

Abstract
Simultaneous profiling of transcriptome and chromatin accessibility within single cells is a powerful approach to dissect gene regulatory programs in complex tissues. However, the current tools are limited by modest throughput. We now describe an ultra high-throughput method, Paired-seq, for parallel analysis of transcriptome and accessible chromatin in millions of single cells. We demonstrate the utility of Paired-seq for analyzing the dynamic and cell-type specific gene regulatory programs in complex tissues, by applying it to mouse adult cerebral cortex and fetal forebrain. The joint profiles of a large number of single cells allowed us to deconvolute the transcriptome and open chromatin landscapes in the major cell types within these brain tissues, infer putative target genes of candidate enhancers, and reconstruct the trajectory of cellular lineages within the developing forebrain.
Introduction
The spatiotemporal gene expression patterns of multi-cellular organisms are driven in large part by the cis-regulatory elements (CREs) in the genome1. Applications of next generation DNA sequencing techniques such as ChIP-seq2, DNase-seq3 and ATAC-seq4 have enabled identification of candidate CREs in the genomes of many species5, 6, but the cellular heterogeneity of primary tissues in these organisms presents a significant challenge for annotation of cell-type specificity of the CREs using the conventional assays. To address this challenge, a variety of single-cell genomic tools have been invented7. In particular, methods have been developed to probe chromatin accessibility8–10, histone modifications, transcription factors11–14, higher-order chromatin conformation15, and DNA methylation16, 17 and its derivatives18–20, in single cells. These techniques have improved our understanding of the heterogeneity of epigenome among cells. More recently, with the advancement in higher throughput single-cell analysis of transcriptome21, 22, chromatin accessibility23, 24, DNA methylome25 and histone modification26, it has been possible to deconvolute cell types from mixed cell populations and dissect the cell-type specific transcriptomic and epigenomic states in primary tissues.
While most of the present single-cell assays profile individual molecular modalities one at a time7, a few do allow for parallel analysis of multiple modalities in the same cells. These include combined analysis of gene expression and genome sequence27, 28, joint transcriptome and DNA methylome profiling29–31, and simultaneous mapping of nucleosome occupancy and DNA methylome32, 33 along with transcriptome34. A method for combined single-cell analysis of chromatin accessibility and transcriptome have also been developed, by physically splitting nuclei and cytoplasm of induvial single cells35. Recently, methods for joint analysis of chromosome architecture and DNA methylation in single cells have also been reported36, 37. Droplet-based systems have also been adopted for simultaneous measurement of protein-epitopes and transcriptome from thousands of single cells38, 39.
To better understand the cell-type specific gene regulatory programs, it is necessary to simultaneously measure the transcriptome and states of the candidate CREs within the same cells. Recently, two methods that allow for co-assay of RNA and accessible chromatin in individual cells were reported40, 41. These strategies can jointly profile chromatin accessibility and transcriptome in tens of thousands individual cells in an experiment. However, for analysis of gene regulatory programs at an organismal scale, a cost- and time-efficient technology of much greater throughput would be desired.
We describe here a strategy for ultra high-throughput joint analysis of transcriptome and accessible chromatin that could be used to study millions of individual cells at once. The method, referred hereafter as Paired-seq, for parallel analysis of individual cells for RNA expression and DNA accessibility by sequencing, adopts a ligation-based combinatorial indexing strategy42 to simultaneously tag both the open chromatin fragments generated by the Tn5 transposases and the cDNA molecules generated from reverse transcription of RNA in millions of cells. We also introduce an amplify-and-split “library-dedicating” strategy to separately amplify the DNA fragments corresponding to the open chromatin and transcriptome, for construction of DNA sequencing libraries. To demonstrate the utility of Paired-seq, we used it to study the gene regulatory programs in forebrain tissues in two stages of mouse fetal development and in cerebral cortex from adult mice. We uncovered major cell types in these brain tissues and revealed the dynamic cellular composition during mouse forebrain development. We also inferred putative target gene for candidate CREs that we mapped, and reconstructed the trajectories for different brain cell lineages.
Results
Joint-analysis of accessible chromatin and transcriptome in single cells
Paired-seq includes the following steps. First, cell-specific combinations of DNA barcodes were introduced to open chromatin fragments and cDNA molecules from the same cells through multiple rounds of ligation reactions coupled with split-and-pooling 42 (Fig. 1a). Specifically, Tn5 tagmentation reaction was first carried out for cells in 8 different wells with Tn5 transposase containing barcoded adaptors. The cells were then subject to reverse transcription (RT) with primers containing the same set of barcodes so that the DNA fragments and cDNA from the same wells were labelled with the same first-round DNA barcodes. Next, the cells were pooled and redistributed to a 96-well plate containing well-specific DNA barcodes, which were ligated to 5’ ends of the DNA fragments released in the tagmentation reaction and the cDNA molecules. Two additional rounds of ligation were then performed in 96-well plates after pooling and splitting, leading to the generation of more than 107 unique barcode combinations (Fig. 1a). Second, the nuclei were split into sub-libraries and lysed, and the DNA was purified from each sub-library, which was subject to amplification by a modified TdT (terminal deoxynucleotidyl transferase)-assisted single-stranded DNA amplification method43. The amplified DNA products were then split into two portions and digested with restriction enzymes targeting the pre-designed sites in Tn5 and RT primers respectively, to give rise to DNA and RNA libraries for sequencing (Fig. 1a, Extended Data Fig. 1a, Supplementary Tables 1, 2 and see Methods).
Fig. 1 |. Paired-seq enables simultaneous profiling of accessible chromatin and gene expression in millions of single cells.
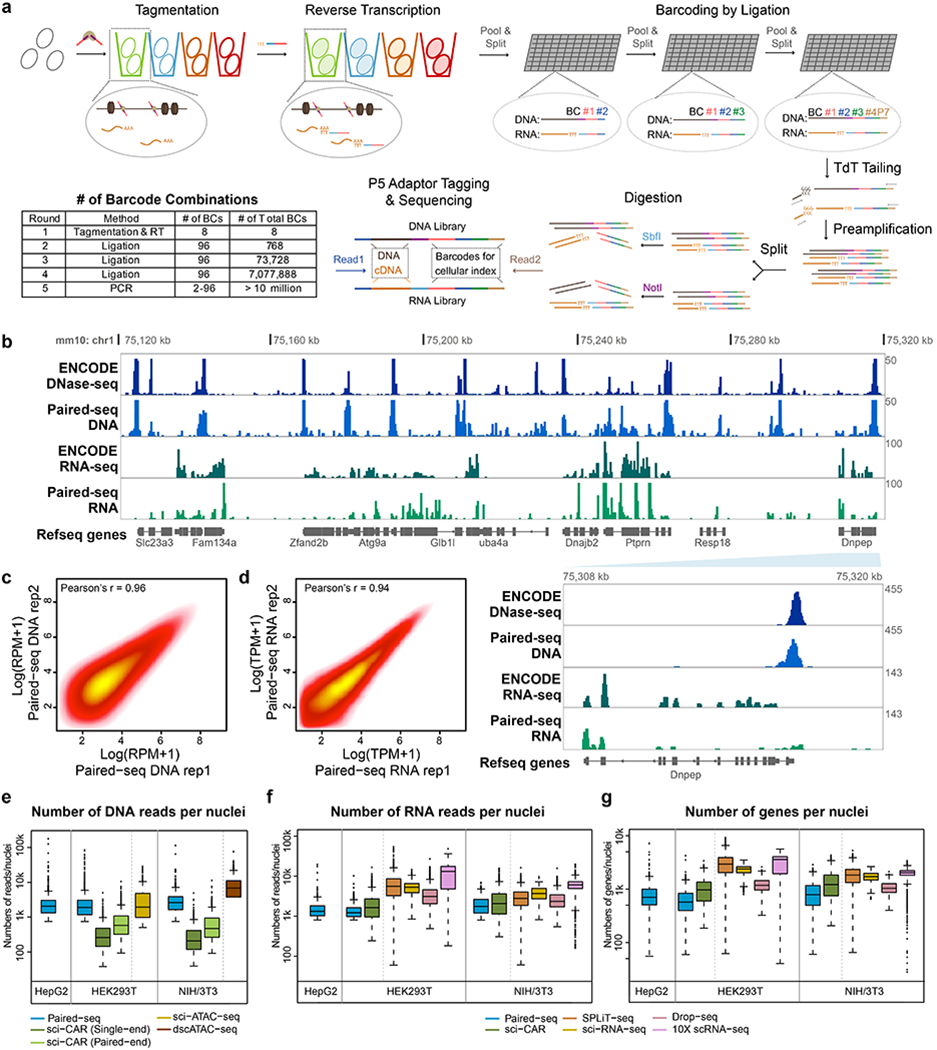
a, Schematic of Paired-seq workflow. Paired-seq includes five rounds of combinatorial barcoding that enables labeling of millions of cells in one single experiment. In the first round, cells are subject to Tn5 transposition followed by reverse transcription in separate tubes. This is followed by three rounds of ligation-mediated barcoding carried out in 96-well plates using a split and pool strategy. In the final round, DNA barcode tags are first added to genomic DNA and cDNA by TdT-assisted DNA tailing. The resulting DNA is PCR amplified with different primers, and subject to restriction digestion to produce separate libraries for detecting chromatin accessibility and RNA transcripts. b, A representative genome browser view of Paired-seq data from NIH/3T3 cells (Mouse genome assembly mm10). Tracks of DNase-seq and RNA-seq data downloaded from ENCODE data portal are also shown. Proportions of DNA and RNA reads in both libraries are shown. A zoomed-in view of Dnpep gene locus were shown in the bottom right panel, indicated by the light blue wedge. Scatter plots show the correlation of read counts from two technical replicates of Paired-seq DNA profiles (c) or RNA profiles (d). Boxplots show (e) the number of uniquely mapped DNA reads, (f) the number of uniquely RNA mapped reads and (g) the number of genes captured per cell from either HEK293T, HepG2 and NIH/3T3 cells. As comparison, the numbers of reads or genes captured per cell by sci-CAR40 (GSE117089), sci-ATAC-seq9 (GSE67446), dscATAC-seq44 (GSE123581), SPLiT-seq42 (GSE110823), sci-RNA-seq45 (GSE98561), Drop-seq21 (GSE63269) and 10X scRNA-seq (1k_hgmm_v3_nextgem dataset) from the same cell types are also shown. All datasets were sequenced or down-sampled to ~15k raw reads per cell. In boxplots center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR). Source data for panels e-g are available online; sample sizes are provided there.
As a proof of principle, we first applied Paired-seq to individual and mixed population of two human cell lines and a mouse cell line, namely NIH/3T3 (murine), HepG2 (human) and HEK293T (human) (Methods). We compared the distribution of mapped reads around transcription start sites (TSS) and transcription termination sites (TTS) from both libraries (Extended Data Fig. 1b). As expected, reads from the DNA library showed a high enrichment around TSS while those from the RNA library were enriched at regions upstream TTS (Fig. 1b). Both DNA and RNA libraries showed high purity, evidenced by high percentage of the restriction enzyme cutting sites in the short-read sequences, suggesting a high efficiency of the restriction enzyme-based “library-dedicating” strategy (Extended Data Fig. 1c). Further, the ensemble signals from the two biological replicates were highly reproducible (Fig. 1c, d), and correlated very well with the published bulk DNase-seq and polyA RNA-seq datasets from the same cell lines5, respectively (Fig. 1b and Extended Data Fig. 1d, e).
The ligation-based combinatorial barcoding strategy used here could tag well over 1 million cells in a single experiment. As a proof of principle, we collected 8.0 million nuclei for barcoding and after 3-round of ligation, we recovered 1.51 million barcoded nuclei (18.9% recovery rate). Without losing generality, we then divided the nuclei into sub-libraries and constructed and sequenced a sub-library corresponding to ~10,000 nuclei (0.66% of the total number of the barcoded nuclei) to a moderate sequencing depth (15 k reads/nuclei and UMI duplication rate ~60%), obtaining median counts of 2,635, 2,066 and 1,641 uniquely mapped DNA reads per nucleus, and median counts of 1,872, 1,337 and 1,236 uniquely mapped RNA reads per nucleus for NIH/3T3, HepG2 and HEK293T, respectively (Fig. 1e, f and Supplementary Table 3). The number of uniquely mapped reads, the fraction of DNA reads around TSS and inside peaks of DNA library, and the numbers of genes captured of RNA library for each cell are similar to those of recently published sci-CAR method40 (Fig. 1e–g and Extended Data Fig. 1f, g). Compared to the stand-alone single-cell methods, Paired-seq DNA have similar coverage to sci-ATAC-seq9 but lower than a recently published dscATAC-seq (droplet single-cell ATAC-seq)44 (Fig. 1e). Paired-seq RNA reads were lower than commonly used single-cell RNA-seq methods21, 42, 45 (Fig. 1f, g), which may result from the sub-optimal buffer conditions and degradation of RNA during the multi-omic barcoding processes. On the other hand, the increased number of barcode combinations reduced the chance of random barcode collision to less than 3%, estimated based on analysis of a mixed population containing human and mouse cells (Extended Data Fig. 1h–j). After filtering out the cells with low sequencing coverages (less than 750 uniquely mapped reads), we recovered 2,053 (out of a total ~6,000 profiled) human cells with both DNA profiles and RNA profiles. Using principal components analysis (PCA) followed by an unsupervised density-based clustering method, these cells were readily clustered into two groups, corresponding to the HepG2 and HEK293T cells, respectively (Extended Data Fig. 1k, l).
Paired-seq recovered the major cell types in the adult mouse cerebral cortex
To demonstrate the utility of Paired-seq to resolve heterogeneity of complex tissues, we applied it to freshly collected adult mouse cerebral cortex. We used 10 million nuclei as input and recovered approximately 2.51 million barcoded nuclei (25.1% recovery rate). We then sequenced a few sub-libraries corresponding to ~30k nuclei (1.20% of total library), obtaining 15,191 nuclei with both DNA and RNA profiles, with median counts of 1,762 uniquely mapped DNA reads and 1,166 uniquely mapped RNA reads per nucleus, respectively, for the sub-library sequenced to ~25k reads/nucleus depth (Supplementary Table 3).
To cluster the cells based on the similarity of the Paired-seq profiles, we adopted a computational software SnapATAC, originally designed for processing of snATAC-seq data46. Cell-to-bins DNA matrix and cell-to-genes RNA matrix were generated from the Paired-seq data and used to calculate the pairwise Jaccard similarity matrices. The resulting two matrices were then combined by computing their Hadamard product. The combined matrix was then subject to dimensionality reduction with the use of PCA, followed by graph-based clustering using Louvain clustering approach as previously described46 (Fig. 2a and see Methods). This analysis revealed nine major cell types in the mouse cerebral cortex, including 3 types of glutamatergic neurons (Snap25+, Neurod6+, Gad1−), 3 types of GABAergic neurons (Snap25+, Neurod6−, Gad1+), and 3 non-neuronal cell types corresponding to astrocytes (Apoe+), microglia (C1qb+) and oligodendrocytes (Mog+)47 (Fig. 2b, c, and Extended Data Fig. 2a–c). These results indicate that Paired-seq can uncover major known cell types in a complex tissue.
Fig. 2 |. Paired-seq identified major cell types in the mouse cerebral cortex.
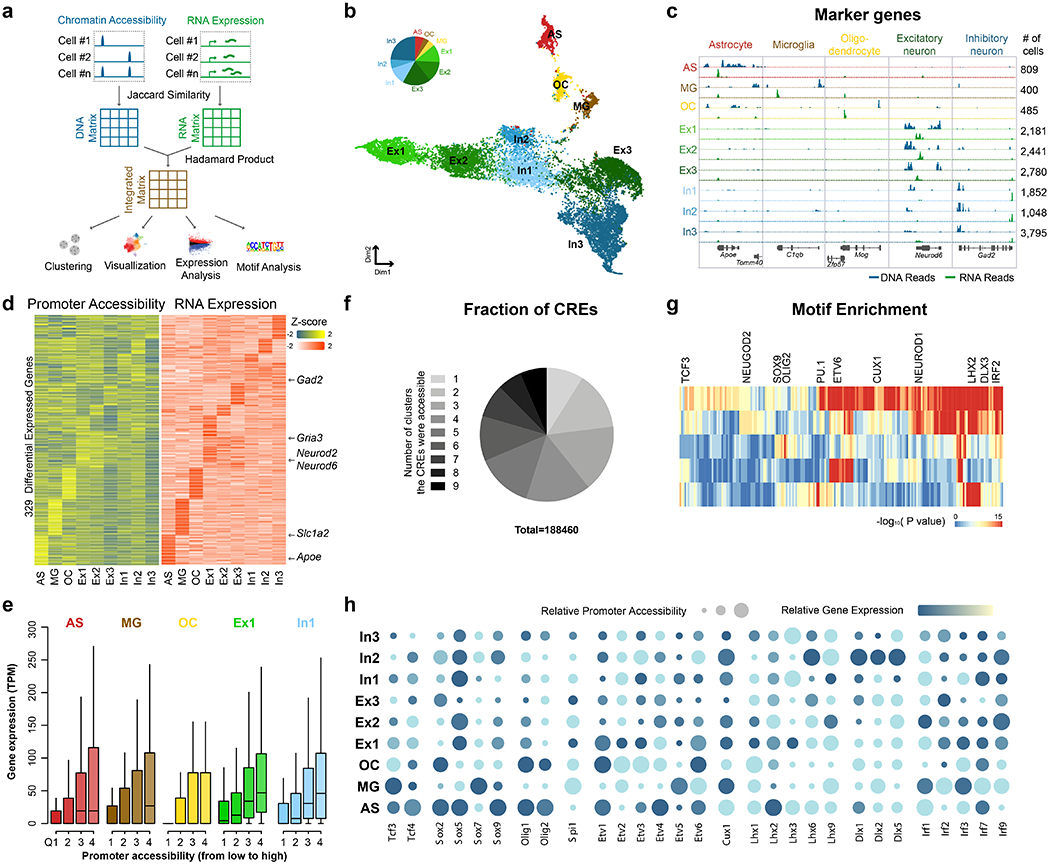
a, Schematic of integrated analysis of Paired-seq DNA and RNA profiles. Pairwise similarity matrices were first constructed from accessible chromatin and expression profiles of the nuclei using the Jaccard similarity index. DNA and RNA matrices are combined into a new matrix by calculating the Hadamard product, which is then processed with SnapATAC to cluster cells and generate both open chromatin and RNA transcript profiles of each cluster. b, Clustering of single nuclei from mouse adult cerebral cortex revealed nine major groups: astrocyte (AS), microglia (MG), oligodendrocyte (OC), Glutamatergic neural cells (Ex1, Ex2 and Ex3) and GABAergic neural cells (In1, In2 and In3). c, Aggregate chromatin accessibility (blue) and gene expression (green) profiles for each cell cluster at several marker gene loci. d, Heatmaps show promoter accessibility and the corresponding gene expression level of differentially expressed genes. e, Expression levels of genes for each cluster are plotted for each quantile of promoter accessibility. In boxplots, center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR). Sample sizes are provided in the Source data available online. f, Pie-chart showing the fractions of CREs accessible in different number of clusters. g, Transcription factor motif enrichment analysis for each major group. h, Promoter accessibility and gene expression of representative TF genes. Relative promoter accessibilities and expression levels of each TF gene are indicated by the size and color of circles. Source data for panels b, e and f are available online.
To identify the genes specifically expressed in each cell population, we performed differential gene expression analysis by comparing the aggregated RNA reads from one cluster with that from all the other clusters, and recovered a combined total of 329 cell-type-specific genes (p < 0.05 by edgeR48, see Methods). The variability of chromatin accessibility at their promoters exhibited good concordance with the variation in gene expression levels (Fig. 2d, e and Extended Data Fig. 2d, e). We also identified 188,460 potential CREs that were accessible in one or more cell types, and found that a majority of them were only accessible in a cell type specific manner (Fig. 2f). Motif enrichment analysis with the JASPAR database49 identified potential transcription factors (TFs) acting in at least one of the major cell groups (Fig. 2g). We further investigated the promoter accessibility and gene expression of individual TF genes across major cell groups, and found members of some TF families exhibit distinct expression patterns in different cell clusters (Fig. 2h). For example, although SOX9 motif was enriched for both neuronal and non-neuronal cells, Sox5 was expressed in multiple cell clusters including astrocyte, excitatory neurons, and inhibitory neurons while expression of Sox9 is more restricted to astrocytes, consistent with previous reports that SOX5 controls generation of multiple neuron subtypes50 and SOX9 regulates astrocyte-specific gene expression in the adult brain51. Hence, combining gene expression with chromatin accessibility analysis is useful for identifying functional regulators.
The dynamic cellular composition of the developing mouse forebrain
We further applied Paired-seq to frozen mouse forebrain samples previously collected from two different stages of fetal development, E12.5 and E16.5, which were analyzed as part of the ENCODE project52 (see Methods). We collected a total of 6.0 million nuclei from these two samples, and recovered 0.56 million barcoded nuclei (9.3% recovery rate). We then constructed and sequenced sub-libraries containing ~20k nuclei (3.57% of total library), obtaining 12,155 nuclei with both DNA and RNA profiles, after removing the nuclei with less than 400 DNA reads and 150 RNA reads (Supplementary Table 3). Using Snap-ATAC46, we classified these nuclear profiles into 8 distinct groups and assigned cell type identity based on marker gene expression (Fig. 3 a, b and Extended Data Fig. 3). In line with previous observations23, the proportion of neuronal progenitor cells decreased from E12.5 to E16.5, together with the expansion of glutamatergic neuron cells. In the adult mouse cerebral cortex, we observed a dramatic increase of astrocytes and mature neurons populations, accompanied by a decrease of neuronal progenitors (dEx1 and dIn1 with accessibility at Hes1 and Ascl1 loci, respectively) (Fig. 3a, Extended Data Fig. 3c). These results also demonstrated the capability of Paired-seq in dissecting heterogeneity of cryo-preserved biospecimens.
Fig. 3 |. Paired-seq links candidate cis-regulatory elements to their putative target genes.
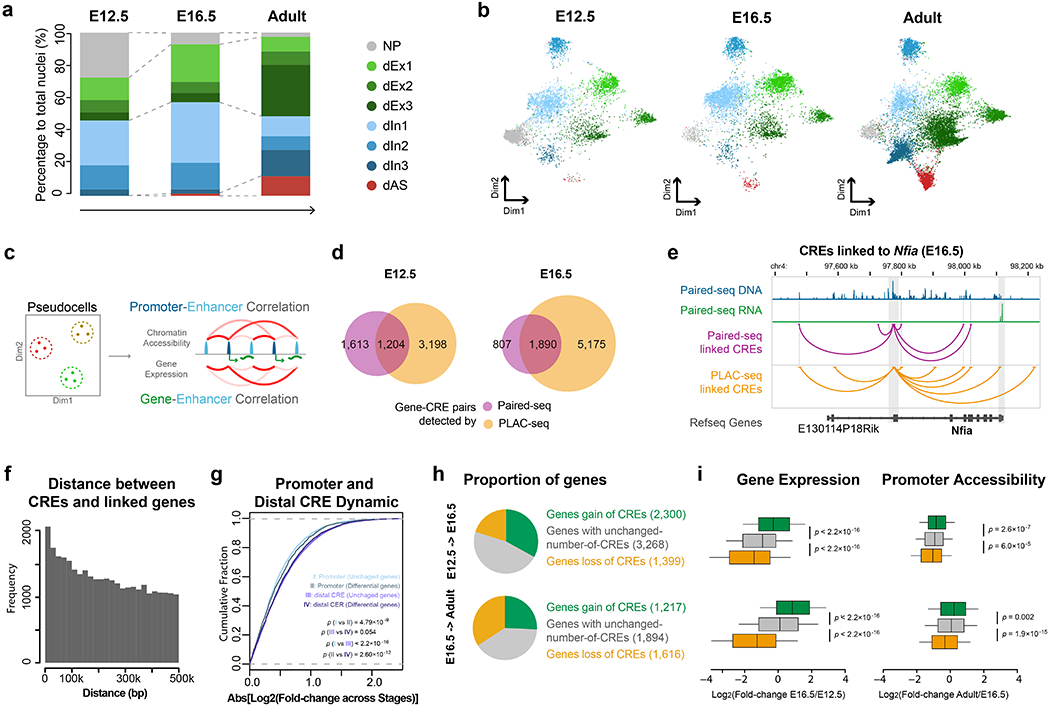
Clustering of single nuclei from mouse E12.5 and E16.5 forebrain samples revealed eight distinct major groups: neuronal progenitors (NP), glutamatergic neural cells (dEx1, dEx2, dEx3), GABAergic neural cells (dIn1, dIn2 and dIn3), and astrocytes (dAS) according to the maker genes (Extended Data Fig. 3c.) a, Stacked bar charts showing the percentages of different cell clusters identified from E12.5 forebrain, E16.5 forebrain and adult cerebral cortex. b, UMAP plot shows the different representation of cell clusters from E12.5 forebrain, E16.5 forebrain and adult cerebral cortex. c, Schematics for identifying potential gene-CRE pairs. d, Venn-diagram showing the fraction of gene-CRE pairs identified from Paired-seq and H3K4me3 PLAC-seq data from mouse E12.5 and E16.5 forebrains. e, Genome browser view of the Nfia locus. Gene-CRE pairs identified by Paired-seq and PLAC-seq data from E16.5 mouse forebrain samples are shown in purple and yellow, respectively. Promoter region and 3’UTR of Nfia gene are highlighted in grey. f, Histogram of the genomic distances between the candidate CREs and their linked genes. g, Cumulative distribution function plot of promoter and CRE dynamics. Genes were grouped into unchanged genes and differentially expressed genes according to the fold-change of the expression level between E12.5 and E16.5 (Log2[Fold-change]>2). The x-axis is the absolute value of fold-change of promoter or CRE accessibility between the two stages. P-value, two sided K-S test, nI and III = 22,923 unchanged genes and nII and IV = 1,776 differentially expressed genes. h, Pie-charts showing genes classified according to changes of candidate CREs linked to them: genes with a gain of linked candidate CREs between stages (Log2[fold-change] > 3), genes with unchanged number of CREs (−1 < Log2[fold-change] < 1) and genes with a loss of linked candidate CREs (Log2[fold-change] < −3). i, Boxplots showing the fold-change of expression and promoter accessibility of genes in the 3 groups. P-value, two-sided K-S test. In boxplots center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR). The sample size of each group is provided in h. Source data for panels a and b are available online.
Paired-seq allowed linking of cis-regulatory elements to their putative target genes
A large number of CREs have been annotated in the mammalian genome, but annotating their target genes remains a challenge due to the fact that many CREs can regulate expression of genes from a large genomic distance53. The knowledge of open chromatin and RNA transcripts from the same cells provides an excellent opportunity to link CREs to potential target genes in specific cell types in the developing mouse brain. The ultra high-throughput nature of Paired-seq can further help to overcome the sparsity of DNA and RNA reads from individual cells: we merged 50 nuclei exhibiting high similarity to each other into pseudocells (based on integrated Jaccard matrix). We then calculated the pairwise Pearson correlation coefficient (PCC) of the normalized gene expression and promoter accessibility with CRE accessibility (within 500-kb range of TSS) across these pseudocells (Fig. 3c and Methods). We first identified 171,551 and 173,694 candidate CREs from E12.5 and E16.5 forebrains, respectively. We found that 10,097 and 6,197 gene-CRE pairs, corresponding to 4,132 and 3,123 genes from these two stages, respectively, showed a significant PCC (FDR < 0.1, Extended Data Fig. 4a and Supplementary Table 4). To validate the predictions, we compared them with enhancer and promoter chromatin contacts mapped in the same tissue samples using Proximity Ligation-Assisted ChIP-seq (PLAC-seq)54 with antibodies against H3K4me3 (Methods). 1,121 (E12.5) and 1,357 (E16.5) genes with linked CREs were detected by both Paired-seq and PLAC-seq, among them 42.7% (E12.5) and 70.1% (E16.5) of gene-CRE pairs from Paired-seq were also supported by PLAC-seq, while by chance 11.0% and 16.4% are expected (Fig. 3d, e and Extended Data Fig. 4 b, c). This result supports the utility of Paired-seq to predict target genes for individual CREs (Extended Data Fig. 4 d–k). We also identified 34,473 gene-CRE pairs for 5,639 genes from Paired-seq data from adult cerebral cortex (FDR < 0.1, median 3 CREs per gene, with most CREs linked to only one gene. Fig. 3f, Extended Data Fig. 4a, l–o and Supplementary Table 5). Only a small proportion of the CREs (17.0%) were linked to the nearest genes, but more than half were linked to the top 5 nearest genes (Extended Data Fig. 4o).
The promoter of differentially expressed genes are generally more dynamic than those of stably expressed genes. For both groups of genes, the linked CREs showed much more dynamics in chromatin accessibility than the corresponding promoters (Fig. 3g). In addition, we found that the changes in gene expression levels between stages are correlated with the numbers of linked CREs, with genes linked to the dynamic CREs tending to be upregulated or downregulated between the two stages, consistent with a role for the candidate CREs in regulation of the linked genes (Fig. 3h, i and Extended Data Fig. 5). Interestingly, many genes with a large number of linked CREs were involved in key cellular processes in neural development (Extended Data Fig. 5c–e).
Reconstruction of cellular trajectory from Paired-seq data
The joint analysis of open chromatin and RNA profiles in individual cells also allowed us to construct cellular trajectories of mouse forebrains during fetal development. As a proof of principle, we used a random-walk-based distance, diffusion pseudotime (DPT)55, to position individual nuclei in neurogenesis or astrogenesis from the common progenitors (Fig. 4a and Extended Data Fig. 6a–d). We then predicted potential transcriptional regulators of cell fate transition by performing motif enrichment analysis using chromVAR56. We further analyzed the expression levels of the corresponding transcription factor encoding genes (Fig. 4b, c and Extended Data Fig. 6e). By plotting the motif enrichment, TF gene expression levels, and chromatin accessibility on the same pseudotime axis, it is possible to uncover potential regulators of mouse brain development. Indeed, Neurog2, known to be involved in neurogenesis57, is found at the starting points of the neurogenesis trajectory, while Neurod2, which play a role in later stages of neurogenesis and in mature neurons23, 47 (Fig. 4c), appears at a later stage after Neurog2 on the same trajectory.
Fig.4 |. Analysis of cellular trajectory in the developing mouse forebrain.
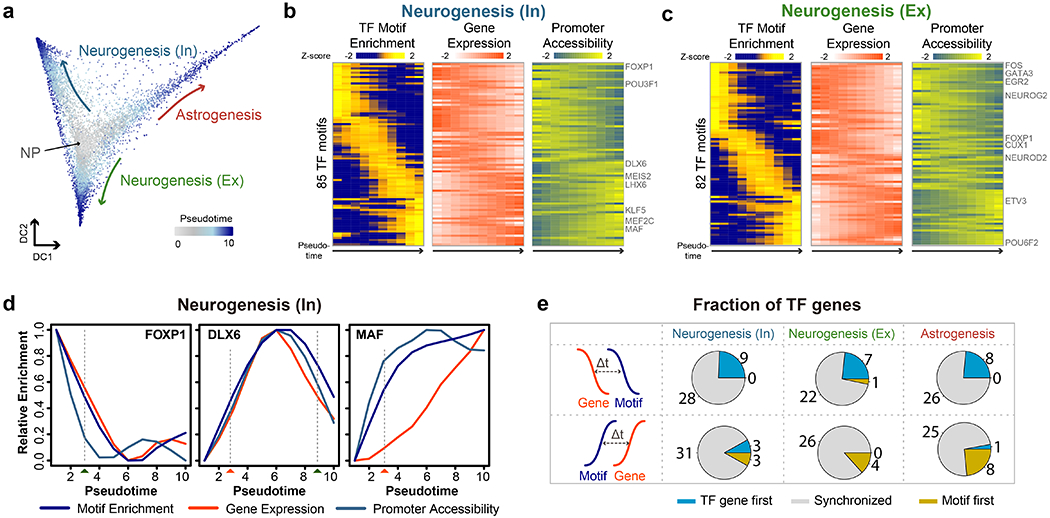
a, Diffusion map showing the trajectories of astrogenesis and neurogenesis. b, c, Heatmaps show the ordering of the chromvAR TF motif enrichments during neurogenesis towards (b) GABAergic neurons and (c) glutamatergic neurons. The relative expression and promoter accessibility of corresponding TF genes are also shown. d, Line plots showing the relative enrichment of TF motifs, gene expression and promoter accessibility for FOXP1, DLX6 and MAF according to the diffusion pesudotime for neurogenesis of GABAergic neurons. The estimated time-of-gain and time-of-loss of TF motif enrichment in open chromatin are indicated by red and green rectangles below. e, Pie-charts of the fraction of TFs showing upregulation of TF genes before (TF gene first), synchronized with, or after (Motif first) the detection of TF motif enrichment in accessible chromatin during neurogenesis towards GABAergic neurons, glutamatergic neurons and astrogenesis. Source data for panels b-d are available online.
To further look into the ordering of TF gene expression and TF motif accessibility, we identified the pseudotime points at which there is a gain or loss of TF motif enrichment in the accessible regions (Fig. 4d, Extended Data Fig. 6f and Methods). We also identified the time of gain or loss based on gene expression and promoter accessibility for TF genes. We then compared the order of activation and inactivation of TF genes and the time of gain or loss of enrichment of their corresponding DNA recognition motifs (Fig. 4e and Extended Data Fig. 6g). We observed that the time of gain or loss of motif in the accessible chromatin are within 20% range of the time of gain or loss of gene expression along the pseudotime trajectories for a majority of the TFs (Fig. 4e and Extended Data Fig. 6g). Only a small portion of TFs do not demonstrate synchronized changes in motif enrichment in the accessible chromatin and gene expression (Fig. 4e). Altogether, these results show that the joint analysis of chromatin accessibility and transcriptome from single nuclei can facilitate the study of gene regulatory programs during development.
Discussion
In summary, we report here an ultra high-throughput method for joint profiling of chromatin accessibility and gene expression in single cells. We demonstrate the utility of this method using both freshly collected and flash frozen brain tissues. Integrated analysis of chromatin accessibility and gene expression from the same cells revealed major cell types during mouse brain development and helped to identify functional regulators involved in this process. We further show that Paired-seq data permitted inference of potential target genes for distal CREs and generation of cell lineage trajectories during forebrain development.
Compared to two previous methods, sci-CAR and SNARE-seq41, Paired-seq dramatically increases the throughput of the analysis by at least two orders of magnitude. Currently, Paired-seq library complexity is comparable to that of both sci-CAR and SNARE-seq, but lower than stand-alone single-cell and single-nucleus ATAC-seq and RNA-seq. Further optimization in experimental conditions58 or reduction of ligation cycles likely could lead to increased coverages (data not shown).
Co-accessibility between promoters and distal candidate CREs, or between gene expression and chromatin accessibility of candidate distal CREs has been used to predict gene-CRE targeting relationships40, 59. Here we took advantages of the joint profiles of transcriptome and chromatin accessibility to identify gene-CRE pairs with higher confidence. It is worth noting that the correlation between chromatin accessibility of candidate CREs and target gene expression levels or between distal CREs and promoter is frequently accompanied with physical proximity of the predicted pairs (Fig. 3d, e). Future studies with increased number of cells from more developmental stages, as well as optimized protocols for better genomic coverage could lead to better delineation of CRE-target gene relationships with higher temporal resolution.
Finally, it is worth noting that the DNA barcoding strategy present here can be further extended to stand-alone or parallel profiling of other molecular biology layers, including DNA methylation, histone modifications, TF binding, and 3D genome organization. The endonuclease-assisted amplify-and-split “library-dedicating” method can also be used to distinguish multiple types of biomolecules in future single-cell multi-omics analyses.
Methods
Cell culture and processing
HEK293T (human, ATCC CRL-11268), HepG2 (human, ATCC HB-8065) and NIH/3T3 (murine, ATCC CRL-1658) cells were cultured according to standard procedures in Dulbecco’s Modified Eagles’ Medium (Thermo Fisher Scientific, 10569010) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, 16000044) and 1% penicillin–streptomycin (Thermo Fisher Scientific, 10378016) at 37 °C with 5% CO2. Cells were not authenticated nor tested for mycoplasma. To prepare nuclei, HepG2 and 3T3 cells were harvested by centrifugation, washed with PBS (Thermo Fisher Scientific, 10010-23) and counted using BioRad TC20 cell counter. The percentage of live cells in the samples were higher than 95%. The cells were then resuspended in cold Lysis Buffer (10 mM Tris-HCl pH 7.4 [Sigma, T4661], 10 mM NaCl [Sigma, S7653], 3 mM MgCl2 [Sigma, 63069], 0.1% IGEPAL CA-630 [Sigma, I8896]) and centrifuged for 15 min at 600 g, 4 °C. For the species mixing experiment, nuclei were then washed with PBS and resuspended, counted using BioRad TC20 cell counter. HepG2, HEK293T and 3T3 nuclei were then mixed in equal proportions and applied to Paired-seq.
Processing of biospecimens
Male C57BL/6J mice were purchased from Jackson laboratories at 8 weeks of age and maintained in the Salk animal barrier facility on 12 hr dark-light cycles with food ad libitum for four weeks before dissection. Cerebral cortex was dissected and snap-frozen in dry ice. All protocols were approved by the Salk Institute’s Institutional Animal Care and Use Committee (IACUC).
Frozen tissues of mouse fetal brains, previously collected as part of the ENCODE project52, were mechanically grinded in liquid nitrogen and weighted. Nuclei were prepared and processed as previously with modifications23. 10-30 mg frozen tissue were transferred to a 1.5 mL Lobind tube (Eppendorf, O22431021) in with 1 mL of NPB (5% BSA [Sigma, A7906], 0.2% IGEPAL-CA630 [Sigma, I8896], 1 mM DTT [Sigma, D9779], 1X cOmplete EDTA-free protease inhibitor [Roche, 05056489001], 0.4 U/μL RNase OUT [Invitrogen, 10777-019] and 0.4 U/μL SUPERase In [Invitrogen, AM2694] in PBS [Thermo Fisher Scientific, 10010-23]) and incubated for 15 min at 4 °C. Nuclei suspension was then filtered over a 30 μm Cell-Tric (Sysmex), counted using BioRad TC20 cell counter and proceed to in situ tagmentation and reverse transcription immediately.
Tn5 Transposomes generation
To generate barcoded Tn5 transposomes, barcoded DNA adaptors oligos were annealed to a common pMENTs oligo (Supplementary Tables 1 and 2) in a thermocycler with the following program: 95 °C for 5min, slowly cooled to 10 °C with a temperature ramp of −0.1 °C/s. The transposons (1μL, 50 μM) were then mixed with 6 μL unloaded transposase Tn5 (0.5 mg/mL), mixed by brief vortex and quickly spin-down, incubated at room temperature for 30 min. 63 μL storage buffer (50% Glycerol [Sigma, G6279], 50 mM Tris-HCl pH 7.4 [Sigma, T4661], 100 mM NaCl [Sigma, S7653], 1 Mm DTT [Sigma, D9779]) was then added and the loaded transposases can be stored at −20 °C for up to 6 months.
To generate DNA barcoded plates, 6 μL 100 μM barcoded oligos (Supplementary Table 2) were distributed to 96-well plates. 44 μL 12.5 μM Linker-R02, Linker-R03 and Linker-R04 oligos (Supplementary Table 1) were then added to each well of the 96-well plates containing corresponding the barcoded oligos. The plates were then sealed and annealed in a thermocycler with the following program: 95 °C for 5 min, slowly cooled to 20 °C with a temperature ramp of −0.1 °C/s.
Paired-seq procedure
In situ tagmentation and reverse transcription
8 of 1.5 mL Maxymum recovery tubes (Axygen, MCT-150-L-C) were pre-washed with 5% BSA in PBS (Sigma, A3311). 250 k of nuclei were transferred to the pre-washed tubes and then centrifuged at 1,000 g for 10 min at 4 °C. The supernatants were aspirated and 45 μL 1.11X TB (36.7 mM Tris-Ac pH 7,8 [Thermo Fisher Scientific, BP-152], 73.3 mM KAc [Sigma, P5708], 12.1 mM MgAc [Sigma, M2545], 17.8 % DMF [EMD Millipore, DX1730]) were used to carefully resuspend the nuclei palleted. 5 μL barcoded Tn5 (BC#1-01 – BC#1-08) were added to the 8 labeled tubes and mixed gently. The tagmentation reaction was carried out in ThermoMixer (Eppendorf) for 30 min at 37 °C and 550 r.p.m. The reaction was terminated by adding 25 μL of 45 mM EDTA (Invitrogen, AM9260G).
The nuclei were then centrifuged at 1,000 g for 10 min at 4 °C. The supernatant was discarded, and nuclei palette were resuspended in 8 μL 0.5 X PBS with RNase Inhibitor Mix (0.5X PBS, 1 U/μL RNase OUT and 1 U/μL SUPERase In), and then transferred to 200 μL tubes with 4 μL corresponding barcoded RT primers (the same order to tagmentation barcodes). 8 μL RT mix (10 pmol dNTPs, 20 U RNaseOUT, 40 U SUPERase In, and 400 U Maxima Reverse Transcriptase [Thermo Fisher Scientific, EP0743] in RT buffer) were then added and reverse transcription were performed using the following program (Step1: 50 °C × 10 min; Step2: 8 °C × 12 s, 15 °C × 45 s, 20 °C × 45 s, 30 °C × 30s, 42 °C × 2 min, 50 °C × 5 min, and repeat Step2 for additional 2 times; Step3: 50 °C × 10 min and hold at 12 °C). After the reaction, the nuclei were transferred to Maxymum recovery tubes pre-washed with 5% BSA in PBS and cooled on ice for 2 min, 0.4 μL of 5% Triton-X100 (Sigma, T9284) were then added. The nuclei from individual reactions were then combined and centrifuged at 1,000 g for 10 min at 4 °C and supernatant were discarded.
Tagging of individual nuclei by ligation-based combinatorial DNA barcoding
Nuclei were resuspended in 1 mL 1X NEBuffer 3.1 and then transferred to Ligation Mix (2,262 μL ultrapure H2O, 500 μL 10X T4 DNA Ligase Buffer, 50 μL 10 mg/mL BSA, 100 μL 10X NEBuffer 3.1 and 100 μL T4 DNA Ligase [NEB, M0202L]). 40 μL of the mix was then distributed to Barcode-plate-R02 and incubate in ThermoMixer (Eppendorf) at 37 °C for 30 min, 300 r.p.m. 10 μL of R02-Blocking-Solution (264 μL of 100 μM Blocker-R02 oligo [Supplementary Table 1], 250 μL of 10X T4 Ligation Buffer, 486 μL ultrapure H2O) was then added to each well and reaction were continued for 30 min. Pool all nuclei together and centrifuge at 1,000 g for 10 min at 4 °C. The 2nd round of ligation was carried out similar to the 1st round of ligation, except using Barcode-plate-R03 and Blocker-R03 oligo instead of the reagents used above. The 3rd round of ligation was carried out similarly with Barcode-plate-R04. After 30 min of the ligation reaction, R04-Termination-Solution (264 μL of 100 μM R04 Terminator oligo [Supplementary Table 1], 250 μL of 0.5 M EDTA and 236 μL ultrapure H2O) was added to quench the reaction.
Typically, between 500,000 to 1,000,000 nuclei could be tagged after 3 rounds of ligation-based barcoding. Nuclei were resuspended in PBS, counted and separated to sub-libraries containing 10k to 100k nuclei (optimal ~25k nuclei per tube) and each sub-library were diluted to 35 μL by PBS. 5 μL 4M NaCl (Sigma, S7653), 5 μL 10% SDS (Invitrogen, 15553-035) and 5 μL 10 mg/mL Protease K (NEB, P8107S) was added and incubated in ThermoMixer (Eppendorf) at 55 °C for 2 hr, 850 r.p.m. The samples were cooled to room temperature, purified with 1X SPRI beads (Beckman coulter, B23319) and eluted in 12.5 μL ultrapure H2O.
TdT-Tailing and pre-amplification
TdT-Tailing and pre-amplification reaction were adopted from TELP with modifications43. 1 μL 10X TdT buffer, 0.5 μL 1 mM dCTP (NEB, N0447S) was added into 12.5 μL purified DNA/cDNA mix. The samples were incubated at 95 °C for 5 min and quickly chilled on ice for 5 min. 1μL of TdT (NEB, M0315S) was then added and tailing reaction was carried out under 37 °C for 30 min followed by inactivation at 75 °C for 20 min. Anchor Mix (6 μL 5X KAPA Buffer, 0.6 μL 10 mM dNTPs, 0.6 μL 10 μM Anchor-Oligo [Supplementary Table 1] and 0.6 μL KAPA HiFi HS [KAPA, KK2502]) were added and the linear amplification was performed with the following program (Step1: 98 °C × 3 min; Step2: 98 °C × 15 s, 47 °C × 60 s, 68 °C × 2 min, 47 °C × 60 s, 68 °C × 2 min and repeat Step2 for additional 14 times; Step3: 72°C × 10 min and hold at 12 °C).
Preamplification Mix (4 μL 5X KAPA buffer, 0.5 μL 10 mM dNTPs, 2 μL of 10 μM PA-F and PA-R [Supplementary Table 1], 0.5 μL KAPA HiFi HS) were then added and preamplification were performed as the following program (Step1: 98 °C × 3 min; Step2: 98 °C × 20 s, 65 °C × 20 s, 72 °C × 2.5 min and repeat Step2 for additional 9 times; Step3: 72°C × 2 min and hold at 12 °C). Amplified products were purified with SPRI double-size selection (10 μL + 32.5 μL) and were eluted in 34 μL ultrapure H2O, use 1 μL for quantification.
2nd adaptor tagging and endonuclease digestion
Divide 33 μL of the purified products into 2 tubes for DNA and RNA libraries construction. Add 2 μL 10X Cutsmart buffer (NEB, M7204S) into each tube. Add 1.5 μL SbfI-HF (NEB, R3642) (per 100 ng amplified product) to DNA-tube and 0.75 μL Notl-HF (R3189) (per 100 ng amplified product) to RNA-tube. The digestion reaction was incubated at 37 °C for 60 min. Add 1 μL 3M NaAc pH 5.4 (Sigma, 71196) and cleaned up using QIAquick PCR purification kit (QIAGEN, 28104) and eluted in 30 μL 0.1X EB (QIAGEN). Add 31 μL 2X TB and 0.5 μL (per 100 ng amplified product) 0.05 mg/mL Tn5-P5 and incubate in ThermoMixer (Eppendorf) at 37 °C for 30 min, 550 r.p.m. Cleaned up using QIAquick PCR purification kit and elute in 30 μL 0.1X EB (QIAGEN).
Indexing PCR
Prepare the PCR mix (30 μL purified P5-tagged product, 10 μL 5X Q5 buffer, 1 μL 10 mM dNTP, 0.5 μL 50 μM N5 primer, 2.5 μL 10 μM P7 primer [Supplementary Table 1], 5 μL H2O and 1 μL NEB Q5 DNA Polymerase [NEB, M0491]) and run the following program (Step1: 72 °C × 5 min, 98 °C × 30 s; Step2: 98 °C × 10 s, 63 °C × 30 s, 72 °C × 1 min and repeat Step2 for additional 10-15 times to reach 10 nM concentration; Step3: 72°C × 1 min and hold at 12 °C). Cleanup the libraries using 0.85X (42.5 μL) SPRI beads. The final libraries were sequenced using a HiSeq 2500 (iliumina) with the following read lengths: PE 53 + 7 + 130 (Read1 + Index1 + Read2).
PLAC-seq procedure
PLAC-seq was performed as previously described54. The frozen tissues were pulverized prior to formaldehyde crosslinking. About 30-50 mg of frozen tissue were crosslinked with 1% formaldehyde at room temperature for 20 min. Dissociation of crosslinked tissues were performed with gentleMACS dissociator. Single-nuclei suspensions prepared from crosslinked tissues were incubated in 50 μL 0.5% of SDS and incubated at 62 °C for 10 min. 25 μL 10% Triton X-100 and 145 μL water were then added, followed by incubation at 37 °C for 15 min. Digestion was performed by Mbol for 2 h 37 °C, followed by inactivation at 62 °C for 20 min. 15 nmol of dCTP, dGTP, dTTP, biotin-14-dATP (Thermo Fisher Scientific) each and 40 unit of Klenow were then added, and incubated at 37°C for 1.5h. Proximity ligation was performed at room temperature in 1X T4 DNA Ligase Buffer, 0.1 mg/ml BSA, 1% Triton X-100 and 4000 unit of T4 DNA Ligase (NEB). The nuclei were harvested at 2,500 g for 5 min and the supernatant was discarded. The nuclei were then resuspended in 130 μl RIPA buffer (10 mM Tris, pH 8.0, 140 mM NaCI, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, proteinase inhibitors) and lysed on ice for 10 min, followed by sonication using Covaris M220. The samples were centrifugation at 14,000 rpm for 20 min and supernatant was collected. The clear cell lysate was incubated with H3K4me3 antibody-coated (04-745, Millipore, 5 μg per sample) Dynabead M-280 Sheep Anti-Rabbit IgG at 4°C overnight. After incubation, the beads were washed with RIPA buffer three times, high-salt RIPA buffer (10 mM Tris, pH 8.0, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate) twice, LiCl buffer (10 mM Tris, pH 8.0, 250 mM LiCI, 1 mM EDTA, 0.5% IGEPAL CA-630, 0.1% sodium deoxycholate) once, TE buffer (10 mM Tris, pH 8.0, 0.1 mM EDTA) twice. To elute DNA, washed beads were first treated with 10 μg RNase A in extraction buffer (10 mM Tris, pH 8.0, 350 mM NaCl, 0.1 mM EDTA, 1% SDS) for 1 h at 37 °C, followed by adding 20 μg proteinase K and incubate at 65 °C 2 h. The fragmented DNA was purified by Zymo DNA Clean&Concentrator kit. Biotinylated DNA was pulled-down by Dynabeads MyOne Streptavidin T1 beads and PCR amplified for sequencing.
Data analysis procedures
Pre-processing of Paired-seq data
Cellular barcodes and the linker sequences are read by Read2. The first base of BC#1, BC#2, BC#3 and BC#4 should locate within 121-124th, 84-87th, 47-50th and 10-13rd base of read2 (see Extended Data Fig. 1a for details). We first compared the sequences adjacent to the locations with linker sequences, only sequences with less than 5 mismatches with linkers for all 4 rounds of barcodes were retained for further analysis. The location of first base for each barcode was identified based on the alignment of the linker sequences. Read1 and Read2 of each library were combined to generate a single new FASTQ file by joining read sequence (sequence of Read1 and UMI from Read2) and quality values into Line1 and joining the 4 rounds of barcodes sequences as well as the quality values into Line 2 and Line 4.
A bowtie2 reference index was generated by combining all possible cellular barcodes combinations. The combined FASTQ file contains barcodes sequences were then mapped to the cellular barcodes reference using Bowtie260 with parameters: -v 3 --norc. The resulting SAM file was then converted to a final FASTQ file by using adding RNAME (of SAM file) into Line1 and extract the original Read1 sequence and quality values from QNAME (of SAM file) into Line2 and Line4 of the final FASTQ file.
Nextera adaptor sequences were trimmed from 3’ of DNA libraries, Poly-dT sequences were trimmed from 3’ of RNA libraries and low-quality reads (length < 30, quality < 20) were excluded for further analysis.
Analysis of Paired-seq data from cultured cells
Reads mapping and evaluation of collision rate:
Reads were first mapped to a reference genome using STAR (version: 2.6.0a61) with the combined reference genome (GRCh37 for human and GRCm38 for mouse). Duplicates were removed based on the mapped position and UMI. For Paired-seq from cultured cells, we used BC#1 for the identification for origin of samples: BC#1-01 to label NIH/3T3 cells, BC#1-02 for HEK293T cells, BC#1-03 for HepG2 cells, BC#1-04 and 05 for mix of NIH/3T3 with HEK293T cells, BC#1-06 and 07 for mix of NIH/3T3 and HepG2 cells, BC#1-08 for mix of all the 3 cell types. For evaluation of the collision rate, we used only cells with BC#1-04 to 08 and nuclei with less than 80% UMIs mapped to one species were classified as mixed cells.
Clustering and quality analysis:
Cells classified as human cells were then used for clustering analysis. DNA accessible peaks were called using MACS262. DNA and RNA alignment files were then converted to a matrix with cells as columns and genes or peaks as rows, DNA matrix was then binarized. Cells with less than 200 peaks or genes and peaks or genes with less than 10 cells were removed from further analysis (Supplementary Table 3). Clustering of both DNA and RNA profiles of cell lines were performed using Seurat63. The read coverages for genomic regions were compared with multiBamSummary of deepTools64 with 10-kb bins. The reads distributions around TSS and TTS were calculated with Homer65.
Analysis of Paired-seq data from adult mouse cerebral cortex and fetal mouse forebrain
Reads mapping:
Reads were first mapped to a reference genome with STAR (version: 2.6.0a61) with mouse GRCm38 genome. Duplicates were removed based on the mapped position and UMI. For Paired-seq from adult mouse cerebral cortex, we used BC#1 for the identification for the origin of samples: BC#1-01 to 04 and BC#1-05 to 08 to label the two replicates. For Paired-seq from archived mouse fetal forebrain, we also used BC#1 for the identification for the origin of samples: BC#1-01 and 02 for the two replicates of E12.5 forebrain, BC#01-03 and 04 for the two replicates of E16.5 forebrain. Libraries were sequenced to 30-70% duplication level. Low coverage nuclei were removed from further analysis (we used different criteria for libraries of different depths, for detailed information see Supplementary Table 3).
Clustering of Paired-seq profiles:
RNA alignment files were converted to a matrix with cells as columns and genes as rows. DNA alignment files were converted to a matrix with cells as columns and 1-kb bins (instead of peaks) as rows. For RNA matrix, cells with less than 200 genes and genes with less than 10 cells were removed from further analysis. For DNA matrix, cells with less than 200 bins and bins with less than 20 cells were removed. DNA matrix was further filtered by removing the 5% highest covered bins. To enable integrated clustering based on both DNA and RNA profiles, we first convert the cell-by-genes matrix of RNA and the cell-by-bins matrix of DNA into cell-by-cell Jaccard similarity matrices with same dimensions (Ncell × Ncell) using snapATAC46. The Jaccard similarity matrices were then normalized by the regression-based normOVE of snapATAC to decrease the effect of read depth. The normalized matrices (O) were further scaled to matrices (S) with all values in between 0 and 1:
The Hadamard product (H) of the scaled DNA matrix (D) and scaled RNA matrix (R) were then calculated:
Dimension reduction (PCA) was then performed on H, followed by construction of k-nearest neighbors (KNN) graph from significant principal components. The Louvain method66 was then used to cluster nuclei with similar Paired-seq profiles (R packages: igraph and FNN). UMAP was used for data visualization (R package: uwot). We found that clustering based on Hadamard product of two matrices is more effective than based on the sum of two matrices. Clustering based on simply appending two matrices biases towards RNA-matrix, necessitating careful weighting between DNA and RNA.
Accessible regions (cis-regulatory elements) were identified by peak-calling of DNA reads using MACS262 with default parameters. To allow the comparison of CREs between different stages, the aggregated DNA profiles from E12.5, E16.5 forebrain and adult cerebral cortex were first down-sampled to the sample depth and merged, peak-calling were then performed with MACS2. Next, CREs were extended to 1-kb bins from the peak summits and RPM of each CRE (of each stage) were calculated. CRE (of each stage) with RPM < 1 were excluded from further analysis. For differential analysis of gene expression and CRE accessibility, RNA and DNA reads were separated according to stages or cell types and then aggerated. The differentially expressed genes, differential accessible promoters and distal accessible sites were identified by edgeR48, by comparing reads of cells from the corresponding cell type or stage with reads of cells from all other clusters, with thresholds of Log2(Fold-change) > 1 and P-value < 0.05 (negative binomial test). Motif enrichment analysis was performed by HOMER65. Only reads located in 3’UTR (or within 1000-bp of TTS for genes with short 3’UTR) were used for differential expression analysis.
Connect cis-regulatory elements with target genes using Paired-seq profiles:
To reduce potential measurement noise, we generated pseudo-cells by merging cells with high-similarity in Paired-seq profiles. We first removed nuclei with less than 500 DNA and 200 RNA reads and randomly selected 5,000 single-nuclei (from E12.5, E16.5 and adult separately), then merging every 50 single-nuclei with highest Jaccard similarity to each other. To estimate the false-positive detection rate (FDR), we randomly selected 100 × 50 single-nuclei to generate 100 shuffled-pseudo-cells; we also permuted the cell IDs, randomly select reads and generated 100 permuted-pseudo-cells with similar read coverages compared to pseudo-cells from high similarity cells and shuffled-pseudo-cells. TPM of each gene and RPM of each cis-regulatory element were calculated and log-transformed (T):
We then calculated the Pearson correlation coefficient between gene expression and cis-regulatory elements accessibility within the 500-kb range from TSS of the corresponding gene. The Pearson correlation between promoter accessibility and cis-regulatory elements accessibility within 500-kb range were also calculated. By comparing with the Pearson correlations coefficient of gene-CRE pairs in pseudo-cells (and shuffled-pseudo-cells) with that in permuted-cell-ID (Δr = (rpseudo-cell or shuffled-pseudo-cell − rpermuted-cell-ID), we defined a correlation threshold (Δr = 0.27 for promoter-CRE and Δr = 0.23 for gene-CRE pairs in mouse adult cerebral cortex, and Δr = 0.09 for promoter-CRE pairs and Δr = 0.11 for gene-CRE pairs in mouse E12.5 and E16.5 forebrains). Only pairs detected by both promoter-CRE and gene-CRE were preserved. Using the same criteria, less than 10% of pairs were identified from shuffled-pseudo-cells; FDR was estimated from dividing the number of pairs identified from shuffled-pseudo-cells by the number of pairs identified from pseudo-cells.
Diffusion pseudotime analysis:
To order cells in pseudotime, we used diffusion map (R package: destiny55) to create a trajectory. First, we selected single cells classified into NP as starting points, single cells of dAC were selected as the endpoint for astrogenesis; for neurogenesis of both GABAergic and glutamatergic neurons, NP cells were also selected as starting points, dIn2 and dEx2 (who present in both fetal and adult mouse brain) were selected as endpoints. Next, we computed the mean coordinates of the aforementioned combined Hadamard product (H) of each cluster, cells of the top 5% Euclidean distance to the mean coordinates were filtered out. We then used diffusion map67 to construct the trajectory projection. The cells were ordered according to the main diffusion coordinate along the direction of astrogenesis or neurogenesis. For visualization, a combined map of NP, dAC, dIn2 and dEx2 were also constructed using the same method. TF motif analyses were performed using chromVAR56, the motif hit counts were smoothed to 10 quantiles according to the cell orders. To find TF-motif pairs more likely to be involved in gene regulation across the diffusion pseudotime, we computed the pairwise Pearson correlation coefficient of log transferred motif hit counts of TFs, expression and promoter accessibility of TF encoding genes and filtered the top 30% TF-motif pairs. Heatmaps were ordered according to the change of row scaled TF motif enrichment, and smoothed using “smooth.spline” with spar=0.5. For the identification of time-of-gain and time-of-loss of TFs, the 10-quantiles smoothed enrichment matrices were used: (1) we first identified the pseudotime point (tmax) with highest enrichment (Emax), we then identified the pseudotime points with lowest enrichment before and after tmax (Emin-1 and Emin-2); (2) for time-of-gain and time-of-loss we searched the pseudotime points with enrichment (E) = (Emax − Emin-1)/2 before tmax (time-of-gain) and E = (Emax − Emin-2)/2 after tmax (time-of-loss), some TFs may have fluctuation with multiple such points and only the one nearest to tmax was considered as time-of-gain or loss; (3) only TFs with time-of-gain later than 2 and time-of-loss earlier than 9 were used for further comparison: we only considered the TF motifs with time-of-gain later than 2 and time-of-loss earlier than 9, as we cannot distinguish unsynchronized from synchronized activation as their relative enrichment was already the highest at the earliest pseudotime point (e.g., FOXP1), and vice versa (e.g., MAF, Fig. 4d). To compare the order of TF motif activation and TF gene upregulation, a cutoff of Δt = 2 was used to classify synchronized and unsynchronized activation.
Analysis of PLAC-seq data
We performed PLAC-seq experiments on mouse E12.5 and E16.5 forebrain tissues, and applied our recently developed MAPS68 for the downstream data analysis. Specifically, we first used “bwa mem” to map the two ends of one paired-end reads to the reference genome mm10 separately, and then kept the valid mapped reads, and removed PCR duplicates by “samtools rmdup”. Next, we divided intra-chromosomal reads into short-range reads (<= 1 kb) and long-range reads (> 1 kb), and used the short-range reads to measure ChIP-enrichment level, and the long-range reads to measure chromatin interactions. We further binned the autosomal chromosomes (chr1-chr19) into consecutive, non-overlapping 10-kb bins, and selected 10-kb bin pairs where at least one bin contains H3K4me3 ChIP-seq peaks (since PLAC-seq is designed to measure protein-mediated chromatin interactions) for the downstream analysis.
We have shown that PLAC-seq data contain multiple layers of systematic biases, including restriction enzyme cutting frequency, GC content, sequence mappability and ChIP-enrichment level68. To normalize PLAC-seq data, we fitted a positive Poisson regression model for the selected 10-kb bin pairs, taking the raw contact frequency as the outcome and the aforementioned systematic biases and 1D genomic distance as predictors. We obtained expected contact frequency and P-value from the fitted positive Poisson regression model, and then converted P-value into false discovery rate (FDR). We defined a 10-kb bin pair as candidate significant interaction if the normalized contact frequency (the ratio between observed contact frequency and the expected contact frequency) is >=2, and the FDR < 1%. We further grouped candidate significant interactions together if they are within 10 kb. If a candidate significant interaction has no other significant interaction in neighborhood region, we defined it as a singleton, otherwise, we defined it as a peak cluster. Since biologically relevant interactions tend to cluster together and singletons are more likely to be false positives, we applied a more stringent FDR threshold 1×10−4 for singletons. The final list of significant interactions consists of peak clusters with FDR < 1% and singletons with FDR < 1×10−4.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Code Availability
MAPS is freely available at https://github.com/ijuric/MAPS. Custom scripts used in this study can be downloaded from https://github.com/cxzhu/paired-seq.
Data Availability
The sequencing data obtained in this study have been deposited to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE130399. Source data for Figure 1e–g, 2b, 2e, 2f, 3a, 3b and 4b–d are available with the paper online. External datasets used in this study are available from GEO: ENCODE DNase-seq (GSE37074), PolyA-RNA-seq (GSE39524) of mouse NIH/3T3 cells, sci-CAR mixed cells datasets (GSE117089), SPLiT-seq (GSE110823), sci-RNA-seq (GSE98561), Drop-seq (GSE63269), sci-ATAC-seq (GSE67446) and dscATAC-seq (GSE123581); or from 10X genomics website: 10X scRNA-seq (https://www.10xgenomics.com, 1k_hgmm_v3_nextgem dataset). All other data are available upon request.
Extended Data
Extended Data Fig. 1. Quality control for Paired-seq libraries.
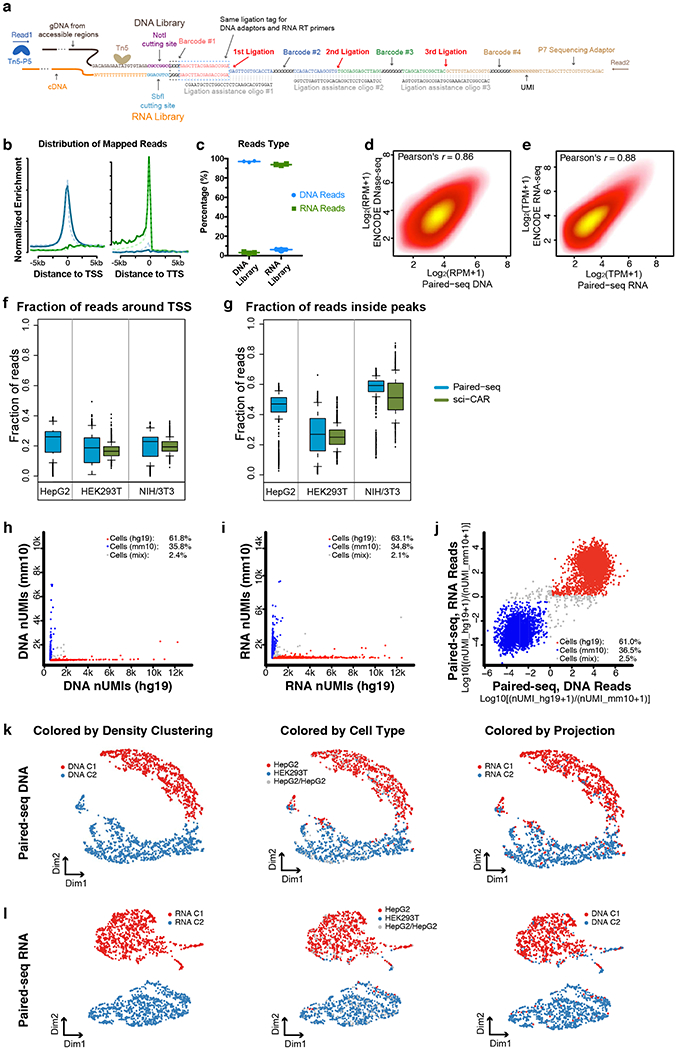
a, Sequence of Paired-seq products illustrating the structure of DNA barcode combinations. b, Paired-seq DNA profiles are enriched around the transcription start sites (TSSs) while (e) RNA profiles are enriched at the transcription termination sites (TTSs) in NIH/3T3 cells. As comparison, DNA and RNA profiles from sci-CAR were also plotted. c, Proportions of DNA and RNA reads in both libraries are shown, n=3 independent experiments. Scatter plots showing the correlation of reads from two replicates of Paired-seq (d) DNA profiles or (e) RNA profiles. Boxplots showing (f) the fraction of reads around TSS (−1000 to +500 bp) and (g) the faction of reads inside known peaks (GSE:49847) of Paired-seq DNA profiles from HEK293T, HepG2 and NIH/3T3 cells. sci-CAR40 datasets (GSE117089) from the same cell types were also used for comparison. Scatter plot showing the proportion of human and mouse reads in each cell in Paired-seq (h) DNA and (i) RNA profiles. j, Scatter plot showing the proportions of both DNA and RNA reads mapped to genomes in the same single cells. Cells with more than 80% reads mapped to human and mouse genome were colored in red and blue, respectively. UMAP visualization of HepG2 and HEK293T cells based on (k) DNA and (l) RNA reads. Cells were colored by density-based clustering from each profile and cell identities. The clustering results were also projected to each other. In boxplots center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR). The sample sizes are provided in the Source Data with this paper online.
Extended Data Fig. 2. Integrative analysis of Paired-seq DNA and RNA profiles from mouse adult cerebral cortex.
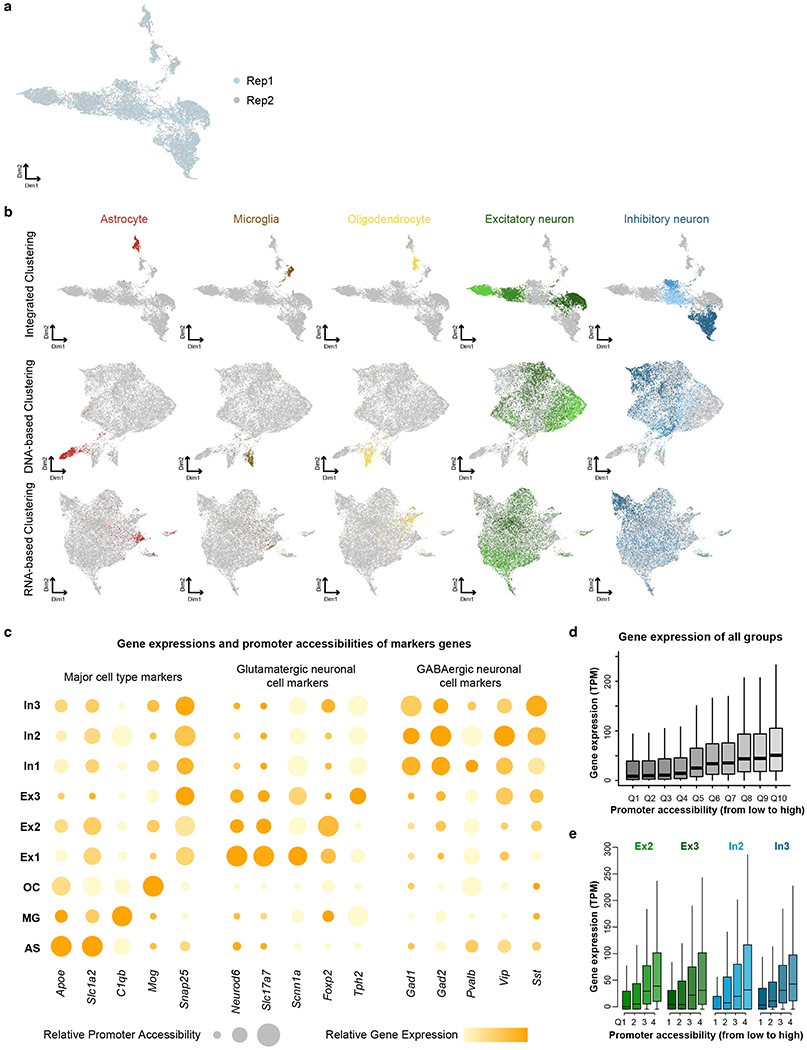
a, UMAP visualization of co-clustering of nuclei from two replicates. b, Comparison of DNA-based, RNA-based and integrated clustering results. Cells were colored based on unsupervised clustering from integrated clustering and colored the same as Fig. 2b. c, Promoter accessibility and gene expression of several marker genes in the nine major groups. Relative promoter accessibilities and gene expressions were indicated in the size and the color of circles. d, Expression levels of genes of all clusters are plotted in a boxplot for each quantile of promoter accessibility. e, For each cell cluster, expression levels of genes are plotted in a boxplot for each quantile of promoter accessibility. In boxplots center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR).
Extended Data Fig. 3. Co-clustering of Paired-seq datasets from mouse E12.5, E16.5 forebrain and adult cerebral cortex.
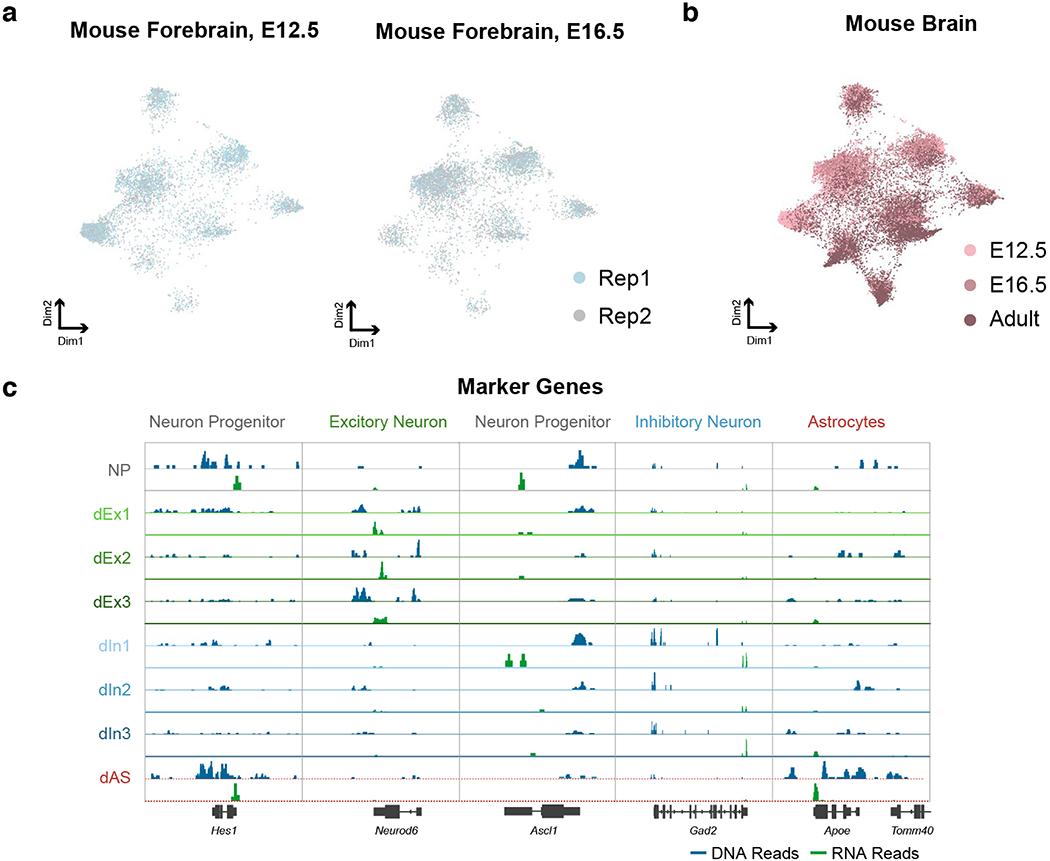
a, UMAP visualization of Paired-seq data from two replicates of both mouse E12.5 and E16.5 forebrains showing clustering of cells based on cell types, not replicates. b, UMAP visualization of Paired-seq data of mouse E12.5, E16.5 forebrains and adult cerebral cortex showing clustering of cells based on cell type, not batches. c, Aggregate chromatin accessibility (blue) and gene expression (green) profiles for each cell clusters at several marker gene loci.
Extended Data Fig. 4. Paired-seq facilitates the linking of candidate CREs to putative target genes in mouse fetal forebrains.
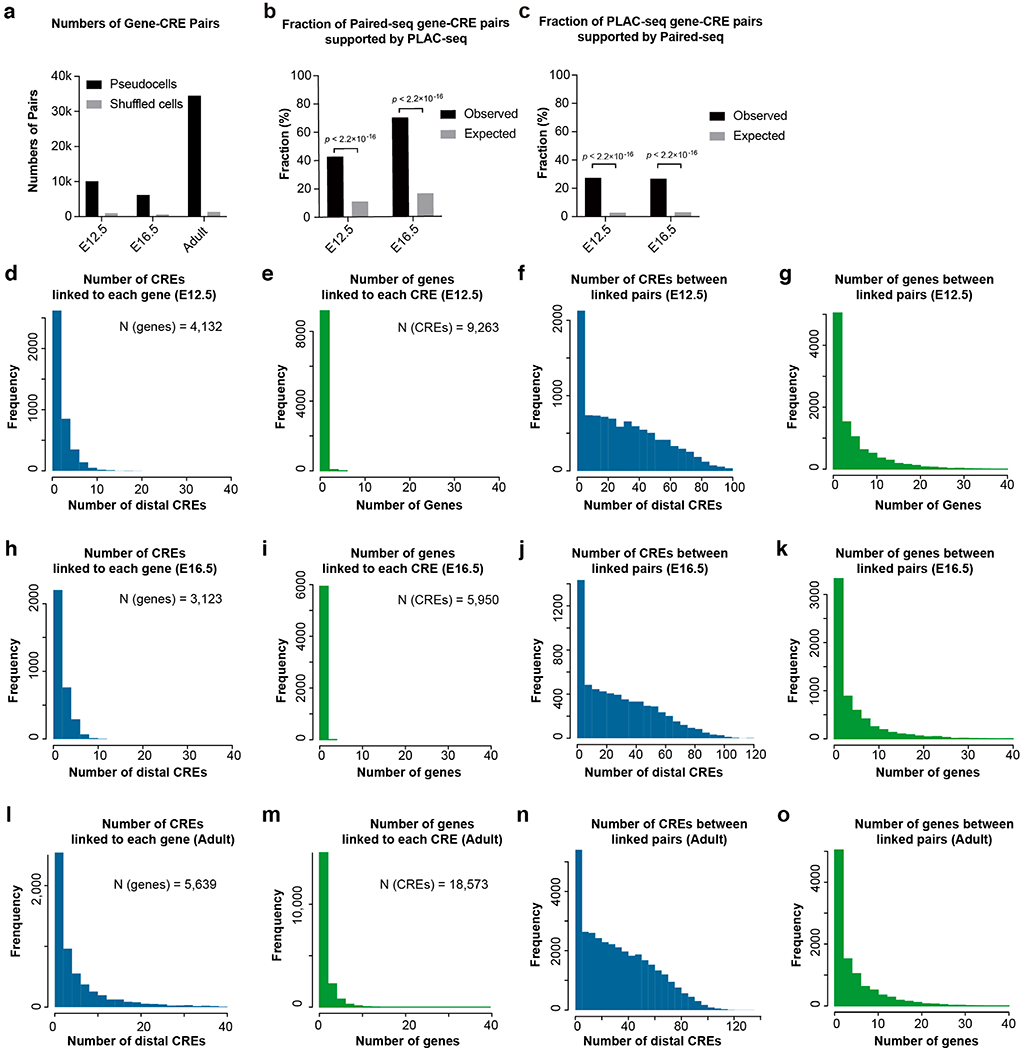
a, Bar charts show the numbers of gene-CRE links identified in mouse E12.5 and E16.5 forebrain, and adult cerebral cortex datasets. b and c, Barcharts show the fractions of gene-CRE pairs (b) identified by Paired-seq and supported by PLAC-seq or (c) identified by PLAC-seq and supported by Paired-seq. P-value, two-sided Fisher’s exact test. d-o, Number of identified CREs linked to each gene, number of identified genes linked to each CRE, number of CREs between CREs and their linked genes, and number of genes between CREs and their linked genes in (d-g) E12.5, (h-k) E16.5 forebrain and (l-o) adult cerebral cortex.
Extended Data Fig. 5. Dynamics of gene-CRE pairing during mouse brain development.
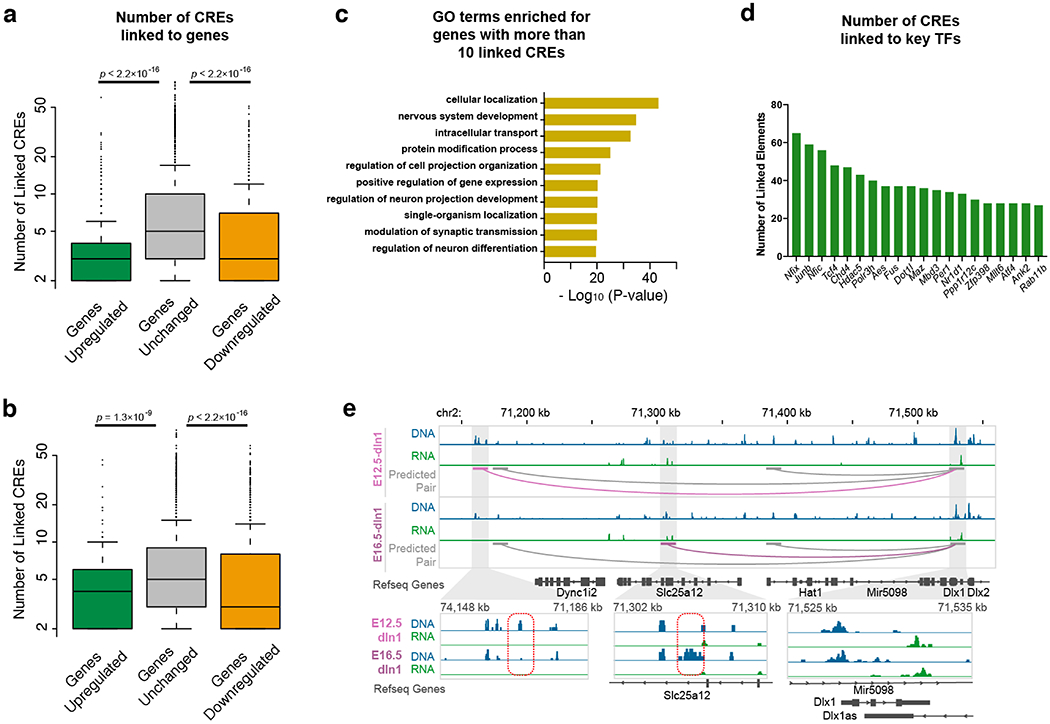
Boxplots showing the number of linked CREs for genes of each group of (a) E12.5 to E16.5 and (b) E16.5 to Adult. P-value, two-sided K-S test. Genes were classified according the number of linked candidate CREs: genes with a gain of CREs (Log2[fold-change] > 3), genes with unchanged number of linked CREs (−1 < Log2[fold-change] < 1) and genes with a loss of linked CREs (Log2[fold-change] < −3). c, DAVID GO analysis of genes with more than 10 linked CREs. d, Top 20 TF genes with the highest number of linked CREs. e, The predicted gene-CRE pair for Dlx1 gene in dIn2 cluster. The common links shared by two stages of development were shown in grey and the stage-specific links were shown in light- and dark-violet red. In the close-up view, the positions of stage-specific CREs were indicated by red dashed box. In boxplots center lines indicate the median, box limits indicate the first and third quartiles and whiskers indicate 1.5x interquartile range (IQR).
Extended Data Fig. 6. Analysis of cellular trajectory of developing mouse forebrain.
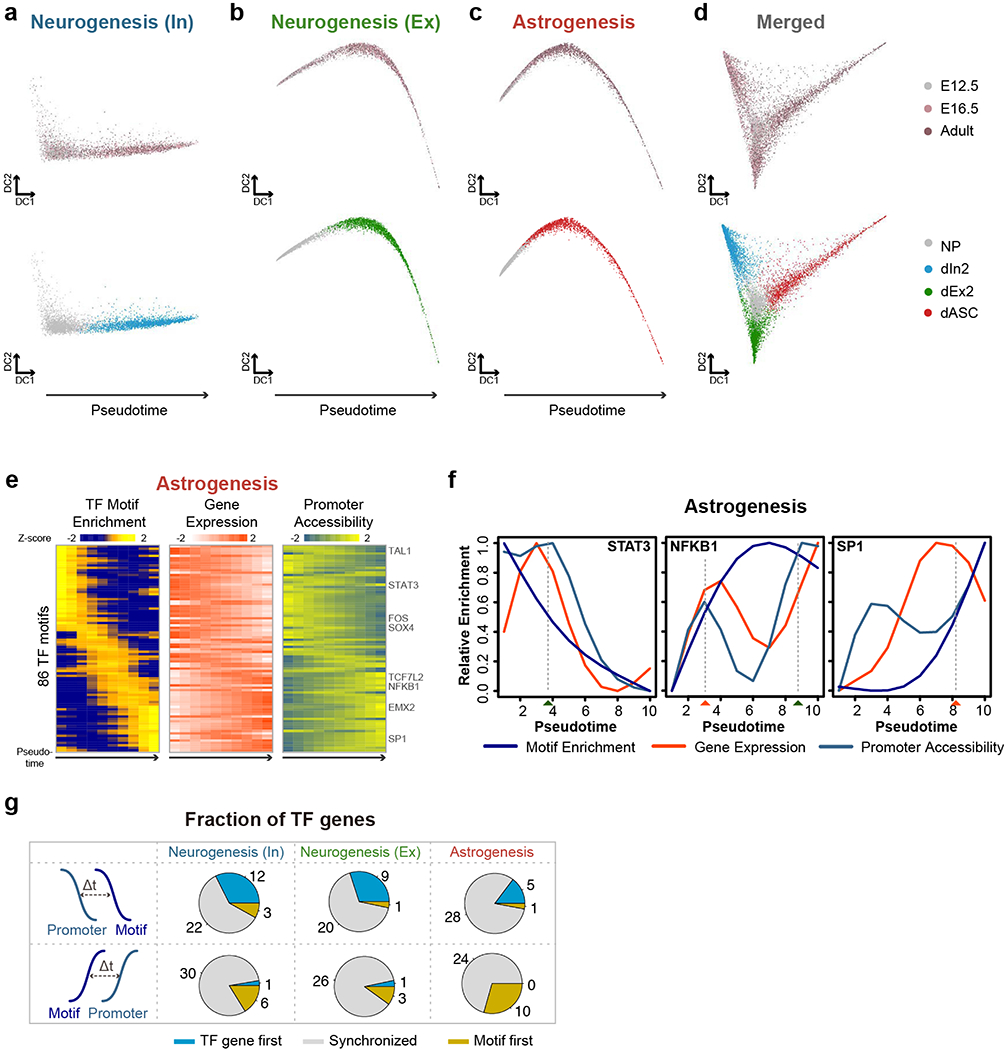
a-c, Diffusion map showing the single-cell trajectories of neurogenesis towards (a) GABAergic neurons, (b) glutamatergic neurons and (c) astrogenesis. d, The combined diffusion map corresponding to Fig. 4a was also shown. The cells were colored by stages and clusters, respectively. e, Heatmap shows the ordering of the chromVAR TF motif enrichments across astrogenesis. The relative expression and promoter accessibility of corresponding TF genes were also shown. f, Line plots showing the relative enrichment of TF motifs, gene expression and promoter accessibility for STAT3, NFKB1 and SP1 according to the diffusion pseudotime for astrogenesis. The estimated time-of-gain and time-of-loss of TF motif were indicated by red and green rectangles below. g, Pie-charts showing the fraction of TFs with the TF gene promoters became accessible before (TF gene first), synchronized with, or after (Motif first) the TF motifs became accessible, for neurogenesis towards GABAergic neurons, glutamatergic neurons and astrogenesis.
Supplementary Material
Acknowledgments
We thank B. Li for bioinformatic support and S. Kuan for sequencing. We thank QB3 MacroLab for purifying the Tn5 enzyme. We thank D. U. Gorkin (UC San Diego) for sharing the frozen archived mouse fetal brain tissues. We thank S. Preissl, R. Fang, X. Hou, J. Song, Y. Li, Y. Zhang and Y. Qiu for discussion. This study was funded by 1U19 MH114831-02, U54 HG006997 and the Ludwig Institute for Cancer Research (to B.R.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.de Laat W & Duboule D Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 502, 499–506 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Crawford GE et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16, 123–131 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yue F et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelsey G, Stegle O & Reik W Single-cell epigenomics: Recording the past and predicting the future. Science 358, 69–75 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin W et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada A et al. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol 21, 287–296 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Hainer SJ, Boskovic A, McCannell KN, Rando OJ & Fazzio TG Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319–1329 e1311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo H et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res 23, 2126–2135 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smallwood SA et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods 11, 817–820 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mooijman D, Dey SS, Boisset JC, Crosetto N & van Oudenaarden A Single-cell 5hmC sequencing reveals chromosome-wide cell-to-cell variability and enables lineage reconstruction. Nat Biotechnol 34, 852–856 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Zhu C et al. Single-Cell 5-Formylcytosine Landscapes of Mammalian Early Embryos and ESCs at Single-Base Resolution. Cell Stem Cell 20, 720–731 e725 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Inoue A, Suzuki T & Zhang Y Simultaneous mapping of active DNA demethylation and sister chromatid exchange in single cells. Genes Dev 31, 511–523 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preissl S et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat Neurosci 21, 432–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo C et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grosselin K et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Dey SS, Kester L, Spanjaard B, Bienko M & van Oudenaarden A Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 33, 285–289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macaulay IC et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods 12, 519–522 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Angermueller C et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 13, 229–232 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou Y et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26, 304–319 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y et al. Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 17, 88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo F et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27, 967–988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pott S Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elite 6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark SJ et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 9, 781 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun 10, 470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li G et al. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat Methods (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee D-S et al. Single-cell multi-omic profiling of chromatin conformation and DNA methylome. bioRxiv, 503235 (2018). [Google Scholar]
- 38.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S, Lake BB & Zhang K Linking transcriptome and chromatin accessibility in nanoliter droplets for single-cell sequencing. bioRxiv, 692608 (2019). [Google Scholar]
- 42.Rosenberg AB et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peng X et al. TELP, a sensitive and versatile library construction method for next-generation sequencing. Nucleic Acids Res 43, e35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lareau CA et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat Biotechnol 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cao J et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang R et al. Fast and Accurate Clustering of Single Cell Epigenomes Reveals <em>Cis</em>-Regulatory Elements in Rare Cell Types. bioRxiv, 615179 (2019). [Google Scholar]
- 47.Tasic B et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCarthy DJ, Chen Y & Smyth GK Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khan A et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46, D260–D266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai T et al. SOX5 controls the sequential generation of distinct corticofugal neuron subtypes. Neuron 57, 232–247 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Sun W et al. SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J Neurosci 37, 4493–4507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gorkin DU et al. An atlas of dynamic chromatin landscapes in the developing mouse fetus. Nature (In Press). [Google Scholar]
- 53.Yu M & Ren B The Three-Dimensional Organization of Mammalian Genomes. Annu Rev Cell Dev Biol 33, 265–289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang R et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 26, 1345–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haghverdi L, Buttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nat Methods 13, 845–848 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martynoga B, Drechsel D & Guillemot F Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mulqueen RM et al. Improved single-cell ATAC-seq reveals chromatin dynamics of <em>in vitro</em> corticogenesis. bioRxiv, 637256 (2019). [Google Scholar]
- 59.Pliner HA et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol Cell 71, 858–871 e858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dobin A & Gingeras TR Mapping RNA-seq Reads with STAR. Curr Protoc Bioinformatics 51, 11 14, 11–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramirez F, Dundar F, Diehl S, Gruning BA & Manke T deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Subelj L & Bajec M Unfolding communities in large complex networks: combining defensive and offensive label propagation for core extraction. Phys Rev E Stat Nonlin Soft Matter Phys 83, 036103 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Angerer P et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016). [DOI] [PubMed] [Google Scholar]
- 68.Juric I et al. MAPS: Model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments. PLoS Comput Biol 15, e1006982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data obtained in this study have been deposited to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE130399. Source data for Figure 1e–g, 2b, 2e, 2f, 3a, 3b and 4b–d are available with the paper online. External datasets used in this study are available from GEO: ENCODE DNase-seq (GSE37074), PolyA-RNA-seq (GSE39524) of mouse NIH/3T3 cells, sci-CAR mixed cells datasets (GSE117089), SPLiT-seq (GSE110823), sci-RNA-seq (GSE98561), Drop-seq (GSE63269), sci-ATAC-seq (GSE67446) and dscATAC-seq (GSE123581); or from 10X genomics website: 10X scRNA-seq (https://www.10xgenomics.com, 1k_hgmm_v3_nextgem dataset). All other data are available upon request.