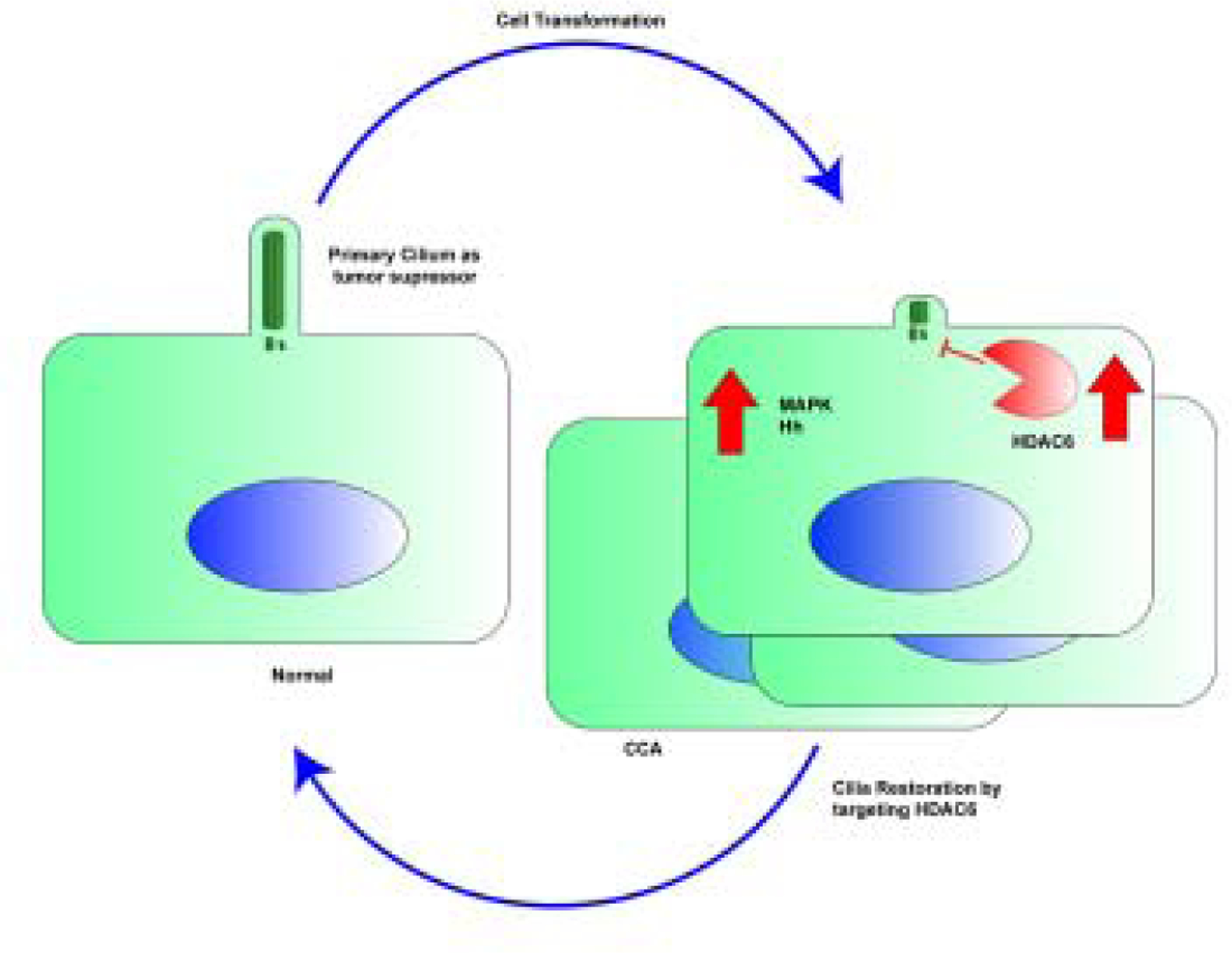
Primary cilia function as a sensor of external stimuli into internal signaling pathways with tumor suppressor characteristics. The cholangiocarcinoma malignant transformation includes loss of cilia and derepression of tumorigenic pathways. The targeting of HDAC6 would be a potential treatment approach or ciliotherapy.
Nevertheless, several non-selective HDAC inhibitors have already been approved by the US Food and Drug Administration (FDA) for cancer treatment including vorinostat, romidepsin, and panobinostat, but they show toxicity to normal tissues during cancer therapy [101]. In contrast with specific HDAC6 targeting, the use of pan-HDAC inhibitors have profound non-desirable effects due to the global epigenetic modification of gene expression. Efforts have been made to increase the specificity of the pharmacological inhibitors of HDAC6, developing compounds based on the structure of its enzymatic active site [101]. Tubacin, tubastatin A, CAY10603 and ACY1215 are among the HDAC6 selective inhibitors that have a protective effect on primary cilia.
Not only may ciliotherapies be useful for cancer but also for ciliopathies such as PKD and PLD. The therapeutic use of Tubastatin-A, Tubacin, or ACY1215, specific HDAC6 inhibitors, reduced cell proliferation and cyst growth in vitro, and ACY-1215 reduced liver cysts and fibrosis in an animal model of PKD in vivo [102]. The inhibition of class I HDACs by valproic acid produced reduction of progression of cyst formation and improved kidney function in a mouse ADPKD model based on Pkd1 [103]. Additionally, on a PKD murine model, the use of the drug fenoldopam, that is a known ciliary DR5 dopaminergic agonist, increased ciliary length and serum nitric oxide (NO) levels, reducing blood pressure [104]. In that regard, recent studies worked on the development of nanoparticles to deliver fenoldopam to the primary cilia as a therapeutic delivery system on PKD2 mice [105]. Similar is the case of nephronophthisis where the treatment with the HDAC6 specific inhibitor ST80 resulted in a rescue of expression of cilia in cells with a downregulation of NPHP2 [106]. Another approach to restore cilia includes the work of Khan et al. [107] where they screened 1600 drugs that may regenerate the cilia and decrease proliferation on tumor cells, and they found that 110 of those compounds increased ciliogenesis by at least a factor 2. Among those, the most potent were clofibrate, gefitinib, sirolimus, imexon and dexamethasone that were able to restore cilia and decrease cell proliferation. Targeting the primary cilium to inhibit tumorigenic pathways such as Sonic Hedgehog or stimulate pathways such as the activation of LKB1 could be other promising approaches. For example, it is possible to activate LKB1, bypassing the need for primary cilia, to inhibit migration, invasion and tumor growth of cholangiocarcinoma by treating with hesperidin methyl chalcone [98].
7. Future Directions
In summary, the primary cilia are linked to multiple diseases including cancer depending the tumor types. The fact that cancer cells lose primary cilia, and experimentally deciliated normal cells acquire malignancy, strongly suggest that these organelles may have a role as tumor suppressors. At least the following two reasons may explain why that is the case: i) the cilium is implicated as a sensor and transducer of different cancer key pathways such as the Hedgehog and tyrosine kinases, and ii) while it is present, the centrosomes needed for cell division are kept as the basal body from which the cilia protrudes, and not as a mitotic spindle organizer during cell division. Nevertheless, it is important to further understand the mechanisms involved in cilia disassembly in order to avoid the loss of this tumor suppressor organelle. Future efforts should be pointed in understanding how the HDAC6-induced ciliary disassembly occurs in order to design better strategies to restore ciliary expression as a potential therapeutic approach for decrease tumor malignancy or ameliorate other genetic ciliopathies such as PKD.
Acknowledgments
This work was supported by National Institutes of Health Grant R01CA183764 (to S.A.G.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Zimmermann KW, Beiträge zur Kenntniss einiger Drüsen und Epithelien, Archiv für mikroskopische Anatomie 52(3) (1898) 552–706. [Google Scholar]
- [2].Sorokin SP, Reconstructions of centriole formation and ciliogenesis in mammalian lungs, J Cell Sci 3(2) (1968) 207–230. [DOI] [PubMed] [Google Scholar]
- [3].Fry AM, Leaper MJ, Bayliss R, The primary cilium: guardian of organ development and homeostasis, Organogenesis 10(1) (2014) 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Venkatesh D, Primary cilia, J Oral Maxillofac Pathol 21(1) (2017) 8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ishikawa T, Axoneme Structure from Motile Cilia, Cold Spring Harb Perspect Biol 9(1) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schou KB, Pedersen LB, Christensen ST, Ins and outs of GPCR signaling in primary cilia, EMBO Rep 16(9) (2015) 1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Masyuk AI, Masyuk TV, LaRusso NF, Cholangiocyte primary cilia in liver health and disease, Dev Dyn 237(8) (2008) 2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Masyuk AI, Gradilone SA, Banales JM, Huang BQ, Masyuk TV, Lee SO, Splinter PL, Stroope AJ, LaRusso NF, Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors, Am J Physiol Gastrointest Liver Physiol 295(4) (2008) G725–G734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Masyuk AI, Masyuk TV, Splinter PL, Huang BQ, Stroope AJ, LaRusso NF, Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling, Gastroenterology 131(3) (2006) 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gradilone SA, Masyuk AI, Splinter PL, Banales JM, Huang BQ, Tietz PS, Masyuk TV, LaRusso NF, Cholangiocyte cilia express TRPV4 and detect changes in luminal tonicity inducing bicarbonate secretion, Proc Natl Acad Sci U S A 104(48) (2007) 19138–19143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fabbri L, Bost F, Mazure NM, Primary Cilium in Cancer Hallmarks, Int J Mol Sci 20(6) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D, Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4, EMBO J 28(3) (2009) 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Verhey KJ, Dishinger J, Kee HL, Kinesin motors and primary cilia, Biochem Soc Trans 39(5) (2011) 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jenkins PM, Hurd TW, Zhang L, McEwen DP, Brown RL, Margolis B, Verhey KJ, Martens JR, Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17, Curr Biol 16(12) (2006) 1211–1216. [DOI] [PubMed] [Google Scholar]
- [15].Milenkovic L, Scott MP, Rohatgi R, Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium, J Cell Biol 187(3) (2009) 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qin H, Wang Z, Diener D, Rosenbaum J, Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control, Curr Biol 17(3) (2007) 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yoshimura S, Egerer J, Fuchs E, Haas AK, Barr FA, Functional dissection of Rab GTPases involved in primary cilium formation, J Cell Biol 178(3) (2007) 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Plotnikova OV, Pugacheva EN, Golemis EA, Primary cilia and the cell cycle, Methods Cell Biol 94 (2009) 137–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Spektor A, Tsang WY, Khoo D, Dynlacht BD, Cep97 and CP110 suppress a cilia assembly program, Cell 130(4) (2007) 678–690. [DOI] [PubMed] [Google Scholar]
- [20].Tsang WY, Bossard C, Khanna H, Peranen J, Swaroop A, Malhotra V, Dynlacht BD, CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease, Dev Cell 15(2) (2008) 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ishikawa H, Marshall WF, Ciliogenesis: building the cell’s antenna, Nat Rev Mol Cell Biol 12(4) (2011) 222–234. [DOI] [PubMed] [Google Scholar]
- [22].Robert A, Margall-Ducos G, Guidotti JE, Bregerie O, Celati C, Brechot C, Desdouets C, The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells, J Cell Sci 120(Pt 4) (2007) 628–637. [DOI] [PubMed] [Google Scholar]
- [23].Mahjoub MR, Qasim Rasi M, Quarmby LM, A NIMA-related kinase, Fa2p, localizes to a novel site in the proximal cilia of Chlamydomonas and mouse kidney cells, Mol Biol Cell 15(11) (2004) 5172–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bradley BA, Quarmby LM, A NIMA-related kinase, Cnk2p, regulates both flagellar length and cell size in Chlamydomonas, J Cell Sci 118(Pt 15) (2005) 3317–3326. [DOI] [PubMed] [Google Scholar]
- [25].Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA, HEF1-dependent Aurora A activation induces disassembly of the primary cilium, Cell 129(7) (2007) 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF, HDAC6 inhibition restores ciliary expression and decreases tumor growth, Cancer Res 73(7) (2013) 2259–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wang G, Chen Q, Zhang X, Zhang B, Zhuo X, Liu J, Jiang Q, Zhang C, PCM1 recruits Plk1 to the pericentriolar matrix to promote primary cilia disassembly before mitotic entry, J Cell Sci 126(Pt 6) (2013) 1355–1365. [DOI] [PubMed] [Google Scholar]
- [28].Kim S, Lee K, Choi JH, Ringstad N, Dynlacht BD, Nek2 activation of Kif24 ensures cilium disassembly during the cell cycle, Nat Commun 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q, Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites, Nature 502(7470) (2013) 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee J, Yi S, Kang YE, Chang JY, Kim JT, Sul HJ, Kim JO, Kim JM, Kim J, Porcelli AM, Kim KS, Shong M, Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy, Oncotarget 7(48) (2016) 79117–79130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pampliega O, Orhon I, Patel B, Sridhar S, Díaz-Carretero A, Beau I, Codogno P, Satir BH, Satir P, Cuervo AM, Functional interaction between autophagy and ciliogenesis, Nature 502 (2013) 194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kasahara K, Kawakami Y, Kiyono T, Yonemura S, Kawamura Y, Era S, Matsuzaki F, Goshima N, Inagaki M, Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension,Nat Commun 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maskey D, Marlin MC, Kim S, Kim S, Ong EC, Li G, Tsiokas L, Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length, EMBO J 34(19) (2015) 2424–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Massa F, Tammaro R, Prado MA, Cesana M, Lee BH, Finley D, Franco B, Morleo M, The deubiquitinating enzyme Usp14 controls ciliogenesis and Hedgehog signaling, Hum Mol Genet 28(5) (2019) 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Johnson CA, Collis SJ, Ciliogenesis and the DNA damage response: a stressful relationship, Cilia 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gerhardt C, Leu T, Lier JM, Rüther U, The cilia-regulated proteasome and its role in the development of ciliopathies and cancer, Cilia 5(1) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Eguether T, Hahne M, Mixed signals from the cell’s antennae: primary cilia in cancer, Embo Reports 19(11) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Katoh Y, Katoh M, Hedgehog Target Genes: Mechanisms of Carcinogenesis Induced by Aberrant Hedgehog Signaling Activation, Curr Mol Med 9(7) (2009) 873–886. [DOI] [PubMed] [Google Scholar]
- [39].Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr., Dlugosz AA, Reiter JF, Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis, Nat Med 15(9) (2009) 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gradilone SA, Lorenzo Pisarello MJ, LaRusso NF, Primary Cilia in Tumor Biology: The Primary Cilium as a Therapeutic Target in Cholangiocarcinoma, Curr Drug Targets (2015) 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Huangfu D, Anderson KV, Cilia and Hedgehog responsiveness in the mouse, Proc Natl Acad Sci U S A 102(32) (2005) 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Larkins CE, Aviles GDG, East MP, Kahn RA, Caspary T, Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins, Molecular Biology of the Cell 22(23) (2011) 4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Han YG, Alvarez-Buylla A, Role of primary cilia in brain development and cancer, Curr Opin Neurobiol 20(1) (2010) 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF, Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms, Nat Cell Biol 10(1) (2008) 70–76. [DOI] [PubMed] [Google Scholar]
- [45].Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E, A role for the primary cilium in Notch signaling and epidermal differentiation during skin development, Cell 145(7) (2011) 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grisanti L, Revenkova E, Gordon RE, Iomini C, Primary cilia maintain corneal epithelial homeostasis by regulation of the Notch signaling pathway, Development 143(12) (2016) 2160–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST, PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts, Curr Biol 15(20) (2005) 1861–1866. [DOI] [PubMed] [Google Scholar]
- [48].Michaud EJ, Yoder BK, The primary cilium in cell signaling and cancer, Cancer Res 66(13) (2006) 6463–6467. [DOI] [PubMed] [Google Scholar]
- [49].Clement CA, Ajbro KD, Koefoed K, Vestergaard ML, Veland IR, Henriques de Jesus MP, Pedersen LB, Benmerah A, Andersen CY, Larsen LA, Christensen ST, TGF-beta signaling is associated with endocytosis at the pocket region of the primary cilium, Cell Rep 3(6) (2013) 1806–1814. [DOI] [PubMed] [Google Scholar]
- [50].Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW, Primary cilia regulate mTORC1 activity and cell size through Lkb1, Nat Cell Biol 12(11) (2010) 1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mansini AP, Peixoto E, Jin S, Richard S, Gradilone SA, The chemosensory function of primary cilia regulates cholangiocyte migration, invasion and tumor growth, Hepatology (2018) 1582–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hansen CG, Moroishi T, Guan KL, YAP and TAZ: a nexus for Hippo signaling and beyond, Trends Cell Biol 25(9) (2015) 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Habbig S, Bartram MP, Sagmuller JG, Griessmann A, Franke M, Muller RU, Schwarz R, Hoehne M, Bergmann C, Tessmer C, Reinhardt HC, Burst V, Benzing T, Schermer B, The ciliopathy disease protein NPHP9 promotes nuclear delivery and activation of the oncogenic transcriptional regulator TAZ, Hum Mol Genet 21(26) (2012) 5528–5538. [DOI] [PubMed] [Google Scholar]
- [54].Kim M, Kim M, Lee MS, Kim CH, Lim DS, The MST1/2-SAV1 complex of the Hippo pathway promotes ciliogenesis, Nat Commun 5 (2014). [DOI] [PubMed] [Google Scholar]
- [55].Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF, Abnormal centrosome amplification in the absence of p53, Science 271(5256) (1996) 1744–1747. [DOI] [PubMed] [Google Scholar]
- [56].Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX, Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells, Mol Cell 3(3) (1999) 389–395. [DOI] [PubMed] [Google Scholar]
- [57].Sivasubramaniam S, Sun XM, Pan YR, Wang SH, Lee EYHP, Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1, Gene Dev 22(5) (2008) 587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Walz G, Role of primary cilia in non-dividing and post-mitotic cells, Cell Tissue Res 369(1) (2017) 11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mansini AP, Peixoto E, Thelen KM, Gaspari C, Jin S, Gradilone SA, The cholangiocyte primary cilium in health and disease, Biochim Biophys Acta Mol Basis Dis 1864(4 Pt B) (2018) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Masyuk TV, Masyuk AI, LaRusso NF, Therapeutic Targets in Polycystic Liver Disease, Curr Drug Targets 18(8) (2017) 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Benzing T, Schermer B, Clinical spectrum and pathogenesis of nephronophthisis, Curr Opin Nephrol Hypertens 21(3) (2012) 272–278. [DOI] [PubMed] [Google Scholar]
- [62].Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H, Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis, Nat Genet 34(4) (2003) 455–459. [DOI] [PubMed] [Google Scholar]
- [63].Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kranzlin B, Nurnberg G, Becker C, Grimm T, Girschick G, Lynch SA, Kelehan P, Senderek J, Neuhaus TJ, Stallmach T, Zentgraf H, Nurnberg P, Gretz N, Lo C, Lienkamp S, Schafer T, Walz G, Benzing T, Zerres K, Omran H, Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia, Am J Hum Genet 82(4) (2008) 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, Hoyng CB, Roepman R, Klevering BJ, Non-syndromic retinitis pigmentosa, Prog Retin Eye Res 66 (2018) 157–186. [DOI] [PubMed] [Google Scholar]
- [65].McCray G, Griffin P, Martinello P, de Iongh R, Ruddle J, Robinson P, Altered airway ciliary orientation in patients with X-linked retinitis pigmentosa, Thorax 74(9) (2019) 914–916. [DOI] [PubMed] [Google Scholar]
- [66].Bai SW, Herrera-Abreu MT, Rohn JL, Racine V, Tajadura V, Suryavanshi N, Bechtel S, Wiemann S, Baum B, Ridley AJ, Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration, BMC Biol 9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, van der Spek RA, Vosa U, de Jong S, Robinson MR, Yang J, Fogh I, van Doormaal PT, Tazelaar GH, Koppers M, Blokhuis AM, Sproviero W, Jones AR, Kenna KP, van Eijk KR, Harschnitz O, Schellevis RD, Brands WJ, Medic J, Menelaou A, Vajda A, Ticozzi N, Lin K, Rogelj B, Vrabec K, Ravnik-Glavac M, Koritnik B, Zidar J, Leonardis L, Groselj LD, Millecamps S, Salachas F, Meininger V, de Carvalho M, Pinto S, Mora JS, Rojas-Garcia R, Polak M, Chandran S, Colville S, Swingler R, Morrison KE, Shaw PJ, Hardy J, Orrell RW, Pittman A, Sidle K, Fratta P, Malaspina A, Topp S, Petri S, Abdulla S, Drepper C, Sendtner M, Meyer T, Ophoff RA, Staats KA, Wiedau-Pazos M, Lomen-Hoerth C, Van Deerlin VM, Trojanowski JQ, Elman L, McCluskey L, Basak AN, Tunca C, Hamzeiy H, Parman Y, Meitinger T, Lichtner P, Radivojkov-Blagojevic M, Andres CR, Maurel C, Bensimon G, Landwehrmeyer B, Brice A, Payan CA, Saker-Delye S, Durr A, Wood NW, Tittmann L, Lieb W, Franke A, Rietschel M, Cichon S, Nothen MM, Amouyel P, Tzourio C, Dartigues JF, Uitterlinden AG, Rivadeneira F, Estrada K, Hofman A, Curtis C, Blauw HM, van der Kooi AJ, de Visser M, Goris A, Weber M, Shaw CE, Smith BN, Pansarasa O, Cereda C, Del Bo R, Comi GP, D’Alfonso S, Bertolin C, Soraru G, Mazzini L, Pensato V, Gellera C, Tiloca C, Ratti A, Calvo A, Moglia C, Brunetti M, Arcuti S, Capozzo R, Zecca C, Lunetta C, Penco S, Riva N, Padovani A, Filosto M, Muller B, Stuit RJ, Registry P, Group S, Registry S, Consortium FS, Consortium S, Group NS, Blair I, Zhang K, McCann EP, Fifita JA, Nicholson GA, Rowe DB, Pamphlett R, Kiernan MC, Grosskreutz J, Witte OW, Ringer T, Prell T, Stubendorff B, Kurth I, Hubner CA, Leigh PN, Casale F, Chio A, Beghi E, Pupillo E, Tortelli R, Logroscino G, Powell J, Ludolph AC, Weishaupt JH, Robberecht W, Van Damme P, Franke L, Pers TH, Brown RH, Glass JD, Landers JE, Hardiman O, Andersen PM, Corcia P, Vourc’h P, Silani V, Wray NR, Visscher PM, de Bakker PI, van Es MA, Pasterkamp RJ, Lewis CM, Breen G, Al-Chalabi A, van den Berg LH, Veldink JH, Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis, Nat Genet 48(9) (2016) 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kurashige T, Morino H, Matsuda Y, Mukai T, Murao T, Toko M, Kume K, Ohsawa R, Torii T, Tokinobu H, Maruyama H, Kawakami H, Retinitis pigmentosa prior to familial ALS caused by a homozygous cilia and flagella-associated protein 410 mutation, J Neurol Neurosurg Psychiatry (2019) 220–222. [DOI] [PubMed] [Google Scholar]
- [69].Huang XF, Xiang L, Fang XL, Liu WQ, Zhuang YY, Chen ZJ, Shen RJ, Cheng W, Han RY, Zheng SS, Chen XJ, Liu XL, Jin ZB, Functional characterization of CEP250 variant identified in nonsyndromic retinitis pigmentosa, Hum Mutat 40(8) (2019) 1039–1045. [DOI] [PubMed] [Google Scholar]
- [70].Gascue C, Tan PL, Cardenas-Rodriguez M, Libisch G, Fernandez-Calero T, Liu YP, Astrada S, Robello C, Naya H, Katsanis N, Badano JL, Direct role of Bardet-Biedl syndrome proteins in transcriptional regulation, J Cell Sci 125(Pt 2) (2012) 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Brancati F, Dallapiccola B, Valente EM, Joubert Syndrome and related disorders, Orphanet J Rare Dis 5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zhou C, Cunningham L, Marcus AI, Li Y, Kahn RA, Arl2 and Arl3 regulate different microtubule-dependent processes, Mol Biol Cell 17(5) (2006) 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Alkanderi S, Molinari E, Shaheen R, Elmaghloob Y, Stephen LA, Sammut V, Ramsbottom SA, Srivastava S, Cairns G, Edwards N, Rice SJ, Ewida N, Alhashem A, White K, Miles CG, Steel DH, Alkuraya FS, Ismail S, Sayer JA, ARL3 Mutations Cause Joubert Syndrome by Disrupting Ciliary Protein Composition, Am J Hum Genet 103(4) (2018) 612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Revenkova E, Liu Q, Gusella GL, Iomini C, The Joubert syndrome protein ARL13B binds tubulin to maintain uniform distribution of proteins along the ciliary membrane, J Cell Sci 131(9) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M, MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome, Nat Genet 38(2) (2006) 155–157. [DOI] [PubMed] [Google Scholar]
- [76].Lewis WR, Bales KL, Revell DZ, Croyle MJ, Engle SE, Song CJ, Malarkey EB, Uytingco CR, Shan D, Antonellis PJ, Nagy TR, Kesterson RA, Mrug MM, Martens JR, Berbari NF, Gross AK, Yoder BK, Mks6 mutations reveal tissue- and cell type-specific roles for the cilia transition zone, FASEB J 33(1) (2019) 1440–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jenks AD, Vyse S, Wong JP, Kostaras E, Keller D, Burgoyne T, Shoemark A, Tsalikis A, de la Roche M, Michaelis M, Cinatl J Jr., Huang PH, Tanos BE, Primary Cilia Mediate Diverse Kinase Inhibitor Resistance Mechanisms in Cancer, Cell Rep 23(10) (2018) 3042–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Sarkisian MR, Siebzehnrubl D, Hoang-Minh L, Deleyrolle L, Silver DJ, Siebzehnrubl FA, Guadiana SM, Srivinasan G, Semple-Rowland S, Harrison JK, Steindler DA, Reynolds BA, Detection of primary cilia in human glioblastoma, J Neurooncol 117(1) (2014) 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Barakat MT, Humke EW, Scott MP, Kif3a is necessary for initiation and maintenance of medulloblastoma, Carcinogenesis 34(6) (2013) 1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A, Dual and opposing roles of primary cilia in medulloblastoma development, Nat Med 15(9) (2009) 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Moser JJ, Fritzler MJ, Rattner JB, Ultrastructural characterization of primary cilia in pathologically characterized human glioblastoma multiforme (GBM) tumors, BMC Clin Pathol 14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kim J, Dabiri S, Seeley ES, Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma, PLoS One 6(11) (2011) e27410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zingg D, Debbache J, Pena-Hernandez R, Antunes AT, Schaefer SM, Cheng PF, Zimmerli D, Haeusel J, Calcada RR, Tuncer E, Zhang Y, Bossart R, Wong KK, Basler K, Dummer R, Santoro R, Levesque MP, Sommer L, EZH2-Mediated Primary Cilium Deconstruction Drives Metastatic Melanoma Formation, Cancer Cell 34(1) (2018) 136–147. [DOI] [PubMed] [Google Scholar]
- [84].Rocha C, Papon L, Cacheux W, Marques Sousa P, Lascano V, Tort O, Giordano T, Vacher S, Lemmers B, Mariani P, Meseure D, Medema JP, Bieche I, Hahne M, Janke C, Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon, EMBO J 33(19) (2014) 2247–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Egeberg DL, Lethan M, Manguso R, Schneider L, Awan A, Jorgensen TS, Byskov AG, Pedersen LB, Christensen ST, Primary cilia and aberrant cell signaling in epithelial ovarian cancer, Cilia 1(1) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, Frost AR, Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues, J Histochem Cytochem 58(10) (2010) 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM, Primary cilia are lost in preinvasive and invasive prostate cancer, PLoS One 8(7) (2013) e68521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Dere R, Perkins AL, Bawa-Khalfe T, Jonasch D, Walker CL, beta-catenin links von Hippel-Lindau to aurora kinase A and loss of primary cilia in renal cell carcinoma, J Am Soc Nephrol 26(3) (2015) 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ding XF, Zhou J, Hu QY, Liu SC, Chen G, The tumor suppressor pVHL down-regulates never-in-mitosis A-related kinase 8 via hypoxia-inducible factors to maintain cilia in human renal cancer cells, J Biol Chem 290(3) (2015) 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Schmidt LS, Linehan WM, FLCN: The causative gene for Birt-Hogg-Dube syndrome, Gene 640 (2018) 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rosengren T, Larsen LJ, Pedersen LB, Christensen ST, Moller LB, TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms, Cell Mol Life Sci 75(14) (2018) 2663–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M, Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia, Cancer Res 69(2) (2009) 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kobayashi T, Nakazono K, Tokuda M, Mashima Y, Dynlacht BD, Itoh H, HDAC2 promotes loss of primary cilia in pancreatic ductal adenocarcinoma, EMBO Rep 18(2) (2017) 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Emoto K, Masugi Y, Yamazaki K, Effendi K, Tsujikawa H, Tanabe M, Kitagawa Y, Sakamoto M, Presence of primary cilia in cancer cells correlates with prognosis of pancreatic ductal adenocarcinoma, Hum Pathol 45(4) (2014) 817–825. [DOI] [PubMed] [Google Scholar]
- [95].Lee J, Yi S, Chang JY, Kim JT, Sul HJ, Park KC, Zhu X, Cheng SY, Kero J, Kim J, Shong M, Loss of Primary Cilia Results in the Development of Cancer in the Murine Thyroid Gland, Mol Cells 42(2) (2019) 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fu W, Asp P, Canter B, Dynlacht BD, Primary cilia control hedgehog signaling during muscle differentiation and are deregulated in rhabdomyosarcoma, Proc Natl Acad Sci U S A 111(25) (2014) 9151–9156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Ho L, Ali SA, Al-Jazrawe M, Kandel R, Wunder JS, Alman BA, Primary cilia attenuate hedgehog signalling in neoplastic chondrocytes, Oncogene 32(47) (2013) 5388–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Mansini AP, Lorenzo Pisarello MJ, Thelen KM, Cruz-Reyes M, Peixoto E, Jin S, Howard BN, Trussoni CE, Gajdos GB, LaRusso NF, Perugorria MJ, Banales JM, Gradilone SA, MiR-433 and miR-22 dysregulations induce HDAC6 overexpression and ciliary loss in cholangiocarcinoma, Hepatology (2018) 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Xiang W, Guo F, Cheng W, Zhang J, Huang J, Wang R, Ma Z, Xu K, HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia, Oncol Rep 38(1) (2017) 229–236. [DOI] [PubMed] [Google Scholar]
- [100].Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P, Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally, Mol Cell Biol 28(5) (2008) 1688–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yu F, Ran J, Zhou J, Ciliopathies: Does HDAC6 Represent a New Therapeutic Target?, Trends Pharmacol Sci 37(2) (2016) 114–119. [DOI] [PubMed] [Google Scholar]
- [102].Gradilone SA, Habringer S, Masyuk TV, Howard BN, Masyuk AI, LaRusso NF, HDAC6 is overexpressed in cystic cholangiocytes and its inhibition reduces cystogenesis, Am J Pathol 184(3) (2014) 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, Sun Z, Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models, Proc Natl Acad Sci U S A 106(51) (2009) 21819–21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Kathem SH, Mohieldin AM, Abdul-Majeed S, Ismail SH, Altaei QH, Alshimmari IK, Alsaidi MM, Khammas H, Nauli AM, Joe B, Nauli SM, Ciliotherapy: a novel intervention in polycystic kidney disease, J Geriatr Cardiol 11(1) (2014) 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Pala R, Mohieldin AM, Sherpa RT, Kathem SH, Shamloo K, Luan Z, Zhou J, Zheng JG, Ahsan A, Nauli SM, Ciliotherapy: Remote Control of Primary Cilia Movement and Function by Magnetic Nanoparticles, ACS Nano 13(3) (2019) 3555–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mergen M, Engel C, Muller B, Follo M, Schafer T, Jung M, Walz G, The nephronophthisis gene product NPHP2/Inversin interacts with Aurora A and interferes with HDAC6-mediated cilia disassembly, Nephrol Dial Transplant 28(11) (2013) 2744–2753. [DOI] [PubMed] [Google Scholar]
- [107].Khan NA, Willemarck N, Talebi A, Marchand A, Binda MM, Dehairs J, Rueda-Rincon N, Daniels VW, Bagadi M, Thimiri Govinda Raj DB, Vanderhoydonc F, Munck S, Chaltin P, Swinnen JV, Identification of drugs that restore primary cilium expression in cancer cells, Oncotarget 7(9) (2016) 9975–9992. [DOI] [PMC free article] [PubMed] [Google Scholar]