

SUMMARY
The mitochondrial calcium uniporter has been proposed to coordinate the organelle’s energetics with calcium signaling. Uniporter current has previously been reported to be extremely high in brown adipose tissue (BAT), yet it remains unknown how the uniporter contributes to BAT physiology. Here, we report the generation and characterization of a mouse model lacking Mcu, the pore forming subunit of the uniporter, specifically in BAT (BAT-Mcu-KO). BAT-Mcu-KO mice lack uniporter-based calcium uptake in BAT mitochondria but exhibit unaffected cold tolerance, diet-induced obesity, and transcriptional response to cold in BAT. Unexpectedly, we found in wild-type animals that cold powerfully activates the ATF4-dependent integrated stress response (ISR) in BAT and up-regulates circulating FGF21 and GDF15, raising the hypothesis that the ISR partly underlies the pleiotropic effects of BAT on systemic metabolism. Our study demonstrates that the uniporter is largely dispensable for BAT thermogenesis and demonstrates activation of the ISR in BAT in response to cold.
In Brief
Flicker et al. generate a mouse lacking mitochondrial calcium uniporter activity in brown fat. They show that the uniporter is dispensable for brown fat bioenergetics. Unexpectedly, they find that in wild type animals, cold stress induces ATF4 signaling in normal brown fat, suggesting a mechanism for cold-induced GDF15 and FGF21 elevation.
Graphical Abstract
INTRODUCTION
For decades, it has been known that calcium ions can enter mitochondria through a highly selective uniporter channel, driven by the electrochemical gradient across the inner mitochondrial membrane (IMM) (Carafoli and Lehninger, 1971; Deluca and Engstrom, 1961; Kirichok et al., 2004; Vasington and Murphy, 1962). Numerous studies have confirmed that calcium uptake in isolated mitochondria leads to transient dissipation of the membrane potential and sustained enhancement of NAD(P)H autofluorescence, oxygen consumption, and ATP phosphorylation (Territo et al., 2000). The effect on membrane potential is readily explained by the fact that the uniporter is electrophoretic, and hence calcium uptake leads to depolarization. A mechanism for the boost in NADH and oxidative phosphorylation was proposed as early as the 1970s, when it was noted that three matrix dehydrogenases are allosterically stimulated by calcium ions in vitro (for reviews, see Denton, 2009; Denton et al., 1996; McCormack et al., 1990).
Because energy-consuming events such as muscle contraction and neurotransmission are triggered by a release of calcium into the cytosol, it is plausible that these calcium boluses could enter mitochondria via the uniporter and trigger ATP production to match the increased cellular demand (Denton, 2009; Territo et al., 2000). This “feed-forward” model has historically been impossible to test, however, due to a lack of tools to selectively modulate the uniporter’s activity in vivo. The molecular identity of the uniporter began to be elucidated in 2010, enabling genetic disruption of its activity for the first time (Perocchi et al., 2010). At present, the mammalian uniporter is known to comprise five primary components, two of which are required to maintain a functional channel: the pore-forming subunit, MCU, and a small transmembrane binding partner, EMRE, both of which localize to the IMM (Baughman et al., 2011; Kovács-Bogdán et al., 2014; Sancak et al., 2013); MICU1 and MICU2, which are soluble subunits in the intermembrane space (IMS) that sense and gate the uniporter in the presence of subthreshold cytosolic calcium levels (Csordás et al., 2013; Kamer and Mootha, 2014; Kamer et al., 2017; Mallilankaraman et al., 2012); and the MCU homolog MCUb, which is thought to negatively regulate the uniporter’s conductance (Raffaello et al., 2013).
The molecular identification of this machinery has provided an unprecedented opportunity to delineate the role of the uniporter in cellular and organismal physiology. Several mouse models have since confirmed a role for the uniporter in supporting tissue bioenergetics, particularly when energy demand is acutely increased (Pan et al., 2013; Kwong et al., 2015; Luongo et al., 2015). In addition, a 2012 electrophysiology study by Fieni et al. (2012) demonstrated that the current density attributable to the uniporter is exceptionally high in mitoplasts isolated from skeletal muscle and brown adipose tissue (BAT) in comparison to liver, kidney, and heart. Skeletal muscle and BAT share a growing list of similarities (Seale et al., 2008; Stanford et al., 2013; Kim et al., 2013; Fisher et al., 2012), and both tissues respond to adrenergic signaling cues by acutely increasing energy consumption (Bachman et al., 2002; Lynch and Ryall, 2008). Because the uniporter has been shown to play a key bioenergetic role in skeletal muscle (Pan et al., 2013), we reasoned that it could serve a similarly important function in BAT; however, no studies to date have examined the role of the uniporter in this tissue.
BAT is a mammalian tissue specialized for metabolic inefficiency (Cannon and Nedergaard, 2004; Townsend and Tseng, 2014). It is densely packed with mitochondria and lipid droplets and is heavily innervated by sympathetic fibers that secrete norepinephrine (NE) in response to stimuli such as cold. NE acts on brown adipocytes primarily through the β3-adrenergic receptor, which signals through the cAMP-PKA pathway to liberate free fatty acids (FFA); these are oxidized to CO2 in the mitochondrial matrix but also activate uncoupling protein 1 (UCP1), a transporter that effectively permeabilizes the IMM to protons (Fedorenko et al., 2012; Wikstrom et al., 2014). NE therefore simultaneously stimulates rapid respiration and uncoupling in BAT, resulting in heat.
Early studies on isolated brown adipocytes found that intracellular calcium pools are mobilized by NE stimulation (Connolly et al., 1984), and subsequent imaging studies have repeatedly confirmed that NE induces a rise in cytosolic calcium in brown adipocytes (Chen et al., 2017; Leaver and Pappone, 2002; Lee et al., 1993; Nakagaki et al., 2005). However, it remains controversial whether this calcium is derived from mitochondria (Leaver and Pappone, 2002), and indeed whether NE stimulation causes a net rise or fall in matrix calcium levels (Hayato et al., 2011; Nakagaki et al., 2005). Although the physiological role of calcium signaling in BAT thermogenesis remains largely unexplored, it has recently been demonstrated that elevated cytosolic calcium can blunt heat production and thermogenic gene expression by repressing cAMP-PKA signaling (Chen et al., 2017; Nam et al., 2017). Elucidating the role of mitochondrial calcium handling in BAT may therefore provide significant insight into the regulation of thermogenesis.
In the present study, we generated and characterized a mouse model harboring BAT-specific loss of MCU (BAT-Mcu-KO). Despite ablated uniporter activity in this tissue, the animals did not exhibit an obvious phenotype: BAT bioenergetics were unimpaired as evidenced by intact cold-tolerance, susceptibility to diet-induced obesity, and thermogenic gene expression. Unexpectedly we found in wild-type animals that the integrated stress response (ISR) triggered by activating transcription factor 4 (ATF4) is strongly induced by cold in BAT.
RESULTS
Uniporter Expression Is Unremarkable in BAT
We first sought to determine whether the high uniporter current density reported in BAT mitochondria could be explained by high expression of MCU (Fieni et al., 2012). Prior large-scale studies found that MCU transcript levels are comparable between BAT, kidney, liver, and heart, and are approximately 2-fold higher in skeletal muscle (Su et al., 2002). Because these measurements do not correlate with the current densities reported in Fieni et al. (2012), we hypothesized that the uniporter components may vary more dramatically at the protein level across this panel of tissues. We therefore measured the abundance of MCU, EMRE, MICU1, and MICU2 in mitochondria isolated from each tissue (Figures S1A and S1B). We confirmed that these four components are highly enriched in skeletal muscle and depleted in heart. Surprisingly, however, the components were similarly expressed in BAT, kidney, and liver, at a level intermediate between that of skeletal muscle and heart. Thus, the current densities reported by Fieni et al. (2012) appear to correlate with uniporter protein levels in skeletal muscle, kidney, liver, and heart, but not in BAT.
Generation of BAT-Specific MCU Knockout Mice
C57BL/6 mice harboring a full-body knockout of Mcu die prior to birth for unknown reasons (Murphy et al., 2014). Therefore, in order to examine the role of the uniporter in BAT, we deleted Mcu in a tissue-specific manner. We first generated a conditional Mcu allele in which the second exon is flanked by LoxP sites (Figure S2A). We then bred mice homozygous for this allele (Mcufl/fl) with a transgenic line expressing Cre recombinase under control of the Ucp1 promoter, which is highly specific to BAT (Kong et al., 2014). Mcufl/fl animals harboring the Ucp1-Cre transgene were born at the expected Mendelian ratio and were grossly indistinguishable from Mcufl/fl animals lacking the Ucp1-Cre transgene.
We first confirmed that Mcufl/fl;Ucp1-Cre (BAT-Mcu-KO) animals exhibited complete loss of MCU in BAT but not in liver, whereas Mcufl/fl (control) animals were unaffected (Figure 1A). EMRE levels were negligible in BAT-Mcu-KO BAT, consistent with reports that this protein is proteolytically degraded when unbound to MCU (Tsai et al., 2017), and MICU1 and MICU2 levels were reduced (Figure 1A). To ensure that uniporter activity was fully ablated, BAT mitochondria were isolated from BAT-Mcu-KO and control animals and energized with L-glycerol-3-phosphate (G3P) plus rotenone in the presence of GDP. As expected, control mitochondria exhibited a robust calcium uptake activity under these conditions, which was completely abrogated in BAT-Mcu-KO mitochondria (Figure 1B).
Figure 1. Selective Ablation of MCU in Brown Fat.

(A) Effect of brown fat-specific Cre recombinase expression on uniporter protein levels on the Mcu[fl/fl] background.
(B) Left: representative Ca2+ uptake traces in isolated brown fat mitochondria energized with glycerol-3-phosphate (G3P) + rotenone in the presence of guanosine diphosphate (GDP). Right: quantification of Ca2+ uptake rates (n = 4).
(C) Oxygen consumption measurements of isolated brown fat mitochondria in a well stirred cuvette at room temperature. Mitochondria, G3P, and GDP were added at indicated time points.
(D) Quantification of oxygen consumption rates (n = 4).
Results are reported as mean + SEM. Statistical significance is indicated as **p < 0.01 (Student’s t test).
Our group previously demonstrated that OXPHOS is intact following MCU knockdown in vivo in liver, as demonstrated by intact State 3 to State 4 transitions in isolated mitochondria (Baughman et al., 2011). Consistently, mitochondria isolated from BAT-Mcu-KO and control BAT exhibited identical rates of oxygen consumption at baseline, in the presence of G3P (State 4u), and in the presence of both G3P and GDP (State 4) (Figures 1C and 1D).
MCU Is Dispensable for BAT Bioenergetics
We next sought to determine the effect of MCU loss on BAT bioenergetics. Mice harboring lesions in mitochondrial bioenergetics in BAT are typically unable to maintain their body temperatures when acutely exposed to 4°C; thus, we speculated that BAT-specific MCU loss would confer increased sensitivity to cold stress (Bachman et al., 2002; Enerbäck et al., 1997; Lowell et al., 1993; Vergnes et al., 2011). Surprisingly, however, both BAT-Mcu-KO and control animals were able to defend their body temperatures to an equal extent when transferred from room temperature (RT) to 4°C (Figures 2A and S2B).
Figure 2. MCU Is Largely Dispensable for Brown Fat Bioenergetics.

(A) Core body temperature of mice transferred from room temperature to 4°C (n = 5–6 per group). Full body temperature data over time is presented in Figure S3A.
(B) Core body temperature of mice transferred to 4°C following 1 week habituation to 30°C (n = 7 per group). Full body temperature data over time is presented in Figure S3B.
(C and D) Body mass of male (C) and female (D) —Cre and +Cre animals fed high-fat diet. Full body mass data over time is presented in Figures S3C and S3D.
(E) Brown fat pyruvate dehydrogenase (PDH) phosphorylation in —Cre or +Cre mice with or without ad libitum access to food at room temperature for 6 h. Animals were starved for 12 h overnight prior to the experiment.
(F) Brown fat pyruvate dehydrogenase (PDH) phosphorylation in —Cre or +Cre mice fasted at room temperature or 4°C for 6 h.
Results are reported as mean + SEM.
The response of BAT to a cold challenge is highly dependent on the temperature to which the animal has been habituated. Mice acclimated to 30°C conditions (thermoneutrality) exhibit an accumulation of unilocular lipid droplets and a mild reduction of mitochondria in interscapular BAT compared to mice housed at room temperature (22°C–26°C), and display a dramatic reduction in BAT oxygen consumption when administered a bolus of norepinephrine (NE) (Cannon and Nedergaard, 2011; Kozak, 2014). Conversely, even mice lacking UCP1 are able to defend their body temperatures at 4°C following 12 days of acclimation to mild cold (18°C) (Keipert et al., 2017). To test whether the role of the uniporter in nonshivering thermogenesis is dependent on the animals’ thermal prehistory, we habituated a cohort of mice to 30°C conditions for 1 week prior to administering a cold challenge. Both BAT-Mcu-KO and control animals were again able to defend their body temperatures to an equal extent (Figures 2B and S2C). We thus conclude that the uniporter is not required for body temperature maintenance via nonshivering thermogenesis.
Lesions in BAT thermogenesis have been shown to confer increased susceptibility to diet-induced obesity (Bachman et al., 2002; Feldmann et al., 2009; Lowell et al., 1993). To determine whether MCU loss constitutes such a lesion, we monitored body mass in BAT-Mcu-KO and control animals maintained on a high-fat diet (HFD) for 16 weeks. Consistent with our observations of cold tolerance, neither male nor female BAT-Mcu-KO animals exhibited a significant difference in body weight from age-matched control animals over the course of the experiment (Figures 2C, 2D, S2D, and S2E). Importantly we note that this study was conducted at room temperature, whereas prior studies have suggested that defects in diet-induced thermogenesis can generally only be observed at thermoneutrality (Enerbäck et al., 1997; Feldmann et al., 2009). It is therefore plausible that a genotype-dependent response to HFD might be evident at 30°C, although this is unlikely given that cold tolerance is unaffected by MCU.
The matrix enzyme pyruvate dehydrogenase phosphatase 1 (PDP1), which dephosphorylates and activates the pyruvate dehydrogenase (PDH) complex, has been shown to be allosterically activated by calcium in vitro (Denton et al., 1996; Huang et al., 1998). Consistently, CD1 total-body MCU knockout mice exhibit a decreased matrix calcium load and increased skeletal muscle PDH phosphorylation following a 16-h fast (Pan et al., 2013). In addition, skin fibroblasts isolated from patients with homozygous MICU1 loss exhibit an increased matrix calcium load and, as reported in a separate study, decreased PDH phosphorylation (Lewis-Smith et al., 2016; Logan et al., 2014). The uniporter has thus emerged as an important regulator of PDH phosphorylation under select conditions.
We speculated that MCU loss might impact PDH phosphorylation in BAT. To characterize the dynamics of PDH phosphorylation in BAT, mice were fasted overnight followed by 6 h of either additional fasting or ad libitum re-feeding. As has previously been demonstrated in skeletal muscle, re-feeding induced robust PDH dephosphorylation in BAT (Gudiksen and Pilegaard, 2017; Sugden et al., 2000). Interestingly, however, MCU loss did not impact refeeding-induced PDH dephosphorylation (Figure 2E).
Multiple studies have shown that adrenergic stimulation elicits a rise in cytosolic calcium in brown adipocytes (Chen et al., 2017; Leaver and Pappone, 2002). Mitochondrial calcium handling has been implicated in this process; however, it remains controversial if mitochondria subsequently take up the cytosolic calcium spike, or if they in fact contribute to it by releasing matrix calcium stores following UCP1-mediated depolarization (Connolly and Nedergaard, 1988; Lee et al., 1993; Nakagaki et al., 2005). We reasoned that according to either of these models, cold might impact PDH phosphorylation in BAT in an MCU-dependent manner. We therefore measured BAT PDH phosphorylation in BAT-Mcu-KO and control mice exposed to either RT or 4°C for 6 h. However, cold exposure did not substantially affect PDH phosphorylation in either genotype; MCU loss also had no effect on PDH phosphorylation at either temperature (Figure 2F).
Taken together, our observations suggest that uniporter function is largely dispensable for BAT bioenergetics, although we cannot exclude subtle effects that might be relevant in more natural environments.
BAT Gene Expression Is Remodeled by Acute Cold Exposure Independently of MCU
Because we were unable to detect a strong in vivo phenotype resulting from MCU loss in BAT, we next aimed to systematically quantify genome-wide gene expression in BAT in control and BAT-Mcu-KO animals. Prior studies of MCU knockout in skeletal muscle and heart revealed that, while no phenotype was evident at baseline, both tissues exhibited a blunted bioenergetic response to physiological stressors (exercise and adrenergic stimulation, respectively) (Kwong et al., 2015; Luongo et al., 2015; Pan et al., 2013). We chose cold as the relevant stressor in the present study and performed global transcriptional profiling on BAT from control and BAT-Mcu-KO animals exposed to RT or 4°C for 6 h. As a positive control for cold exposure, we confirmed that UCP1, PGC1A, and DIO2 were strongly upregulated in 4°C exposed mice in a genotype-independent manner (Ye et al., 2013); in contrast, MCU was strongly reduced in BAT-Mcu-KO animals but was transcriptionally unaffected by temperature (Figures 3A and 3B).
Figure 3. Transcriptional Response to Cold Exposure.

(A) qPCR of MCU and cold-induced transcripts in brown fat (n = 5–6 mice per group).
(B) Normalized counts of transcripts in (A) measured by RNA-seq.
(C) Principal component analysis of gene expression data.
(D) Volcano plots comparing —Cre with +Cre gene expression at two temperatures, and comparing RT with 4°C gene expression in both genotypes (blue points denote transcripts achieving an adjusted p value < 0.05).
(E) Top 10 genes enriched by genotype at either RT or 4°C (genes ranked by adjusted p value).
(F) Top 10 genes enriched by temperature in either —Cre or +Cre mice.
(G) MSigDB hallmark genesets enriched at 4°C in —Cre or +Cre mice.
(H) DNA motifs enriched at 4°C in —Cre or +Cre mice. Red points indicate motifs annotated as ATF3 or ATF4 targets. Genes were rank ordered using the Student’s t test metric. Samples were permuted 1,000 times to evaluate significance.
Results are reported as mean + SEM. Statistical significance is indicated as *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
We observed relatively few significant transcriptional changes resulting from MCU loss at either RT or 4°C (Figures 3D and 3E). Besides Mcu, we identified three genes that were differentially expressed by genotype at both RT and 4°C: ELMOD2, TBC1D9, and GSTA3, all of which are upregulated in BAT-Mcu-KO BAT (STAR Methods). Elmod2 and Tbc1d9 are located in close proximity to Ucp1 in the mouse genome and are likely overexpressed due to their presence in the UCP1-Cre transgene. Gsta3 encodes a cytosolic enzyme that participates in the biosynthesis of steroid hormones (Johansson and Mannervik, 2001; Raffalli-Mathieu et al., 2008). Its significance in brown fat and its connection to mitochondrial calcium homeostasis are unclear.
In stark contrast to the mild effect of genotype, we found that over 15% of detected genes were significantly up or downregulated by at least 2-fold between RT and 4°C (Figures 3D and 3F). Principal component analysis (PCA) yielded robust clustering of samples by temperature along the first principal component axis, confirming just how pervasive and strong the thermogenic gene expression program is (Figure 3C). Pgc1a, Dio2, and Ucp1 all scored within the top 100 genes in the loading of principal component 1 (STAR Methods).
When we sorted the genes enriched at 4°C in order of significance, we found that the transcription factor ATF4 was among the top 10 most significantly enriched genes in control and Mcu-KO BAT (Figure 3F) (Han et al., 2013). ATF4 is a highly studied molecule that orchestrates the integrated stress response (ISR), a gene expression program activated in response to a wide variety of perturbations including endoplasmic reticulum (ER) unfolded protein stress, amino acid or glucose deprivation, and severe hypoxia (Han et al., 2013; Harding et al., 2003; Pakos-Zebrucka et al., 2016). To investigate whether the canonical ISR was induced by cold in our experiment, we performed gene set enrichment analysis (GSEA) to identify hallmark gene sets and promoter motifs enriched at 4°C compared to RT (Mootha et al., 2003; Subramanian et al., 2005). The hypoxia and unfolded protein response hallmark gene sets, both of which contain reported ATF4 targets (Bao et al., 2016; Han et al., 2013), were significantly enriched in both control and BAT-Mcu-KO mice (Figure 3G), as were all four motifs annotated as binding to ATF4 or its downstream target ATF3 (Figure 3H).
Cold Induces the ATF4-Associated Integrated Stress Response in BAT
In order to validate that the canonical ISR is operative in BAT at 4°C, we first confirmed that several ATF4 target genes were robustly upregulated by cold in an independent cohort of mice (Figure 4A). We additionally found that treatment of differentiated immortalized brown adipocytes with NE induced both uncoupled respiration and upregulation of ATF3 within 4 hours, suggesting that adrenergic stimuli may induce the ISR in a cell-autonomous manner (Figures S3A–S3D).
Figure 4. Cold Challenge Activates the Integrated Stress Response in Brown Fat.

(A) qPCR of transcripts corresponding to the thermogenic gene expression or the integrated stress response (ISR) in wild type mice housed for 6 h at RT or 4°C (n = 6 mice per group).
(B) BAT ATF4 protein levels in fasting mice housed for 6 h at RT or 4°C.
(C) Circulating levels of FGF21 and GDF15 in mice starved for 6 h at RT or 4°C (n = 7 mice per group).
Results are reported as mean + SEM. Statistical significance is indicated as *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
ATF4 is acutely regulated at the translational level by the initiation factor eIF2α, which is itself activated upon phosphorylation by one of four protein kinases (for review, see Pakos-Zebrucka et al., 2016). Although the phosphorylated form of eIF2α broadly represses translation initiation, it also promotes skipping of a uORF in the ATF4 mRNA, leading to increased translation of the main coding region and rapid accumulation of ATF4 protein (Dey et al., 2010; Vattem and Wek, 2004). Consistently, we found that cold exposure led to accumulation of ATF4 at the protein level (Figure 4B). Interestingly, ATF4 accumulation was suppressed when mice were given ad libitum access to food over the course of the cold exposure period, regardless of whether animals were fasted beforehand (Figures S4A and S4B).
What purpose does cold induction of the ISR serve in BAT? To address this question, we noted that two genes known to be induced as part of the ISR are the cytokines FGF21 and GDF15 (Jousse et al., 2007; Khan et al., 2017; Kim et al., 2013). Both cytokines have been demonstrated to promote glucose tolerance, insulin sensitivity, resistance to diet-induced obesity, and upregulated expression of lipolysis and beta-oxidation genes in white and beige fat (Chrysovergis et al., 2014; Chung et al., 2017; Kharitonenkov et al., 2005; Kim et al., 2013; Fisher et al., 2012; Xiong et al., 2017). Furthermore, cold exposure has been shown to induce FGF21 transcription in BAT after 4–6 h and a 2-fold rise in circulating FGF21 levels after 24 h, raising the possibility that FGF21 may mediate the pleiotropic effects of BAT on systemic metabolism (Chartoumpekis et al., 2011; Hondares et al., 2011). We reasoned that, if ISR activation in BAT serves such a purpose, then both circulating FGF21 and GDF15 should be raised after 6 h of cold exposure. Indeed, we found that circulating FGF21 and GDF15 levels increased by nearly 5-fold and 2-fold, respectively (Figure 4C). To our knowledge, cold-induction of circulating GDF15 has not previously been reported. In order to determine whether any tissues besides BAT secrete significant quantities of FGF21 and GDF15 during cold exposure, we measured transcript levels of these and other ISR target genes in a panel of tissues known to be physiological sources of either hormone (Estall et al., 2009; Kempf et al., 2006; Khan et al., 2017) (Figure S4C). Among the tissues examined, BAT demonstrated by far the strongest transcriptional upregulation of FGF21 and GDF15. While we cannot rule out the possibility that other tissues contribute to the circulating FGF21 and GDF15 pools during cold exposure, these results suggest that the increase in both hormones may be primarily attributable to their production in BAT. Collectively, our observations indicate that the ISR may support many of the endocrine functions of BAT implicated in modulating systemic metabolism; furthermore, in addition to FGF21, thermogenic BAT may serve as an important physiological source of GDF15.
DISCUSSION
The current study indicates that the mitochondrial calcium uniporter is largely dispensable for BAT bioenergetics: the mouse model we used completely lacks uniporter activity in BAT, but displays no obvious abnormalities in cold tolerance, diet-induced obesity, BAT PDH phosphorylation, or BAT transcriptome-wide gene expression at baseline or in response to acute cold challenge. Although not the goal of the current study, examining the global gene expression response to cold led us to identify a robust induction of the ISR in BAT in wild type animals. While determining the full functionality of the ISR in thermogenic BAT is beyond the scope of this study, we speculate that it may support the endocrine role of BAT by strongly inducing secretion of cytokines such as FGF21 and GDF15.
The uniporter has already been demonstrated to support exercise tolerance in skeletal muscle and sympathetic contractility stimulation in heart, both of which are highly energy-demanding processes linked to adrenergic signaling (Kwong et al., 2015; Luongo et al., 2015; Pan et al., 2013). Given that non-shivering thermogenesis is another such process, combined with the fact that a previous study identified the uniporter current to be extremely high in BAT (Fieni et al., 2012), we were surprised to discover the minimal role of the uniporter in BAT bioenergetics.
The study of various MCU knockout models has implicated a crucial role for genetic background in unmasking uniporter-associated phenotypes. The most dramatic example is, of course, the embryonic lethality of inbred full-body MCU knockout mice compared with the mild phenotype of the CD1 MCU knockout model (Murphy et al., 2014). Because we observed that MCU knockout results in complete ablation of uniporter activity in BAT, it is clear that the function of MCU is non-essential in this tissue on the C57BL/6 background.
While the uniporter is thought to mediate the only rapid mitochondrial calcium uptake pathway, it is certainly not the only means by which mitochondrial calcium influx can occur. For example, a mechanism that can theoretically sustain mitochondrial calcium homeostasis in the absence of uniporter function is reversal of the IMM sodium-calcium exchanger, NCLX, which ordinarily serves as the primary mitochondrial calcium efflux pathway (De Marchi et al., 2014; Palty et al., 2010). NCLX reversal may therefore play a greater role in mitochondrial calcium homeostasis than the uniporter in thermogenic BAT.
It is noteworthy that PDH is acted upon by two phosphatases: PDP1 and PDP2, the latter of which is calcium-insensitive (Huang et al., 1998). Previous studies (Su et al., 2002) and our own RNA sequencing (RNA-seq) data indicate that in BAT, PDP2 is over 10-fold more abundant than PDP1 at RT, and over 20-fold more abundant at 4°C (STAR Methods). It is therefore reasonable to expect that calcium exerts relatively little control over the PDH phosphorylation state in BAT, consistently with our results (Figures 2E and 2F).
Given that BAT mitochondria contain substantially less MCU and EMRE protein than skeletal muscle mitochondria (Figures S1A and S1B), it is unclear why Fieni et al. (2012) observed that mitoplasts from these two tissues harbor equally high current densities. Notably, the BAT mitoplasts used for patch clamp experiments in this study were isolated from mice lacking UCP1, suggesting that the high uniporter current density in BAT may be specific to this genotype; indeed, a recent quantitative proteomics study demonstrated that MCU, EMRE, MICU1, and MICU2 are all upregulated by ~2-fold in UCP1 knockout BAT (Kazak et al., 2017).
Importantly, an additional use for the BAT-Mcu-KO model that we have not yet explored is to examine the role of MCU in beige fat, a UCP1-expressing cell type dispersed primarily throughout the inguinal fat pad (Wu et al., 2012). Unlike interscapular BAT, the abundance of beige fat is relatively low at room temperature and is strongly increased following multi-day cold exposure (Young et al., 1984) or chronic, repeated administration of a β3-adrenergic agonist (Bertholet et al., 2017). Recent studies have identified multiple UCP1-independent pathways by which beige, but not brown, adipocytes can generate heat (Bertholet et al., 2017; Kazak et al., 2015), including futile cycling of calcium across the endoplasmic reticulum membrane (Ikeda et al., 2017); it is thus conceivable that MCU plays a substantive role in this tissue’s bioenergetics despite its dispensability in BAT.
An unexpected finding from our work is that cold powerfully induces the ATF4-dependent ISR in BAT. Relatively few studies have addressed the dynamics and functional significance of the ISR in this tissue (Bettaieb et al., 2012; Liu et al., 2017; Mamady and Storey, 2008; Sekine et al., 2016; Seo et al., 2009; Wang et al., 2010, 2013), and the interplay between the ISR and the thermogenic gene program is poorly understood. One recent study showed that mice lacking the mitochondrial Ser/Thr-specific protein phosphatase PGAM5 have increased levels of phospho-eIF2α and FGF21 in BAT following a 12-h fast and 3–6 h cold exposure; cold-induction of FGF21 mRNA was also blunted by treatment with ISRIB, which blocks the downstream effects of eIF2α phosphorylation (Anders and Huber, 2010; Sekine et al., 2016). Interestingly, while fasting plus cold also induced FGF21 transcription in wild type BAT, only an ~2-fold change in FGF21 mRNA was observed relative to fasted mice at RT. Our study validates and extends this finding by showing that acute cold exposure induces ATF4 protein accumulation, reproducibly engages the full ISR at a genome-wide level, and leads to a rapid and substantial increase of FGF21 and GDF15 in the circulation. We have also shown that fasting animals prior to cold exposure is not necessary for induction of the ISR in BAT, although feeding during cold exposure suppresses ATF4 accumulation; this suggests that the ISR is likely to be operative in BAT under normal physiological circumstances, and not just in the controlled setting of a 12-h fast. Furthermore, in our hands, a 6-h cold exposure increases FGF21 mRNA levels in BAT by up to 50-fold (STAR Methods).
Three other studies have directly explored the connection between ATF4 and BAT thermogenesis, all utilizing a full-body Atf4 knockout mouse model (Seo et al., 2009; Wang et al., 2010, 2013). Surprisingly, these mice are smaller and have a lower body fat percentage than wild type mice; they are more insulin-sensitive and have higher resting energy expenditure both on chow and high-fat diets; they better maintain core body temperature during a 3-h cold challenge; and they exhibit mild upregulation of UCP1, PGC1A, select lipolysis genes, and select b-oxidation genes in BAT. One of these studies examined gene expression in wild type mice in response to a 7-h cold challenge and surprisingly found ATF4 levels to slightly decrease at 4°C (Wang et al., 2013). The same study proposed a model in which ATF4 displaces the cAMP-responsive transcription factor CREB from the PGC1A promoter, thereby repressing cold-induced PGC1A upregulation. Our results show that ISR activation in response to cold is highly dependent on the presence or absence of food (Figures S4A and S4B); this or other environmental factors may explain the discrepancy in ATF4 dynamics between the aforementioned study and ours.
Further work will be required to fully understand the interplay between the ISR, ATF4, FGF21, GDF15, and the thermogenic gene program in BAT. Additional studies will also be required to precisely delineate the conditions under which the ISR is activated by cold in BAT, particularly given that this effect is highly dependent on feeding status (Figures S4A and S4B). Notably, ATF4 signaling is also upregulated in multiple cellular and animal models of mitochondrial dysfunction (Bao et al., 2016; Quirós et al., 2017; Tyynismaa et al., 2010), including forced uncoupling via FCCP treatment (Quirós et al., 2017) or ectopic UCP1 expression (Keipert et al., 2014; Ost et al., 2015). Consistently, both FGF21 and GDF15 have emerged as promising blood biomarkers for human mitochondrial disorders (Davis et al., 2013; Lehtonen et al., 2016; Montero et al., 2016; Yatsuga et al., 2015). Future efforts to decipher the mechanism and physiological consequences of cold-induced ATF4 signaling in BAT may therefore help elucidate the role of the ISR in mitochondrial disease.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, V.K.M. (vamsi@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mouse experiments were performed according to procedures approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee. All mice were housed in groups of up to 5 animals in a room temperature facility with 12-hour light and dark cycles, and were provided access to a standard rodent chow diet. B6.FVB-Tg(Ucp1-cre)1Evdr/J (UCP1-Cre) mice described in Kong et al. (2014) were obtained from The Jackson Laboratory. All experimental animals were obtained by breeding MCU[fl/fl] mice with MCU[fl/fl];UCP1-Cre mice to yield a 1:1 ratio of progeny harboring zero or one copy of the UCP1-Cre transgene. Each experiment was performed using sex- and age-matched mice; animals used for cold tolerance and RNA-seq experiments were males at 8–12 weeks of age.
METHOD DETAILS
Generation of MCUfl/fl Mouse Line
BAC RP23–371B1 was purchased from BacPac Resources and was modified to flank exon 2 of Mcu with LoxP sites using recombineering as described (Sharan et al., 2009). ES cells from a mixed genetic background (129/BL6) were injected with the modified BAC (Figure S2) in the Beth Israel Deaconess Medical Center Transgenic Core Facility. Neomycin resistant ES cells were further screened with PCR and Southern blotting. ES cells with correct targeting were used to generate MCUfl/fl mice. MCUfl/fl mice were then crossed with wild-type C57BL/6J until ~95% background homogeneity was achieved by single nucleotide polymorphism (SNP) analysis (The Jackson Laboratory).
Isolation of Mouse Tissue Mitochondria
Crude mitochondria isolation was performed essentially as described by Fieni et al. (2012), with all steps performed at 0–4°C and BAT harvested from the interscapular region. Briefly, mice were sacrificed by CO2 asphyxiation followed by cervical dislocation. Tissues were dissected immediately, rinsed briefly in ice-cold PBS, and immersed in 10mL ice-cold isolation buffer (250mM sucrose, 10mM HEPES, 1mM EGTA, pH 7.25 with KOH). Tissues were then finely minced and homogenized with six slow strokes of a Potter-Elvehjem homogenizer rotating at 280 (liver) or 600 (brown adipose tissue, skeletal muscle, kidney, heart) rpm. To increase mitochondrial yield, the homogenate was centrifuged at 700g for 5 mins, and the resulting nuclear/unbroken cell pellet was resuspended in the same supernatant and homogenized again as above. The homogenate was then centrifuged at 700g for 10 mins, and the supernatant was collected and centrifuged at 8,500g for 10 mins. The resulting mitochondrial pellet was rinsed with 1mL isolation buffer and resuspended in 5mL isolation buffer. The 700g and 8,500g centrifugation steps were then repeated, and the resulting mitochondrial pellet was resuspended in approx. 200 μL isolation buffer. Mitochondrial protein content was measured by lysing a small sample in RIPA buffer and performing a Bradford assay.
Cold Exposure and Body Temperature Measurements
Age and sex-matched animals were individually housed at 4°C for up to six hours in pre-cooled cages without bedding, with ad libitum access to pre-cooled water. Animals at 4°C did not have access to food unless otherwise indicated; if food was provided, it was pre-cooled to 4°C overnight. Body temperature was measured rectally at indicated time points using a Physitemp BAT-12 thermometer outfitted with a RET-3 probe. All animals used for cold tolerance and RNA-seq experiments were males at 8–12 weeks of age.
High Fat Diet
Age and sex-matched animals at 6–10 weeks of age were housed with 1–2 animals per cage, and standard chow was replaced with a diet containing 60% kcal from fat (Research Diets formula D12492). Animal weights and average food intake were measured by hand twice weekly at approx. the same time of day.
BAT Mitochondrial Calcium Uptake Measurements
BAT mitochondrial calcium uptake measurements were performed according to Al-Shaikhaly et al. (1979). 30 mg of crude BAT mitochondria in isolation buffer was centrifuged at 8,500g for 10 mins at 4°C, and the resulting pellet was resuspended in 150 μL calcium uptake buffer (125mM sucrose, 20mM Tris-HCl, pH 7.2 with Tris base) supplemented with 2 μM rotenone, 1mM GDP, 0.1% bovine serum albumin (fatty acid free), and 1 μM membrane impermeable Oregon Green BAPTA-6F. Immediately prior to calcium uptake measurement, the medium was supplemented with 5mM L-glycerol-3-phosphate and mixed by gentle agitation. Fluorescence was monitored with a PerkinElmer Envision plate reader before and after injection of 50 μM CaCl2 using FITC filter sets (Ex485/Em535), with a 0.5 s measuring interval. Calcium uptake rates were calculated using the linear fit of uptake curves between 40–60 s.
BAT Mitochondrial Oxygen Consumption Measurements
30 μg of BAT crude mitochondria in isolation buffer was centrifuged at 8,500g for 10 mins at 4°C, and the resulting pellet was resuspended in calcium uptake buffer (125mM sucrose, 20mM Tris-HCl, pH 7.2 with Tris base) supplemented with 0.1% bovine serum albumin (fatty acid free) and 0.5uM tetramethylrhodamine methyl ester. Mitochondria were then added to a well-stirred cuvette at 25°C to reach 500 μL total volume. At indicated time points 1mM L-glycerol-3-phosphate and 1mM GDP were added. O2 consumption and membrane potential were measured simultaneously using a custom spectrophotometer outfitted with an Ocean Optics benchtop NeoFox-GT phase fluorimeter, as previously described (Gohil et al., 2013).
Western Blotting
Animals were sacrificed by CO2 asphyxiation followed by cervical dislocation. Tissues were immediately harvested and snap frozen in liquid N2. For preparation of protein lysates, 1 BAT depot (or equivalent volume of another tissue) was immersed in approx. 300 μL ice-cold RIPA buffer supplemented with either complete EDTA-free protease inhibitor cocktail or Protease/Phosphatase Inhibitor Cocktail. The tissue was then lysed with two 5mm stainless steel beads using a QIAGEN TissueLyser for 2 mins at 25 Hz. The resulting homogenate was centrifuged for 10 mins at maximum speed at 4°C, and the supernatant was centrifuged a second time to remove residual insoluble material. Protein content of the resulting clarified lysate was determined using a Bradford assay. Appropriate volumes of lysate were boiled for 5 mins in the presence of SDS sample buffer, resolved on Tris-Glycine SDS-PAGE gels, and transferred to PVDF membranes for western blotting. All antibodies used are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal against MCU | Cell Signaling Technologies | Cat#14997; RRID: AB_2721812 |
| Rabbit monoclonal against MICU1 | Cell Signaling Technologies | Cat#12524; RRID: AB_2797943 |
| Rabbit polyclonal against MICU2 | Bethyl | Custom synthesized |
| Rabbit polyclonal against EMRE | Bethyl | Custom synthesized |
| Mouse monoclonal against ATP5A | Abcam | Cat#AB14748; RRID: AB_301447 |
| Rabbit polyclonal against LRPPRC | Sigma | Cat#SAB2700419 |
| Rabbit polyclonal against VDAC | Cell Signaling Technologies | Cat#4866; RRID: AB_2272627 |
| Rabbit polyclonal against Actin | Cell Signaling Technologies | Cat#4967 |
| Rabbit monoclonal against Beta-Tubulin | Cell Signaling Technologies | Cat#2128 |
| Mouse monoclonal against PDH | Thermo Fisher | Cat#459400 |
| Rabbit polyclonal against Phospho-PDH (S293) | EMD Millipore | Cat#ABS204 |
| Rabbit polyclonal against Phospho-PDH (S300) | Calbiochem | Cat#AP1064 |
| Rabbit monoclonal against ATF-4 | Cell Signaling Technologies | Cat#11815; RRID: AB_2616025 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phosphate buffered saline (PBS) | Thermo Fisher | Cat# 10010023 |
| Sucrose | Sigma | Cat# S0389 |
| HEPES | Sigma | Cat# H3375 |
| EGTA | Sigma | Cat# E3889 |
| KOH | Sigma | Cat# P5958 |
| RIPA Buffer with EDTA and EGTA | Boston BioProducts | Cat# BP-115DG |
| Quick Start Bradford 1x Dye Reagent | Bio-Rad | Cat# 5000205 |
| Rodent High Fat Diet | Research Diets | D12492 |
| Trizma® hydrochloride (Tris-HCl) | Sigma | Cat# T3253 |
| Trizma® BASE (Tris base) | Sigma | Cat# 93362 |
| Rotenone | Sigma | Cat #R8875 |
| Guanosine 5′-diphosphate [GDP] disodium salt | Abcam | Cat# 7415-69-2 |
| Bovine serum albumin (fatty acid free) | Sigma | Cat# A8806 |
| Oregon Green 488 BAPTA-6F, hexapotassium salt | Thermo Fisher | Cat# O23990 |
| sn-Glycerol 3-phosphate bis(cyclohexylammonium) salt | Sigma | Cat# G7886 |
| Calcium chloride | Sigma | Cat# C1016 |
| Tetramethylrhodamine methyl ester (TMRM) | Thermo Fisher | Cat# T668 |
| cOmplete ETDA-free protease inhibitor cocktail | Sigma | Cat# 11873580001 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technology | Cat# 5872S |
| SDS sample buffer (Laemmli) | Boston BioProducts | Cat# BP-111R |
| QIAzol Lysis Reagent | QIAGEN | Cat# 79306 |
| Chloroform | Sigma | Cat# 288306 |
| QIAGEN buffer RLT | QIAGEN | Included in RNeasy Mini Kit |
| 100% Ethanol | Decon Labs | Cat# 2716 |
| GIBCO DMEM, high glucose | Thermo Fisher | Cat# 11965092 |
| GIBCO Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher | Cat# 15140122 |
| GIBCO GlutaMAX Supplement | Thermo Fisher | Cat# 35050061 |
| Insulin | Sigma | Cat# I5500 |
| 3,3′,5-Triiodo-L-thyronine sodium salt (T3) | Sigma | Cat# T6397 |
| Dexamethasone | Sigma | Cat# D4902 |
| 3-Isobutyl-1-methylxanthine (IBMX) | Sigma | Cat# I5879 |
| Indomethacin | Sigma | Cat# I7378 |
| Dulbecco’s Phosphate-Buffered Saline (DPBS) | Thermo Fisher | Cat# 14190136 |
| Formalin solution, neutral buffered, 10% | Sigma | Cat# HT501128 |
| Isopropanol | Sigma | Cat# I9516 |
| Oil Red O | Sigma | Cat# O0625 |
| Hematoxylin Solution, Harris Modified | Sigma | Cat# HHS16 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR | Thermo Fisher | Cat# 11752050 |
| TaqMan Gene Expression Master Mix | Thermo Fisher | Cat# 4369016 |
| Mouse/Rat FGF-21 Quantikine ELISA Kit | R&D Systems | Cat# MF2100 |
| Mouse/Rat GDF-15 Quantikine ELISA Kit | R&D Systems | Cat# MGD150 |
| Seahorse XF24 Extracellular Flux Analyzer | Seahorse Bioscience | https://www.agilent.com/en/products/cell-analysis/seahorse-analyzers |
| Deposited Data | ||
| RNA-seq results generated in this manuscript, raw and processed | NCBI Gene Expression Omnibus | http://www.ncbi.nlm.nih.gov/geo, query GEO: GSE119964 |
| Experimental Models: Cell Lines | ||
| DE-2-3 immortalized brown adipocytes | Gift from Dr. Bruce Spiegelman | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Lab | Cat# 000664 |
| Mouse: B6FVB-Tg(UCP1-Cre)1Evdr/J | The Jackson Lab | Cat# 024670 |
| Oligonucleotides | ||
| MCU TaqMan Probe | Thermo Fisher | Mm01168773_m1 |
| UCP1 TaqMan Probe | Thermo Fisher | Mm01244861_m1 |
| PPARGC1A (PGC1A) TaqMan Probe | Thermo Fisher | Mm01208835_m1 |
| DIO2 TaqMan Probe | Thermo Fisher | Mm00515664_m1 |
| ATF4 TaqMan Probe | Thermo Fisher | Mm00515325_g1 |
| ATF3 TaqMan Probe | Thermo Fisher | Mm00476033_m1 |
| MTHFD2 TaqMan Probe | Thermo Fisher | Mm00485276_m1 |
| CBS TaqMan Probe | Thermo Fisher | Mm00460654_m1 |
| CTH TaqMan Probe | Thermo Fisher | Mm00461247_m1 |
| TRIB3 TaqMan Probe | Thermo Fisher | Mm00454879_m1 |
| VEGFA TaqMan Probe | Thermo Fisher | Mm01281449_m1 |
| ASNS TaqMan Probe | Thermo Fisher | Mm00803785_m1 |
| HPRT TaqMan Probe | Thermo Fisher | Mm03024075_m1 |
| Recombinant DNA | ||
| BAC RP23-371B1 (LB Stab) | BACPAC Resources | RP23-371B1 |
| Software and Algorithms | ||
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| STAR 2.4.0j | Picelli et al. (2014) | https://github.com/alexdobin/STAR |
| HTSeq | Dobin et al. (2013) | https://github.com/simon-anders/htseq |
| DESeq2 | Anders et al. (2015) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Gene Set Enrichment Analysis (GSEA) | Mootha et al. (2003); Subramanian et al. (2005) | http://software.broadinstitute.org/gsea/index.jsp |
RNA isolation from BAT
Animals were sacrificed by CO2 asphyxiation followed by cervical dislocation, and tissues were immediately harvested and snap frozen in liquid N2. Frozen BAT samples were homogenized in 1mL Qiazol per 100mg tissue using the QIAGEN TissueRuptor II. The homogenate was mixed thoroughly with chloroform (1:5 chloroform:homogenate), incubated for 3 mins at room temperature, and centrifuged at 12,000g for 15 mins at 4°C, and 100 μL of the resulting aqueous phase was added to 350 μL QIAGEN buffer RLT plus 250 μL of 100% ethanol. The resulting mixture was transferred to a column from the QIAGEN RNeasy Mini Kit, and RNA was purified according to the manufacturer’s protocol.
Quantitative Real-Time PCR
RNA was reverse transcribed using the SuperScript III First-Strand Synthesis SuperMix Kit according to the manufacturer’s protocol. Quantitative real-time PCR (qPCR) was performed using TaqMan assays; all probe IDs are listed in the key resources table. Relative gene expression was calculated as 2−ΔΔCT, where HPRT was used as the housekeeping gene for normalization.
RNA-seq
RNA-sequencing libraries were prepared by the Broad Technology Labs at the Broad Institute based on the Smart-seq2 protocol (Picelli et al., 2014) and sequenced on an Illumina NextSeq 500 instrument to generate 2×25bp paired-end reads. The reads were aligned to the mouse genome (mm10, with gencode M7 annotations) using STAR 2.4.0j (default parameters) (Dobin et al., 2013). Counts of reads uniquely mapping within exonic regions of annotated genes (irrespective of strand) were collated using HTSeq (Anders et al., 2015). 14,651 genes with at least 8 reads mapping to them in at least 6 of the samples were retained for differential expression analysis. This was performed in R using DESeq2 (Love et al., 2014) and the design formula ~Genotype + Temperature + Genotype:Temperature. Where gene expression levels are reported, they represent normalized read counts following application of the estimateSizeFactors function, which implements the median ratio method. Where log2 fold-changes and p values are reported, they represent the result of the Wald test. P values are adjusted with the method of Benjamini-Hochberg. Further data analysis based on the normalized read counts was performed in MATLAB. Gene Set Enrichment Analysis (GSEA) was performed as described in Mootha et al. (2003) and Subramanian et al. (2005). All sequencing results have been deposited in NCBI GEO with the accession number GSE119964.
Cytokine assays
Mice were sacrificed by CO2 asphyxiation and blood was immediately drawn from the inferior vena cava. To obtain plasma, blood was incubated in EDTA-treated tubes and centrifuged at 14,000rpm for 10 mins at 4°C. FGF21 and GDF15 were measured from plasma samples using R&D Systems Quantikine ELISA Kits MF2100 and MGD150, respectively, according to the manufacturer’s protocols.
DE-2–3 cell culture and differentiation
DE-2–3 cells were cultured and differentiated essentially as described in Pan et al. (2009) and Shen et al. (2017). Briefly, cells were passaged in growth media consisting of high glucose DMEM supplemented with 10% fetal bovine serum, penicillin and streptomycin (100 U/mL), and GlutaMAX (2mM). For differentiation, cells were seeded at day −2 in differentiation medium (DM, growth media supplemented with 20nM insulin and 1nM T3) to reach confluence on day 0. On day 0, media was switched to induction media (DM supplemented with 0.5mM IBMX, 0.5 μM dexamethasone, and 0.125mM indomethacin). Media was changed to DM on day 2 and day 4. Cells were considered to be fully differentiated on day 6, and experiments were performed on day 6–9. For oxygen consumption measurements, DE-2–3 cells were differentiated in Seahorse XF24 Cell Culture Microplates, and oxygen consumption rates were measured using a Seahorse XF24 Extracellular Flux Analyzer instrument.
Oil Red O Stain
Oil Red O staining was performed as described in Shen et al. (2017), according to the protocol by Lonza. Briefly, 300mg of Oil Red O was dissolved in 100mL of 99% isopropanol to prepare Oil Red O stock solution. 30mL Oil Red O Stock solution was then mixed with 20mL deionized water, incubated for 10 mins at room temperature, and passed through a 0.45 mm filter to yield Oil Red O working solution. Fully differentiated DE-2–3 cells were gently rinsed with sterile DPBS and fixed for 30–60 mins in 10% formalin. The fixed cells were then rinsed with sterile water, incubated in 60% isopropanol for 2–5 mins, and incubated in Oil Red O working solution for 5 mins. The cells were rinsed with water until excess stain was removed, and then incubated for 1 min with hematoxylin counterstain. The cells were then rinsed with water until excess stain was removed, and kept under water prior to and during imaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical comparisons for low-throughput data were performed as described in the appropriate figure legends using Microsoft Excel. Significance for Student’s t tests was evaluated using a two-tailed test assuming unequal variances. Statistical analysis of the RNA-seq data was performed as described in the RNA-seq section of the STAR methods.
DATA AND SOFTWARE AVAILABILITY
Software availability
Source codes for the STAR 2.4.0j (Dobin et al., 2013), HTSeq (Anders et al., 2015), and DEseq (Love et al., 2014) software packages are freely available for download through the corresponding references. GSEA and MSigDB can be freely accessed through the Broad Institute website, and is implemented via a graphical user interface. MATLAB is accessible through MathWorks, Inc. on a subscription basis.
Data availability
The accession number for all the sequencing results reported in this paper is GEO: GSE119964. Additional data is available by request to the lead contact.
Supplementary Material
Highlights.
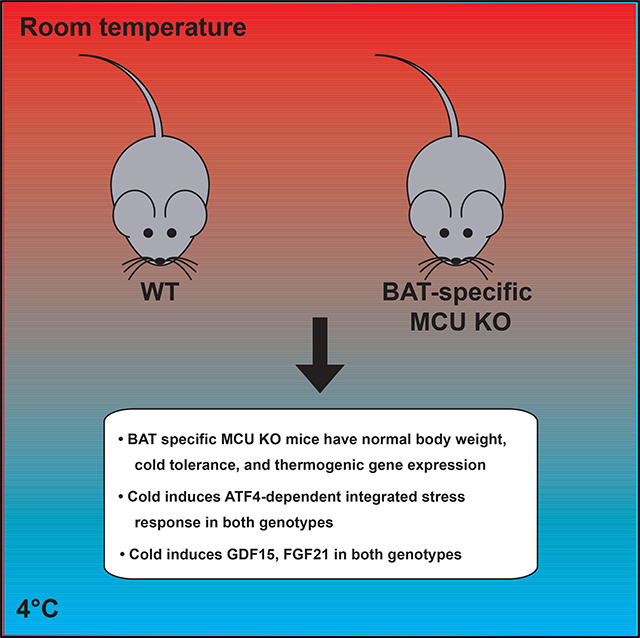
BAT-specific MCU KO mice have normal body weight and cold tolerance
MCU KO does not impact the gene expression response to acute cold stress in BAT
Cold induces ATF4-dependent integrates stress response in both genotypes
Circulating GDF15 and FGF21 rise in both genotypes in response to cold
ACKNOWLEDGMENTS
We thank B. Spiegelman for providing the immortalized BAT cell line and for technical feedback; B. Lowell and D. Kong for technical help and guidance in generation of MCUfl/fl mouse line; J. Avruch, L. Zhao, and A. Soukas for assistance with mouse metabolic studies and harvesting BAT; B. Reinstadler for help on statistical analysis; and other members of the Mootha laboratory for discussions and advice. Embryonic stem cell injections to generate the MCUfl/fl mouse line were performed by the Beth Israel Deaconess Medical Center Transgenic Core. This work was supported in part by grants from the National Institutes of Health R01HL130143 to V.K.M and F31DK107187 to D.F. E.M. was supported by a graduate research fellowship from the Howard Hughes Medical Institute. Y.S. received support from the Helen Hay Whitney Foundation. V.K.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
DECLARATION OF INTERESTS
V.K.M. is a paid advisor to Janssen Pharmaceuticals and 5am Ventures and is a founder and owns equity in Raze Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.013.
REFERENCES
- Al-Shaikhaly MH, Nedergaard J, and Cannon B (1979). Sodium-induced calcium release from mitochondria in brown adipose tissue. Proc. Natl. Acad. Sci. USA 76, 2350–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, and Lowell BB (2002). betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science 297, 843–845. [DOI] [PubMed] [Google Scholar]
- Bao XR, Ong S-E, Goldberger O, Peng J, Sharma R, Thompson DA, Vafai SB, Cox AG, Marutani E, Ichinose F, et al. (2016). Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Kazak L, Chouchani ET, Bogaczyńska MG, Paranjpe I, Wainwright GL, Bétourné A, Kajimura S, Spiegelman BM, and Kirichok Y (2017). Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1- Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab. 25, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A, Matsuo K, Matsuo I, Wang S, Melhem R, Koromilas AE, and Haj FG (2012). Protein tyrosine phosphatase 1B deficiency potentiates PERK/eIF2α signaling in brown adipocytes. PLoS ONE 7, e34412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2011). Nonshivering thermogenesis and its adequate measurement in metabolic studies. J. Exp. Biol 214, 242–253. [DOI] [PubMed] [Google Scholar]
- Carafoli E, and Lehninger AL (1971). A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem. J 122, 681–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartoumpekis DV, Habeos IG, Ziros PG, Psyrogiannis AI, Kyriazopoulou VE, and Papavassiliou AG (2011). Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol. Med 17, 736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zeng X, Huang X, Serag S, Woolf CJ, and Spiegelman BM (2017). Crosstalk between KCNK3-Mediated Ion Current and Adrenergic Signaling Regulates Adipose Thermogenesis and Obesity. Cell 171, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrysovergis K, Wang X, Kosak J, Lee SH, Kim JS, Foley JF, Travlos G, Singh S, Baek SJ, and Eling TE (2014). NAG-1/GDF-15 prevents obesity by increasing thermogenesis, lipolysis and oxidative metabolism. Int. J. Obes 38, 1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, et al. (2017). Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol 216, 149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E, and Nedergaard J (1988). Beta-adrenergic modulation of Ca2+ uptake by isolated brown adipocytes. Possible involvement of mitochondria. J. Biol. Chem 263, 10574–10582. [PubMed] [Google Scholar]
- Connolly E, Nånberg E, and Nedergaard J (1984). Na+-dependent, alpha-adrenergic mobilization of intracellular (mitochondrial) Ca2+ in brown adipocytes. Eur. J. Biochem 141, 187–193. [DOI] [PubMed] [Google Scholar]
- Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 17, 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Liang C, Edema-Hildebrand F, Riley C, Needham M, and Sue CM (2013). Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology 81, 1819–1826. [DOI] [PubMed] [Google Scholar]
- De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, and Demaurex N (2014). NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem 289, 20377–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca HF, and Engstrom GW (1961). Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 47, 1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Denton RM, McCormack JG, Rutter GA, Burnett P, Edgell NJ, Moule SK, and Diggle TA (1996). The hormonal regulation of pyruvate dehydrogenase complex. Adv. Enzyme Regul 36, 183–198. [DOI] [PubMed] [Google Scholar]
- Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, and Wek RC (2010). Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J. Biol. Chem 285, 33165–33174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper M-E, and Kozak LP (1997). Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94. [DOI] [PubMed] [Google Scholar]
- Estall JL, Ruas JL, Choi CS, Laznik D, Badman M, Maratos-Flier E, Shulman GI, and Spiegelman BM (2009). PGC-1alpha negatively regulates hepatic FGF21 expression by modulating the heme/Rev-Erb(alpha) axis. Proc. Natl. Acad. Sci. USA 106, 22510–22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko A, Lishko PV, and Kirichok Y (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, and Nedergaard J (2009). UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209. [DOI] [PubMed] [Google Scholar]
- Fieni F, Lee SB, Jan YN, and Kirichok Y (2012). Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat. Commun 3, 1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, and Spiegelman BM (2012). FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Zhu L, Baker CD, Cracan V, Yaseen A, Jain M, Clish CB, Brookes PS, Bakovic M, and Mootha VK (2013). Meclizine inhibits mitochondrial respiration through direct targeting of cytosolic phosphoethanolamine metabolism. J. Biol. Chem 288, 35387–35395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudiksen A, and Pilegaard H (2017). PGC-1α and fasting-induced PDH regulation in mouse skeletal muscle. Physiol. Rep. 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Hayato R, Higure Y, Kuba M, Nagai H, Yamashita H, and Kuba K (2011). β3-Adrenergic activation of sequential Ca(2+) release from mitochondria and the endoplasmic reticulum and the subsequent Ca(2+) entry in rodent brown adipocytes. Cell Calcium 49, 400–414. [DOI] [PubMed] [Google Scholar]
- Hondares E, Iglesias R, Giralt A, Gonzalez FJ, Giralt M, Mampel T, and Villarroya F (2011). Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J. Biol. Chem 286, 12983–12990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Gudi R, Wu P, Harris RA, Hamilton J, and Popov KM (1998). Isoenzymes of pyruvate dehydrogenase phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem 273, 17680–17688. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M, Shinoda K, Chen Y, Lu X, Maretich P, et al. (2017). UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med 23, 1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson AS, and Mannervik B (2001). Human glutathione transferase A3–3, a highly efficient catalyst of double-bond isomerization in the biosynthetic pathway of steroid hormones. J. Biol. Chem 276, 33061–33065. [DOI] [PubMed] [Google Scholar]
- Jousse C, Deval C, Maurin AC, Parry L, Chérasse Y, Chaveroux C, Lefloch R, Lenormand P, Bruhat A, and Fafournoux P (2007). TRB3 inhibits the transcriptional activation of stress-regulated genes by a negative feedback on the ATF4 pathway. J. Biol. Chem 282, 15851–15861. [DOI] [PubMed] [Google Scholar]
- Kamer KJ, and Mootha VK (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Grabarek Z, and Mootha VK (2017). High-affinity cooperative Ca2+ binding by MICU1-MICU2 serves as an on-off switch for the uniporter. EMBO Rep. 18, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, Cohen P, Vetrivelan R, Lu GZ, Laznik-Bogoslavski D, Hasenfuss SC, et al. (2015). A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163, 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazak L, Chouchani ET, Stavrovskaya IG, Lu GZ, Jedrychowski MP, Egan DF, Kumari M, Kong X, Erickson BK, Szpyt J, et al. (2017). UCP1 deficiency causes brown fat respiratory chain depletion and sensitizes mitochondria to calcium overload-induced dysfunction. Proc. Natl. Acad. Sci. USA 114, 7981–7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keipert S, Ost M, Johann K, Imber F, Jastroch M, van Schothorst EM, Keijer J, and Klaus S (2014). Skeletal muscle mitochondrial uncoupling drives endocrine cross-talk through the induction of FGF21 as a myokine. Am. J. Physiol. Endocrinol. Metab 306, E469–E482. [DOI] [PubMed] [Google Scholar]
- Keipert S, Kutschke M, Ost M, Schwarzmayr T, van Schothorst EM, Lamp D, Brachthäuser L, Hamp I, Mazibuko SE, Hartwig S, et al. (2017). Long-Term Cold Adaptation Does Not Require FGF21 or UCP1. Cell Metab. 26, 437–446. [DOI] [PubMed] [Google Scholar]
- Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, Heineke J, Kotlarz D, Xu J, Molkentin JD, et al. (2006). The transforming growth factor-β superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ. Res 98, 351–360. [DOI] [PubMed] [Google Scholar]
- Khan NA, Nikkanen J, Yatsuga S, Jackson C, Wang L, Pradhan S, Kivelä R, Pessia A, Velagapudi V, and Suomalainen A (2017). mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 26, 419–428. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest 115, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al. (2013). Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med 19, 83–92. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Kong X, Banks A, Liu T, Kazak L, Rao RR, Cohen P, Wang X, Yu S, Lo JC, Tseng YH, et al. (2014). IRF4 is a key thermogenic transcriptional partner of PGC-1α. Cell 158, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs-Bogdan E, Sancak Y, Kamer KJ, Plovanich M, Jambhekar A, Huber RJ, Myre MA, Blower MD, and Mootha VK (2014). Reconstitution of the mitochondrial calcium uniporter in yeast. Proc. Natl. Acad. Sci. USA 111, 8985–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak LP (2014). Genetic variation in brown fat activity and body weight regulation in mice: lessons for human studies. Biochim. Biophys. Acta 1842, 370–376. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, and Molkentin JD (2015). The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart. Cell Rep. 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver EV, and Pappone PA (2002). Beta-adrenergic potentiation of endoplasmic reticulum Ca(2+) release in brown fat cells. Am. J. Physiol. Cell Physiol 282, C1016–C1024. [DOI] [PubMed] [Google Scholar]
- Lee SC, Nuccitelli R, and Pappone PA (1993). Adrenergically activated Ca2+ increases in brown fat cells: effects of Ca2+, K+, and K channel block. Am. J. Physiol 264, C217–C228. [DOI] [PubMed] [Google Scholar]
- Lehtonen JM, Forsström S, Bottani E, Viscomi C, Baris OR, Isoniemi H, Höckerstedt K, Österlund P, Hurme M, Jylhävä J, et al. (2016). FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology 87, 2290–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis-Smith D, Kamer KJ, Griffin H, Childs A-M, Pysden K, Titov D, Duff J, Pyle A, Taylor RW, Yu-Wai-Man P, et al. (2016). Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet 2, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gu H, Gan L, Xu Y, Feng F, Saeed M, and Sun C (2017). Reducing Smad3/ATF4 was essential for Sirt1 inhibiting ER stress-induced apoptosis in mice brown adipose tissue. Oncotarget 8, 9267–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CV, Szabadkai G, Sharpe JA, Parry DA, Torelli S, Childs AM, Kriek M, Phadke R, Johnson CA, Roberts NY, et al. ; UK10K Consortium (2014). Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet 46, 188–193. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, and Flier JS (1993). Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 366, 740–742. [DOI] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. (2015). The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch GS, and Ryall JG (2008). Role of beta-adrenoceptor signaling in skeletal muscle: implications for muscle wasting and disease. Physiol. Rev 88, 729–767. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamady H, and Storey KB (2008). Coping with the stress: expression of ATF4, ATF6, and downstream targets in organs of hibernating ground squirrels. Arch. Biochem. Biophys 477, 77–85. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, and Denton RM (1990). Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev 70, 391–425. [DOI] [PubMed] [Google Scholar]
- Montero R, Yubero D, Villarroya J, Henares D, Jou C, Rodríguez MA, Ramos F, Nascimento A, Ortez CI, Campistol J, et al. (2016). GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS ONE 11, e0148709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, and Finkel T (2014). Unresolved questions from the analysis of mice lacking MCU expression. Biochem. Biophys. Res. Commun 449, 384–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagaki I, Sasaki S, Yahata T, Takasaki H, and Hori S (2005). Cytoplasmic and mitochondrial Ca levels in brown adipocytes. Acta Physiol. Scand 183, 89–97. [DOI] [PubMed] [Google Scholar]
- Nam M, Akie TE, Sanosaka M, Craige SM, Kant S, Keaney JF Jr., and Cooper MP (2017). Mitochondrial retrograde signaling connects respiratory capacity to thermogenic gene expression. Sci. Rep 7, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ost M, Keipert S, van Schothorst EM, Donner V, van der Stelt I, Kipp AP, Petzke KJ, Jove M, Pamplona R, Portero-Otin M, et al. (2015). Muscle mitohormesis promotes cellular survival via serine/glycine pathway flux. FASEB J. 29, 1314–1328. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep. 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 107, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Fujimoto M, Lopes A, and Wang YX (2009). Twist-1 is a PPAR-delta-inducible, negative-feedback regulator of PGC-1α in brown fat metabolism. Cell 137, 73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, and Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 467, 291–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Quirós PM, Prado MA, Zamboni N, D’Amico D, Williams RW, Finley D, Gygi SP, and Auwerx J (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, and Rizzuto R (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffalli-Mathieu F, Orre C, Stridsberg M, Hansson Edalat M, and Mannervik B (2008). Targeting human glutathione transferase A3–3 attenuates progesterone production in human steroidogenic cells. Biochem. J 414, 103–109. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. (2008). PRDM16 controls a brown fat/skeletal muscle switch. Nature 454, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Yao A, Hattori K, Sugawara S, Naguro I, Koike M, Uchiyama Y, Takeda K, and Ichijo H (2016). The Ablation of Mitochondrial Protein Phosphatase Pgam5 Confers Resistance Against Metabolic Stress. EBioMedicine 5, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, and Graff JM (2009). Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 58, 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan SK, Thomason LC, Kuznetsov SG, and Court DL (2009). Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protoc 4, 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Campanello GC, Flicker D, Grabarek Z, Hu J, Luo C, Banerjee R, and Mootha VK (2017). The Human Knockout Gene CLYBL Connects Itaconate to Vitamin B12. Cell 171, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford KI, Middelbeek RJ, Townsend KL, An D, Nygaard EB, Hitchcox KM, Markan KR, Nakano K, Hirshman MF, Tseng YH, et al. (2013). Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Invest 123, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. (2002). Large-scale analysis of the human and mouse transcriptomes. Proc. Natl. Acad. Sci. USA 99, 4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Kraus A, Harris RA, and Holness MJ (2000). Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem. J. 346, 651–657. [PMC free article] [PubMed] [Google Scholar]
- Territo PR, Mootha VK, French SA, and Balaban RS (2000). Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am. J. Physiol. Cell Physiol 278, C423–C435. [DOI] [PubMed] [Google Scholar]
- Townsend KL, and Tseng YH (2014). Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab. 25, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CW, Wu Y, Pao PC, Phillips CB, Williams C, Miller C, Ranaghan M, and Tsai M-F (2017). Proteolytic control of the mitochondrial calcium uniporter complex. Proc. Natl. Acad. Sci. USA 114, 4388–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjøsund KE, Tulkki V, et al. (2010). Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet 19, 3948–3958. [DOI] [PubMed] [Google Scholar]
- Vasington FD, and Murphy JV (1962). Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem 237, 2670–2677. [PubMed] [Google Scholar]
- Vattem KM, and Wek RC (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 101, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes L, Chin R, Young SG, and Reue K (2011). Heart-type fatty acidbinding protein is essential for efficient brown adipose tissue fatty acid oxidation and cold tolerance. J. Biol. Chem 286, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Huang Z, Du Y, Cheng Y, Chen S, and Guo F (2010). ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 20, 174–184. [DOI] [PubMed] [Google Scholar]
- Wang C, Xia T, Du Y, Meng Q, Li H, Liu B, Chen S, and Guo F (2013). Effects of ATF4 on PGC1α expression in brown adipose tissue and metabolic responses to cold stress. Metabolism 62, 282–289. [DOI] [PubMed] [Google Scholar]
- Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, et al. (2014). Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J 33, 418–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Boström P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, et al. (2012). Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Walker K, Min X, Hale C, Tran T, Komorowski R, Yang J, Davda J, Nuanmanee N, Kemp D, et al. (2017). Long-acting MIC-1/GDF15 molecules to treat obesity: Evidence from mice to monkeys. Sci. Transl. Med 9, 1–12. [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, Kojima T, Ito M, Tanaka M, Saiki R, and Koga Y (2015). Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol 78, 814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Wu J, Cohen P, Kazak L, Khandekar MJ, Jedrychowski MP, Zeng X, Gygi SP, and Spiegelman BM (2013). Fat cells directly sense temperature to activate thermogenesis. Proc. Natl. Acad. Sci. USA 110, 12480–12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Arch JRS, and Ashwell M (1984). Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett. 167, 10–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software availability
Source codes for the STAR 2.4.0j (Dobin et al., 2013), HTSeq (Anders et al., 2015), and DEseq (Love et al., 2014) software packages are freely available for download through the corresponding references. GSEA and MSigDB can be freely accessed through the Broad Institute website, and is implemented via a graphical user interface. MATLAB is accessible through MathWorks, Inc. on a subscription basis.
Data availability
The accession number for all the sequencing results reported in this paper is GEO: GSE119964. Additional data is available by request to the lead contact.
The accession number for all the sequencing results reported in this paper is GEO: GSE119964. Additional data is available by request to the lead contact.