

Abstract
Naked and protein-blocked DNA ends occur naturally during immune cell development, meiosis, and at telomeres as well as from aborted topoisomerase reactions, collapsed replication forks, and other stressors. Damaged DNA ends are dangerous in cells and if left unrepaired can lead to genomic rearrangement, loss of genetic information, and eventually cancer. Mre11 is part of the Mre11-Rad50-Nbs1 complex that recognizes DNA double-strand breaks and has exonuclease and endonuclease activities that help to initiate the repair processes to resolve these broken DNA ends. In fact, these activities are crucial for proper DNA damage repair pathway choice. Here, using Pyrococcus furiosus Mre11, we question how two Mre11 separation of function mutants – one previously described but the second first described here – maintain endonuclease activity in the absence of exonuclease activity. To start, we performed solution state NMR experiments to assign the side chain methyl groups of the 64 kDa Mre11 nuclease and capping domains, which allowed us to describe the structural differences between Mre11 bound to exo- and endonuclease substrates. Then, through biochemical and biophysical characterization, including NMR structural and dynamics studies, we compared the two mutants and determined that both affect the dynamic features and double-stranded DNA binding properties of Mre11, but in different ways. In total, our results illuminate the structural and dynamic landscape of Mre11 nuclease function.
Keywords: NMR, side chain methyl group, DNA double strand break repair, Mre11-Rad50, separation of function
Introduction
DNA double strand breaks (DSBs) are dangerous to the integrity of the genomes of all organisms because they can lead to the loss of genetic information or gross chromosomal rearrangements [1]. These potentially damaging events occur during normal cellular processes (e.g., V(D)J or meiotic recombination) or from internal (e.g., reactive oxygen species) or external (e.g., radiation or genotoxic chemicals) stressors. Given the highly cytotoxic nature of DNA DSBs, the cell has developed several strategies for repairing these lesions with non-homologous end joining (NHEJ) and homologous recombination (HR) functioning as the two main repair pathways. The Mre11-Rad50-Nbs1/Xrs2 (MRN/X) complex is an essential player in both of these repair pathways [2–5].
The protective properties of the MRN/X complex are achieved through its various catalytic functions as well as other protein-protein or protein-DNA binding activities and include detecting, signaling, and processing DNA DSBs [3,6]. Mutations in each member of the complex are associated with disease states or have been found in a number of cancer types. Specifically, hypomorphic mutations in Mre11 have been described that give rise to ataxia telangiextasia-like disorder (ATLD) [7,8]. Patients with these mutations generally suffer from immunodeficiency, developmental and neurodegenerative disorders, and a predisposition to certain cancers. Additionally, spontaneous mutations in Mre11 have been noted in colorectal, breast, and ovarian cancers with the hallmark of chromosomal instabilities [9]. Many of these mutations occur in regions of Mre11 that are important for Nbs1 binding [10–12].
Mre11, which functions as a dimer, is a member of the calcineurin-like metallophosphoesterase superfamily of enzymes and has Mn2+-dependent double stranded DNA (dsDNA) 3’-to-5’ exonuclease and single stranded DNA (ssDNA) endonuclease activities [13–16]. Mre11 endonuclease activity can also remove DNA-protein adducts that are formed from aborted topoisomerase reactions in replicating cells, Spo11-linked DNA products formed during meiotic recombination, and Ku70/80 stably associated to DNA DSBs [17–20]. Bound to each Mre11 protomer is a Rad50, a member of the ATP-binding cassette (ABC) ATPase family of proteins [21,22]. Rad50 folds back onto itself to form a functional nucleotide binding domain (NBD), which binds and hydrolyzes ATP, and a ~600–800 Å long coiled-coil and apical Zn-hook domain, which holds two Rad50s together and aids in tethering the two ends of the broken DNA [23,24]. Together, the Mre112-Rad502 (M2R2 or MR) complex constitutes the evolutionarily conserved DNA DSB sensing and processing machine. Finally, Nbs1 (or Xrs2 in budding yeast), found only in eukaryotes, binds to Mre11 via a domain in its C-terminus and acts as a scaffold for bringing downstream effectors such as Mdc1, ATM kinase, and CtIP to sites of DNA damage [25–28].
The nuclease domain of Mre11 contains five conserved motifs (I-V), highlighted on the structure of P. furiosus (Pf) Mre11 in Fig. 1a [13,16]. These motifs are primarily responsible for octahedrally coordinating the two catalytic Mn2+ ions (Fig. 1b). Motif III contains a conserved histidine residue (P. furiosus His85, S. cerevisiae His125, H. sapiens His129) that is required for catalytic activity. This conserved histidine does not directly coordinate the Mn2+ ions, but instead stabilizes the more negatively charged transition state [16,29]. In addition to these catalytic motifs, several recognition loops (RL, Fig. 1a) that are important for substrate DNA binding have been identified based on X-ray crystal structures of Mre11 in complex with various DNAs [29]. Within one of these recognition loops (RL2) is another histidine (P. furiosus His52, S. cerevisiae His59, H. sapiens His63) previously shown to be necessary for Mre11 exonuclease activity. In fact, Pf Mre11 H52S, which is the last residue in motif II, is a separation of function mutant that was shown to be inactive as an exonuclease but active as an endonuclease [29]. This is a surprising phenotype given that both activities should originate at the same di-Mn2+ active site. A similar phenotype was also observed with a series of mirin-based inhibitors. Using these small molecules in human cell lines, it was found that selectively inhibiting endonuclease activity stimulated NHEJ, whereas a lack of exonuclease activity led to a DNA DSB repair defect [30]. However, a detailed understanding for how these inhibitors work or more generally how the two different and separable Mre11 nuclease activities arise from the same active site is lacking.
Fig. 1.

(a) Schematic of full-length P. furiosus Mre11 and X-ray crystal structure of P. furiosus Mre11ND (PDB ID: 1II7) highlighting conserved catalytic motifs (right protomer: I – V) and DNA recognition loops (left protomer: RL1 – 5) [29]. (b) Stereo-view of the Mre11 active site with catalytic motifs I – V colored red, orange, yellow, green, and blue, respectively. H52 (motif II), H85 (motif III), Y187, and dAMP are shown in stick representation. Mn2+ ions are shown as purple spheres. (c) Mre11 exonuclease activity for Pf MRNBD complexes with various Mre11 mutants in the absence and presence of Mn2+ (grey and green bars, respectively). (d) Mre11 endonuclease activity for Pf MRNBD complexes with various Mre11 mutants in the absence and presence of Mn2+ (grey and blue bars, respectively). Data are presented as the mean (bars) and standard deviation (error bars) of at least three measurements.
We were therefore interested in identifying additional separation of function mutants to learn about the underlying mechanism of Mre11 nuclease activities. Pf Mre11 Tyr187 was previously identified through X-ray crystallographic structural studies as a residue important for interacting with the nucleotide monophosphate product of cleavage – dAMP in this case (Fig. 1b) [16]. In this structure, the nucleobase is positioned to form a π-π stacking interaction with the tyrosine residue and the monophosphate interacts with the Mn2+ ions. The importance of this interaction was strengthened by structure-based alignments suggesting that an aromatic residue is conserved at this position (S. cerevisiae F224, H. sapiens F227) [16]. At this time, no disease-associated or natural variant of this residue has been reported. Here, we describe the Y187C mutant of Pf Mre11, which is also inactive as an exonuclease but active as an endonuclease. We have compared Y187C and H52S using a variety of biochemical and biophysical assays and structural studies via methyl-based solution state nuclear magnetic resonance (NMR) spectroscopy. Along the way, we characterized for the first time the solution state dynamics and DNA binding properties of Pf Mre11, which provided a novel site-specific view of ssDNA bound to Mre11. The data show that both separation of function mutants alter the dynamic landscape of Mre11, but in different ways, ultimately affecting the interactions between dsDNA and the protein. This suggests another avenue, besides the mirin-based inhibitors, for chemically separating Mre11 nuclease functions to affect pathway choice in DNA DSB repair.
Results
Pf Mre11 Y187C is exonuclease inactive but endonuclease active
Pf Mre11 Tyr187 was mutated to various polar and non-polar amino acids. The exonuclease activity of these mutants was measured in Pf Mre11-Rad50 NBD complexes (M2R2NBD or simply MRNBD) using a 2-aminopurine based fluorescence assay (Fig. 1c). Wild type MRNBD showed typical Mn2+-dependent exonuclease activity, whereas the H85S negative control did not cleave the DNA under any condition. Y187F substitutes the amino acid at this position for the one found in some eukaryotic Mre11s [16,29], and displayed wild type levels of exonuclease activity. Substitution to the bulkier tryptophan (Y187W) also yielded wild type levels of exonuclease activity. Conversely, non-polar (e.g., alanine or leucine), polar charged (e.g., glutamate or lysine), or polar neutral (e.g., cysteine or glutamine) substitutions all resulted in no exonuclease activity (Fig. 1c). We also expressed several of the Tyr187 mutants as a truncated Pf Mre11 nuclease and capping domains (Mre11ND – amino acids 1–342) construct. These gave the same exonuclease results as the MRNBD mutants (Fig. S1a).
We next questioned if the Tyr187 mutants had a similar effect on Mre11 ssDNA endonuclease activity. Endonucleolytic cleavage of a Cy3/BHQ2-labeled 17 nucleotide ssDNA substrate resulted in Cy3 fluorescence signal (Fig. 1d). Pf MRNBD H85S showed no endonuclease activity as expected, and Y187F and Y187W gave greater than wild type levels of endonuclease activity. Surprisingly, we also observed Mn2+-dependent endonuclease activity for Y187C (Fig. 1d). To confirm this result, we monitored the endonuclease function of the Pf MRNBD complexes over time against a ssDNA virion plasmid as the substrate (Fig. S1c). Indeed, endonucleolytic cleavage of the plasmid by wild type, Y187F, and Y187C produced smaller DNA fragments that migrated faster through the agarose gel. The results for the mutants in the Mre11ND construct mimic the observations for the MRNBD complexes in both endonuclease assays (Figs. S1b and S1d). Thus, the mutation of an aromatic residue at position 187 in Pf Mre11 serves to separate exo- and endonuclease functions.
Pf Mre11 H52, a conserved residue in motif II of the nuclease domain (Figs. 1a and 1b), was previously shown to be required for exonuclease, but not endonuclease activity [29]. We confirmed those results here in our nuclease assays using H52S (Figs. 1 and S1). Thus, the separation of nuclease functions for Y187C is the second example of this phenotype in Mre11.
Separation of Mre11 nuclease activities is not due to difference in substrate affinity
The decoupling of nuclease activities noted above could occur if mutations at Tyr187 and His52 alter the affinity for dsDNA substrates, while leaving the affinity for ssDNA unaffected. To test this hypothesis, we measured the binding affinity (KD) of mutant Pf MRNBD complexes and Mre11ND for substrate DNAs using the change in fluorescence polarization of a labeled DNA molecule upon protein binding (Figs. 2a and 2b and S1e and S1f). For MRNBD, binding reactions were performed in the absence of ATP to ensure that DNA binding originated from Mre11 and not the ATP-bound ‘closed’ form of Rad50, which also binds DNA. Similar KDs for ssDNA of 0.17 – 0.21 μM and Hill coefficients of 2 were obtained for wild type, Y187C, Y187F, and H52S MRNBDs (Fig. 2a and Table 1). ssDNA binding to Mre11ND mutants gave very similar KD and Hill values (Fig. S1e and Table 1) as determined for the MRNBD complexes. A Hill coefficient of 2 suggests that two ssDNAs bind to the Mre11 dimer in a highly cooperative manner.
Fig. 2.

(a) Plots of the change in the fluorescence polarization of a fluorescein-labeled ssDNA as a function of Pf MRNBD concentration. Data for wild type, Y187C, H52S, and Y187F are shown in blue, green, orange, and red points, respectively. The solid line represents the fit of the data to the Hill binding model. (b) Plots of the change in fluorescence polarization of a fluorescein-labeled dsDNA as a function of Pf MRNBD concentration. Data points are colored as in (a). The solid line represents the fit of the data to the Hill binding model. (c) Plots of the change in fluorescence polarization of BODIPY FL-labeled AMP as a function of Pf Mre11ND dimer concentration. Data points (connected with lines) are colored as in (a) with the addition of H85S (grey) and wild type in the absence of divalent cations (black). In all panels, data points and their associated error bars represent the average and standard deviation of at least three measurements.
Table 1.
Mre11 DNA binding constants
| ssDNA Binding | ||||
|---|---|---|---|---|
| Pf Mre11 Mutant | MRNBD | Mre11ND | ||
| KD (μM) | n | KD (μM) | n | |
| WT | 0.18 ± 0.01 | 2.1 ± 0.1 | 0.13 ± 0.01 | 2.1 ± 0.1 |
| Y187C | 0.21 ± 0.01 | 1.9 ± 0.1 | 0.24 ± 0.01 | 1.8 ± 0.1 |
| H52S | 0.17 ± 0.01 | 2.1 ± 0.1 | 0.17 ± 0.01 | 1.9 ± 0.1 |
| Y187F | 0.19 ± 0.01 | 2.0 ± 0.1 | 0.11 ± 0.01 | 2.1 ± 0.1 |
| dsDNA Binding | ||||
| Pf Mre11 Mutant | MRNBD | Mre11ND | ||
| KD (μM) | n | KD (μM) | n | |
| WT | 1.41 ± 0.11 | 0.9 ± 0.1 | 1.30 ± 0.04 | 1.5 ± 0.1 |
| Y187C | 1.80 ± 0.31 | 0.8 ± 0.1 | 1.98 ± 0.05 | 1.5 ± 0.1 |
| H52S | 4.17 ± 0.44 | 1.0 ± 0.1 | 3.17 ± 0.17 | 1.6 ± 0.1 |
| Y187F | 1.32 ± 0.09 | 0.9 ± 0.1 | 0.71 ± 0.01 | 2.4 ± 0.1 |
n is the Hill coefficient.
Values are the average ± the standard deviation of at least three replicates.
For dsDNA binding, similar KDs of ~1.3 – 1.8 μM were measured for wild type, Y187F, and Y187C MRNBD, whereas H52S showed ~3-fold lower affinity (Fig. 2b and Table 1). In contrast to ssDNA binding, the MRNBD dsDNA binding data fit to Hill coefficients of ~1 suggesting that only one dsDNA binds to these MRNBD complexes. Although binding of mutant Mre11ND constructs to dsDNA substrates produced very similar KDs as MRNBD, the Hill coefficients were ~1.5 instead of 1 (Fig. S1f and Table 1). The notable exception was Y187F Mre11ND where the Hill coefficient for dsDNA binding was 2.4. A Hill coefficient of ~1.5 implies that more than one dsDNA can bind to Mre11ND. Altogether, our DNA binding data indicate that ssDNA binding is not affected by these mutants and that dsDNA binding is slightly diminished by H52S; it also suggests that the presence of Rad50NBD changes Mre11 dsDNA binding such that one dsDNA (or two non-cooperative dsDNAs) is bound in MRNBD but two are bound in Mre11ND alone.
The first X-ray crystal structure of Pf Mre11ND had a dAMP bound at the active site (Fig. 1b) to mimic a nucleotide monophosphate nuclease product [16]. The dAMP was stacked against Tyr187 and led to the hypothesis that this residue was important for product release. Using the change in fluorescence polarization of a fluorescently labeled AMP, we measured the affinity of AMP for the mutant Pf Mre11ND constructs, which ensured that binding was to Mre11 and not Rad50. Because of the low binding affinity of AMP (KD > 50 μM) and the decreased stability of Pf Mre11 H52S (protein precipitation above 200 μM dimer concentration), we were unable to calculate reliable KDs for nucleotide binding. However, analysis of the binding trajectories suggests that Y187C binds AMP with the same approximate affinity as wild type, Y187F, and H85S Mre11ND (Fig. 2c). Therefore, mutation at Tyr187 does not disrupt product release, suggesting that the observed interaction of the monophosphate with the Mn2+ ions is more important than base stacking. Interestingly, we saw a lower binding trajectory for H52S, signifying a lower affinity for AMP. In fact, when we omitted the divalent cations from the wild type Mre11ND reaction, we observed AMP binding data that overlaid that of H52S with divalent cations. From that, we infer that H52S, which is relatively far away from the two Mn2+ ion co-factors (~7 and ~9 Å), has somehow altered the cation coordination and/or occupancy within the active site. It is important to note that this effect is not mediated through a charge relay with His85 (~4.5 Å from the Mn2+ ions), as H85S has the same apparent AMP affinity as wild type.
Solution state NMR on Pf Mre11ND
To examine the effect of Y187C and H52S on the structure and dynamics of Pf Mre11, we used NMR spectroscopy on uniformly deuterated, side chain methyl group Ileδ1-[13CH]; Leuδ/Valγ-[13CH3/12CD2]; and Metε-[13CH3] – labeled (hereafter referred to as ILVM-labeled) wild type, Y187C, and H52S Mre11ND constructs. Since solution state NMR has not been undertaken with Mre11 before, we began by obtaining side chain methyl group assignments for the enzyme. Nearly complete stereospecific methyl group side chain assignments (~98%) of Pf Mre11ND were obtained by comparing through-space NMR data with existing crystal structures of the protein and by making point mutations designed to help remove ambiguities (Figs. S2a and S2b).
Methyl group assignments were initially determined on the monomeric construct of Pf Mre11ND L97D [29] and then transferred to wild type Mre11ND. We noted significant chemical shift perturbations between the two constructs for the methyl groups located in the dimerization interface (Fig. S2c). Moreover, those peak positions are unchanged between the Mre11ND and MRNBD spectra (61Leu, 67Leu, 98Leu, and 103Leu in Fig. S2c), confirming that the presence of Rad50NBD does not alter any Pf Mre11 monomerdimer equilibrium. We also measured the translational diffusion of monomeric Mre11ND L97D, dimeric Mre11ND, and the MRNBD complex (Fig. S2d) [31], and we observed decreasing diffusion times indicative of increasing size from monomer to dimer to MRNBD complex. Therefore, in solution at the nearly physiological Pf temperature of 50 °C, Pf Mre11 is a dimer.
ssDNA and dsDNA have different binding interactions with Mre11
We next characterized DNA binding to Pf Mre11ND by NMR to understand how DNA DSB substrates affect the structure of Mre11 and how Mre11 affects the DNA. For the first approach, we employed 13CH3 ILVM-labeled samples of Pf Mre11ND for NMR-observed DNA binding experiments. We collected three different methyl-TROSY 13C,1H correlation spectra: protein alone (no DNA), protein with 1:4 ratio of ssDNA, and protein with 1:4 ratio of dsDNA. Fig. 3a highlights an overlay of a region from the methyl-TROSY spectra for wild type Pf Mre11ND in the apo, ssDNA-, and dsDNA-bound conditions. The data showed that in this region 46Ileδ1 and 146Metε are sensitive to dsDNA but not to ssDNA. On the other hand, 107Ileδ1, 306Ileδ1, and 333Ileδ1 were reporters of ssDNA binding but not dsDNA. Our experimental NMR data therefore show, for the first time, that ssDNA and dsDNA bind to Mre11ND differently. Furthermore, we observed only one set of peaks in each of the DNA-bound samples (Fig. 3a). This indicates that either two DNAs stably bind to the Mre11ND dimer (one to each protomer), that one DNA is rapidly sampling both binding sites in fast exchange on the NMR chemical shift timescale, or that a single DNA interacts “symmetrically” with the Mre11ND dimer. Two DNAs bound to Mre11ND is consistent with the Hill coefficients of ~1.5 for dsDNA binding and 2 for ssDNA binding to Mre11ND (Fig. S1 and Table 1). If one DNA bound to the Mre11ND dimer asymmetrically, two sets of peaks would have been observed in our NMR data (i.e., a peak for the methyl group within the protomer interacting with the DNA and a separate peak for the protomer not interacting with the DNA).
Fig. 3.
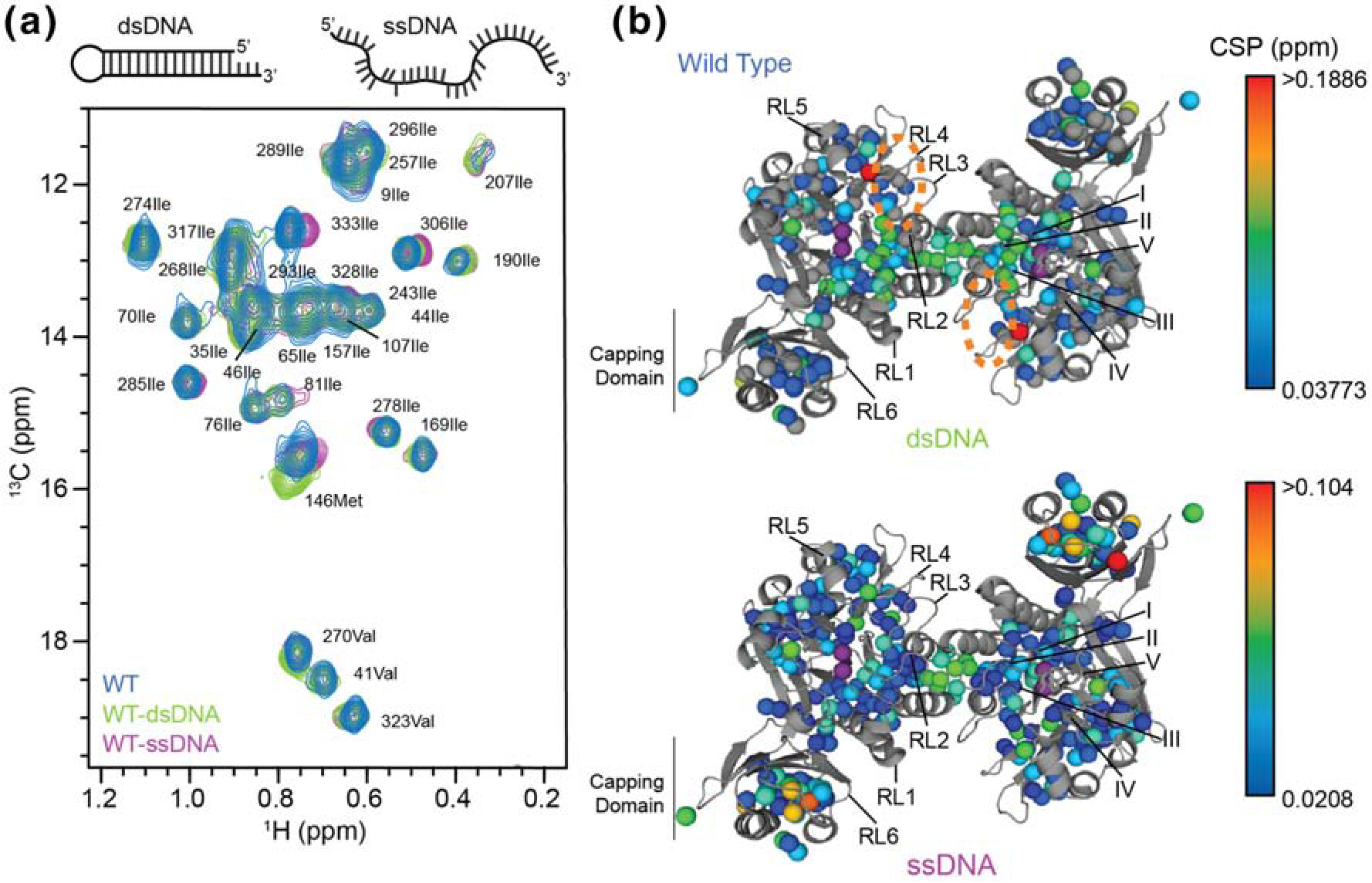
(a) Overlay of a region from the 2D 13C,1H HMQC spectra for wild type Mre11ND in the apo form (blue contours) or bound to dsDNA (green) or ssDNA (purple). (b) CSPs for wild type Mre11ND between the apo and dsDNA-bound (top) or ssDNA-bound (bottom) are shown on the structure of Pf Mre11ND (PDB ID: 1II7) [29]. Conserved catalytic motifs (I – V) and DNA recognition loops (RL1 – 5) are indicated. Side chain methyl groups experiencing CSPs are colored according to the associated gradient. The orange circle indicates the conserved basic patch.
The chemical shift perturbations (CSPs) we observed upon binding of DNA substrate mimics were mapped onto the structure of Pf Mre11ND (Fig. 3b) and revealed distinct binding surfaces for ss- and dsDNA substrates. As predicted by the crystal structures of Pf and Mj Mre11ND bound to synaptic dsDNA DSB mimics [29,32], we saw CSPs to the layer of methyl groups under RL2–4 and motifs I-III upon the addition of dsDNA (Fig. 3b, top). 146Metε, which changed the most upon dsDNA binding, is the N-terminal residue of the α-helix that contains RL5 and is also adjacent to a conserved basic patch (indicated by the dashed ovals in Fig. 3b, top) initially identified in Mj Mre11ND. Based on these perturbations, we hypothesize that one end of our dsDNA DSB mimic is bound to this region of the protein.
Fig. 3b, bottom highlights the CSPs that were observed for ssDNA binding to Pf Mre11ND. Again, we noted changes in the chemical environment of the methyl groups in the layer of residues beneath RL2–4, motifs I-III, and the four helix bundle dimerization interface, as was all observed for Mre11ND bound to dsDNA. However, the changes arising from ssDNA binding were not as large in magnitude as those we observed for the dsDNA substrate. Interestingly, the largest CSPs upon ssDNA-binding were seen in the capping domain. In fact, the largest CSP belonged to 333Ileδ1, which is at the C-terminal end of RL6 and is the C-terminal residue of our nuclease/capping domain construct. In total, our CSP data suggest that RL2–5 are important for dsDNA binding, whereas RL2–4 and RL6 are important for ssDNA binding to Mre11ND.
Y187C and H52S mutants affect Mre11 protein structure and dynamics
We next analyzed the effects that the two separation of functions mutants had on apo Mre11. Comparison of the side chain methyl group resonances in the two-dimensional methyl-TROSY 13C,1H correlation spectra for Pf Mre11ND wild type and Y187C revealed that the vast majority of the peaks overlay for the two constructs (Figs. 4a and 4b, top) indicating that the global fold of Mre11ND is unchanged. The greatest change was observed for 207Ileδ1 and 179Valγ1 (Fig. 4a), which pack against Tyr187 and motif V in the crystal structure of the wild type enzyme; these larger peak movements were expected due to the loss of the aromatic residue in Y187C dramatically changing the chemical environment. Other, smaller CSPs were observed in the nuclease domain (Fig. 4b, top). Interestingly, there is a network of CSPs that extends from the area around motif III in the active site to the four helix bundle responsible for Mre11 dimerization.
Fig. 4.

(a) Overlay of two regions for the 2D 13C,1H HMQC spectra for wild type (blue), Y187C (green), and H52S (orange) Pf Mre11ND. Assignments are provided. (b) Side chain methyl group CSPs between wild type and Y187C (top) and H52S (bottom) are colored according to the associated gradient on the structure of Pf Mre11ND (PDB ID: 1II7) [29]. (c) Representative 1H-1H ‘forbidden’ triple quantum coherence relaxation curves. The data for wild type and the mutants are colored as in (a). Error in the data points are derived from the noise in the spectra. The solid line represents the fit of the data (see Methods). Calculated η rates are given in the lower right corner. (d) Δη are mapped on the structure of Pf Mre11ND (PDB ID: 1II7) [29] and are shaded according to the associated gradient.
Unlike Y187C, the methyl-TROSY 13C,1H correlation spectrum for H52S (Fig. 4a) generally shows more widespread perturbations from wild type (Fig. 4b, bottom). Here, the perturbations encompass much of the nuclease domain, including most of the motifs (I, II, III, and IV) responsible for coordinating the catalytic Mn2+ ions as well as the dimerization interface. Interestingly, we observed CSPs for several residues in the capping domain. Neither Y187C nor H52S appears to alter the chemical environment of the RLs that are responsible for DNA binding, generally consistent with our DNA binding results presented above.
Next, we used the ILVM-labeled Pf Mre11ND samples to examine the effect of the mutations on the amplitude of fast timescale (i.e., picosecond – nanosecond) side chain methyl group motions at 50 °C. Side chain dynamics on this timescale are involved in key protein functions such as allostery, catalysis, and molecular recognition [33]. These motions can readily be characterized in ILVM-labeled protein samples by measuring the relaxation rates (η) of methyl 1H-1H “forbidden” triple quantum coherences, which report on the amplitude of side chain methyl group motion and the timescale of global tumbling [34]. As these methyl 1H relaxation measurements are made on dimeric Mre11ND, we interpreted changes in η rates only as differences in side chain motion. Fig. 4c presents representative relaxation curves for individual side chain methyl groups. The four methyl groups selected here highlight the various trends of the η rates upon mutation of Pf Mre11ND: for 270ValCγ2, wild type has a different rate compared to the mutants; for 278IleCδ1, the η rate for Y187C is different than the others; for 204LeuCδ2, all three have η rates that are within the same range; and for 130LeuCδ2, H52S has the different η rate.
Fig. 4d shows the differences in the η relaxation rates (Δη = ηMUT - ηWT) on the crystal structure of Pf Mre11ND. The data showed both increases and decreases in Δη for Y187C and H52S; however, on average, both mutants generally led to Δη > 0, which indicates increased side chain methyl group flexibility throughout the entire structure – the average Δη rates were 16.3 and 18.5 s−1 for Y187C and H52S, respectively. Collectively, we observed that the mutants are more dynamically similar to each other than to the more rigid wild type Mre11ND (i.e., the graph for 270ValCγ2 in Fig. 4c is representative of the majority of the data: 44 out of 78 methyl groups analyzed). Thus, we conclude that wild type Pf Mre11 has been optimized to a specific level of rigidity that may aid in its function (see Discussion). For both mutants, we could see changes in dynamics around the nuclease motifs and dimerization region. These mirror the changes observed in the CSPs. The largest Δη values for Y187C are predominantly to side chain methyl groups in the nuclease domain, whereas H52S had additional effects on the dynamics in the capping domain, further mirroring the differences observed for the CSPs between the two mutants. Our NMR data therefore show that the structural and dynamic basis for the differences in exo- and endonuclease activities in these two mutants could be different as Y187C predominantly affects the immediate area around the mutation, whereas H52S perturbs the chemical environment of methyl groups throughout the structure. However, both mutants appear to perturb the structure and dynamics of the nuclease motifs and dimerization interface, which are distal to the site of the mutations and implies allostery within Mre11.
Since η reports on local dynamics and global tumbling, an alternative explanation for the overall positive Δη in the mutants could be because of faster macromolecular tumbling (i.e., from disrupting the Mre11ND dimer) for Y187C and H52S. However, a comparison of η rates for mutant versus wild type (Fig. S2e) suggests at most only a ~17% difference in global tumbling time for the two mutants (from the slope of the correlation lines through the data in Fig. S2e). This difference is within the average error in η (~10 – 16%) and is not significant enough for the difference between dimer and monomer global tumbling times. Moreover, the η rates for the N-terminal −3Metε and 1Metε residues are unchanged across the constructs (32.7 – 37.1 sec−1 and 7.2 – 8.0 sec−1, respectively). A different tumbling time, resulting from a different size, would have been clearly seen as different η rates for these residues.
Y187C and H52S alter the structure of dsDNA-bound Mre11
We next added dsDNA and ssDNA to 13CH3 ILVM-labeled samples of Y187C and H52S Mre11ND (Fig. 5a). To identify differences in the DNA-bound structures of the mutants, we calculated the absolute deviation of the chemical shift perturbations between the wild type and mutant DNA-bound forms (ΔCSP = |CSPWT – CSPmutant|). The significant ΔCSPs (i.e. those greater than 1.5x the standard deviation of the median ΔCSPs) are mapped onto the structure of Pf Mre11ND in Fig. 5b. Generally speaking, more significant ΔCSPs were observed when the mutants were bound to the dsDNA DSB mimic as opposed to ssDNA (compare the number of light vs dark spheres in each structure in Fig. 5b). For dsDNA (Fig. 5b, light green and light orange spheres for Y187C and H52S, respectively), we observed differences in the vicinity of the catalytic motifs (I and II for Y187C and II and III for H52S), the dimerization region, RL5, and the conserved basic patch. In the case of Y187C, a group of significant ΔCSPs were observed around the site of the mutation, whereas for H52S, ΔCSPs were seen near the site of that mutation and extended toward the dimer interface. This analysis suggests that although these mutants can bind dsDNA with a similar affinity as wild type Pf Mre11 (Fig. 2b and S1f), structural differences exist in key catalytic and substrate binding regions that likely lead to the loss of exonuclease function.
Fig. 5.

(a) Overlay of a region from 2D 13C,1H HMQC spectra for Mre11ND. Wild type (blue contours), Y187C (green), and H52S (orange) Mre11ND bound to dsDNA (right) or ssDNA (left). (b) Significant ΔCSPs between the wild type and Y187C (left) and H52S (right) DNA-bound forms of Mre11ND are shown on the structure of Pf Mre11ND (PDB ID: 1II7) [29]. For each, the left protomer highlights the differences from the dsDNA-bound form of wild type, whereas the right protomer highlights the differences from the ssDNA-bound form of wild type. Conserved catalytic motifs (I – V) and DNA recognition loops (RL1 – 5) are indicated. The orange circle indicates the conserved basic patch.
In contrast to the dsDNA-bound forms, ssDNA-bound mutants showed relatively few significant deviations from ssDNA-bound wild type Mre11ND (Fig. 5b, dark green and dark orange spheres for Y187C and H52S, respectively). ssDNA-bound Y187C (dark green spheres) showed a patch of ΔCSPs around motifs II and III and the site of mutation, whereas ssDNA-bound H52S had only small, structurally dispersed perturbations. Overall, the methyl-based NMR data on wild type and mutant Mre11ND bound to the two DNA substrates provide insight for understanding the mechanism of separation of Mre11 nuclease function in these mutants – Y187C and H52S have a problem forming the correct dsDNA-bound complex.
Y187C and H52S destabilize the bound dsDNA substrate
For the second approach toward understanding DNA binding to Mre11, we utilized NMR spectroscopy to monitor the stability of the DNA DSB mimic bound to unlabeled Pf Mre11ND. The imino protons of guanosine and thymidine nucleobases have characteristic chemical shifts that are downfield of other biological proton resonances. These peaks are only observed when a stable base pair is formed in a duplex DNA; otherwise, rapid exchange with solvent broadens the signal beyond detection. The addition of Pf Mre11ND to the hairpin dsDNA DSB mimic (1.2:1 Mre11ND monomer:dsDNA) shifted several imino resonances of the DNA indicating that Mre11ND bound to the DNA (Fig. S3a). We then monitored the intensity of the imino proton signals as a function of increasing temperature. As expected, a shift in resonance position and loss of signal occurred as the base pairs of the hairpin DNA opened (Figs. 6a and S3b). Subsequent integration of the imino 1H signals gave typical melting curves, which were used to extract the melting temperature (Tm; temperature where 50% of the duplex is unfolded/folded) and ΔH (change in enthalpy for DNA duplex formation) (Fig. 6b). As expected, when bound to protein the DNA is more stable and thus melts at a higher temperature – Tm for apo dsDNA, grey curve, ~52 °C vs 55 – 59 °C for Mre11ND bound (Fig. 6b and Table 2). Interestingly, when the hairpin dsDNA was bound to Y187C and H52S the increase in Tm was slightly less than for wild type and Y187F (Fig. 6b and Table 2). The difference in Tms (55 °C for H52S and 57 °C for Y187C vs 59 °C for WT and Y187F) indicates that the dsDNA DSB mimic was not as stable when bound to these mutants. The ΔH values were used to calculate ΔS, using the known enthalpy/entropy relationship for DNA melting/hybridization [35], and ΔG at 50 °C (Table 2). We then calculated ΔΔG values between apo and Mre11ND-bound hairpin DNA. The negative ΔΔG values (−1.1 – −3.7 kJ mol−1; Table 2) again show that the dsDNA is slightly stabilized when bound to Mre11ND. Finally, we determined ΔΔG values between wild type and mutant Mre11ND-bound dsDNA. Positive ΔΔG values were obtained for Y187C and H52S indicating that the dsDNA is somewhat less stable than when bound to wild type Mre11ND (Table 2). These results were consistent with the changes in side chain methyl group dynamics in the mutants that we observed in Fig. 4d and suggest that the more dynamic Mre11ND mutants either transferred some of their motion to the dsDNA or were incapable of specifically binding the dsDNA like wild type, which destabilized the duplex causing it to melt at a lower temperature.
Fig. 6.

(a) Stacked 1D 1H NMR spectra of the thymine and guanine imino proton region for the hairpin dsDNA DSB mimic in the presence of wild type Pf Mre11ND. Spectra collected from 30 to 70 °C are colored from blue to red according to the gradient. (b) van’t Hoff analysis of dsDNA DSB mimic stability in the absence and presence of wild type and mutant Mre11ND. Data points for the integrated imino intensity, which were converted to the fraction folded, are shown for dsDNA in the presence of wild type (blue), Y187C (green), Y187F (red), and H52S (orange). Data for dsDNA in the absence of Mre11ND is in grey. The solid line represents the best fit of the data to the van’t Hoff equation. The inset shows the calculated ΔH values with error bars derived from the covariance of the fit.
Table 2.
Thermodynamics of dsDNA melting in the presence and absence of Mre11ND
| Rpa | Tm (°C)b | ΔH (kJ moi−1)b | ΔS (kJ mol−1 K−1)c | ΔG (kJ moi−1)d | ΔΔGe | ΔΔGf | |
|---|---|---|---|---|---|---|---|
| apo | 0.995 | 52.4 ± 0.7 | −98.1 ± 6.2 | −0.36 ± 0.03 | 18.1 | ||
| WT | 0.998 | 58.7 ± 0.4 | −117.7 ± 5.3 | −0.43 ± 0.03 | 21.8 | −3.7 | |
| Y187C | 0.998 | 57.5 ± 0.4 | −103.8 ± 4.4 | −0.38 ± 0.02 | 19.2 | −1.1 | 2.6 |
| H52S | 0.993 | 54.9 ± 0.9 | −106.3 ± 9.0 | −0.39 ± 0.05 | 19.7 | −1.5 | 2.1 |
| Y187F | 0.997 | 59.2 ± 0.7 | −117.7 ± 6.6 | −0.43 ± 0.03 | 21.9 | −3.7 | 0.0 |
Pearson correlation coefficient
Errors are from the covariance of the fits.
Calculated with Eq. 5 of Petruska and Goodman [41]. Errors propagated from ΔH.
Calculated with ΔG = ΔH – TΔS at 50 °C, which is the temperature used for NMR experiments.
ΔGMre11ND, bound - ΔGapo
ΔGwt - ΔGmutant
Given this site-specific view for the effects that these separation of function mutants have on substrate DNA binding and stability, we can now propose that His52 and Tyr187 in Pf Mre11 are critical for the proper positioning of the 3’-end across the two Mn2+ cofactors for exonuclease activity. In fact, Pf Mre11 Tyr187 likely recognizes the 3’-end of dsDNA substrates through π-π stacking between the nucleobase and aromatic side chain or a potential π−3’-OH hydrogen bond. As previously proposed, H52S stabilizes the dsDNA substrate on the other side of the active site. Interestingly, failure to stabilize the 3’-end leads to improper binding of the rest of the dsDNA substrate as well, which we detect as CSPs of 143Metε near a conserved basic pocket. These interactions are critical for holding dsDNA in the stable conformation necessary for the exonuclease reaction. Since ssDNA substrates bind to Mre11 differently compared to dsDNAs, mutations to these two conserved residues do not affect endonuclease activity like they do exonuclease activity.
DNA DSB repair in vivo is unaffected by the separation of function mutants
Finally, we tested the ability of analogous mutants in S. cerevisiae (Sc) Mre11 to repair DNA DSBs in vivo. Unfortunately, there is currently no crystal structure of Sc Mre11 for direct comparison, so the analogous amino acid to Pf Mre11 Y187 in budding yeast was identified by two different means. First, overlays of crystal structures of P. furiosus, S. pombe, and C. thermophilum Mre11ND (PDB IDs 1II7, 4FCX, and 4YKE, respectively; Fig. S4a) combined with sequence alignment (T-Coffee [36]) indicated that Sc Mre11 T217 was the structurally analogous residue [12,37]. Second, other structure-based sequence alignments showed that Sc Mre11 F224 was equivalent to Pf Y187 [16,29]. Therefore, both mutations were introduced into budding yeast. Myc-tagged mre11, mre11-H59S (Pf Mre11 H52S), mre11-H125S (Pf Mre11 H85S), mre11-T217C, and mre11-F224C were stably integrated into the endogenous mre11 locus of S. cerevisiae. A mre11Δ strain was generated by replacing the endogenous mre11 with the kanMX gene. Western blots confirmed that mutant Mre11 expression was equal to wild type (Fig. S4b), and growth curves showed that only the mre11Δ experienced slower growth rates (Fig. 7a).
Fig. 7.

(a) Growth curves for S. cerevisiae at 30 °C harboring wild type, mre11Δ, or single-site mutations. Each data point, representing the construct indicated by the legend, is the average of four independent measurements. (b) S. cerevisiae survival assays for serial dilutions of various yeast strains on No Drug, 1 μg/mL CPT, and 0.03% MMS containing XY plates. (c) Box plot, showing the median, high, and low values for the efficiency of the plasmid-based NHEJ assay. Data were normalized relative to the wild type mre11 strain. At least three independent measurements were used. (d) Chromosomal end resection assay results showing the fraction of cells from the various yeast strains that underwent DNA end resection after a galactose-induced HO endonuclease cut. The points and associated error bars are the average and standard deviation of at least three independent measurements.
We first examined if the separation of function mutants are sensitive to the genotoxic agents camptothecin (CPT), a topoisomerase I inhibitor which results in DNA DSB at replication forks, and methyl methanesulfonate (MMS), a DNA methylating agent which results in stalled replication forks. As previously observed, mutations to the conserved histidines in motifs II and III (mre11-H59S and mre11-H125S, respectively) show little-to-no sensitivity to these DNA damaging agents (Fig. 7b). Likewise, the new separation of function mutant (mre11-T217C and mre11-F224C) does not affect repair of DNA lesions generated by CPT or MMS.
Next, we tested the effect of the mutants on NHEJ in budding yeast. The ability to perform NHEJ was assessed by transforming cells with a linearized plasmid containing a yeast origin of replication and complementary nutritional marker (HIS3) [38,39]. The plasmid was linearized with EcoRI at a site lacking homology to the yeast genome; therefore, repair of the plasmid and growth on histidine drop-out media requires NHEJ at the site of the cut. As previously shown, the presence of Mre11, but not its nuclease activities, is required for NHEJ (Fig. 7c) [40]. Interestingly, mre11-T217C and mre11-F224C both show greater growth on drop-out media as compared to wild type and the two conserved histidine mutants, indicating the possibility of more efficient DNA damage repair by NHEJ.
Finally, we assessed the ability of the Sc Mre11 mutants to perform DNA end resection following a single HO endonuclease-mediated cut in the yeast genome [28,41]. Kinetic analysis (Fig. 7d) revealed that all of the Mre11 mutants were capable of performing end resection, whereas the mre11Δ strain could not. We observed a slight lag in the fraction resected at the 0.5 and 1 hour time points in all of the mre11 mutants compared to wild type. Only mre11-H125S still showed a slightly lower fraction after 2 hours. By 4 and 6 hours, all of the mutants produced similar levels of resection as wild type. Overall, this result is consistent with previous data demonstrating that although Mre11 is necessary for DNA DSB repair in budding yeast, its catalytic activities are not [42].
Discussion
Herein, we presented the first ever NMR data on Mre11 and then explored the origins for the separation of Mre11 exo- and endonuclease functions. Previous structural and biochemical studies have described a mutant and small molecule inhibitors that uncouple Mre11 exo- and endonuclease activities [29,30]. Pf Mre11 H52S was originally identified as an exonuclease inactive, endonuclease active mutant during structural studies of Mre11ND bound to two different DNA DSB mimics [29]. This conserved motif II histidine is thought to rotate the scissile phosphate bond over the catalytic Mn2+ ions for hydrolysis. Subsequently, rational design of second generation mirin-based inhibitors identified a new small molecule that only inhibited exonuclease activity (PFM39) and two new molecules that only inhibited endonuclease activity (PFM01 and PFM03) [30]. It was hypothesized that these small molecule inhibitors differentially prevented exonuclease activity (mirin and PFM39) by limiting the ability of H52 (T. maritima H61) to rotate the scissile phosphate for hydrolysis or inhibited endonuclease activity (PFM01 and PFM03) by limiting ssDNA binding [30]. In this study, we described a new mutant of Pf Mre11, Y187C, that is also inactive as an exonuclease but active as an endonuclease. Through a series of biochemical and structural biology experiments, we were able to trace the origins of the Y187C exo-dead/endo-active phenotype to the improper alignment of the dsDNA substrate, both structurally and dynamically, for catalysis. Our NMR data suggest that the conserved aromatic ring at residue 187 interacts with and recognizes the 3’-end of the DNA DSB, either through base stacking or potentially a π-OH hydrogen bond. Failure to recognize the 3’-end results in alterations to the architecture and side chain dynamics of the active site and conserved basic patch of the dsDNA-bound structure.
Interestingly, Pf Mre11 Tyr187 is in a loop that was not visible in the electron density of the recent cryo-electron microscopy (cryo-EM) structure of full-length E. coli (Ec) MR (SbcCD) in an ADP-bound ‘closed’ state associated with dsDNA [44]. Käshammer et al referred to this loop as the ‘sealing loop’, and the deletion of this loop from Ec MR altered nuclease activity. Intriguingly, Pf Mre11 207IleCδ1 is at the C-terminus of the homologous loop, and this residue has the weakest peak intensity in the isoleucine region of our HMQC data (Figs. 3a, 4a, and 5a), which is indicative of motions on the millisecond timescale. This peak intensifies, signifying rigidification of millisecond timescale motions, upon DNA binding in wild type and Y187C but not in H52S (Fig. 5a). Thus, Y187C, in a loop on the opposite side of the catalytic Mn2+ ions, allowed us to further define the position, orientation, and stability of DNA substrates for the Mre11 nuclease reactions.
Our NMR results for H52S compare favorably with the crystal structures of T. maritima (Tm) Mre11ND bound to the exonuclease inhibitors mirin and PFM39 [30]. In those structures, the small molecule inhibitors bind in between the motif II and III loops that contain His52 (Tm His61) and His85 (Tm His94), and we observe side chain methyl group CSPs in this region for the H52S mutation (Fig. 4). Moreover, these inhibitors led to a very slight shift (~1.15 Å) in the positions of the two catalytic Mn2+ ions [30]. Similarly, we observed that H52S bound AMP in the presence of divalent cations with a similar affinity as wild type Mre11ND in the absence of metal ions (Fig. 2c), suggesting that this mutant, like mirin and PFM39, likely alters the metal coordination. Surprisingly, we also noted CSPs and differences in dynamics in the methyl groups of the H52S Mre11 capping domain (Figs. 4b and 4d). These effects distant to the mutation site are reminiscent of perturbations that we observed in a set of mutants in Rad50 NBD, which illuminated an allosteric network in that protein [43]. H52S may likewise affect an allosteric network between the Mre11 active sites and capping domain, a domain that we (Fig. 3b) and others have shown to be important for ssDNA binding [29].
It was recently shown for the FAN1 endonuclease that aromatic residues acted as a wedge to split dsDNA into two ssDNA strands and to guide one of these strands to the active site [45]. Other wedge motifs have been noted in the flap endonuclease FEN1 and in the alkylated base binding Atl1 [46,47]. We hypothesized that Pf Mre11 Tyr187 might play such a role in an extended wedge motif with His52. This idea is consistent with a proposed role of the motif II histidine in rotating the dsDNA ribose-phosphate backbone into the correct orientation for the nuclease reaction. Presumedly, the loss of the wedge or guide would cause a failure of exonuclease activity while retaining endonuclease activity, since a wedge is not needed to open the ssDNA substrate. We tested this hypothesis by determining the stability of a dsDNA substrate bound to Mre11ND assuming that a wedge would have already destabilized the dsDNA by partially unwinding it. Thus, the dsDNA would melt at a lower temperature for wild type and Y187F (wedge motif intact) compared to Y187C and H52S (wedge motif disrupted). However, both Y187C and H52S led to less stable dsDNA compared to wild type (Fig. 6). Therefore, the Tyr187 and His52 residues do not act as an extended wedge motif, as originally hypothesized. The decreased stability of the dsDNA however is consistent with our dynamics measurements demonstrating that the mutants are generally more flexible than wild type. Additionally, the stability/dynamics of the DNA substrate was recently proposed to be important for differences in endo- and exonuclease activities [48]. Indeed, we observed that the loss of exonuclease activity in Y187C and H52S is correlated with lower stability for the dsDNA substrate when bound to Mre11ND (Fig. 6b and Table 2).
We can compare our solution state NMR data on Pf Mre11ND with other DNA-bound Mre11ND structures. Like the crystal structures of DNA-bound Pf and Mj Mre11ND, we observed the dsDNA interacting with a conserved basic patch opposite of the capping domain [29,32]. Furthermore, in agreement with the recent cryo-EM structure of the complete Ec MR-dsDNA complex [44], our NMR data placed the 3’-end of the dsDNA substrate in-line with the catalytic Mn2+ ions. We also observed CSPs in the capping domain for ssDNA, which is in agreement with similar interactions observed in the Pf Mre11ND crystal structure with a branched DNA [29]. On the other hand, three of the four existing DNA-bound Mre11ND crystal structures and the Ec MR cryo-EM structure had only one DNA in or near the Mre11 active site [29,32,44], yet our NMR data is consistent with Pf Mre11ND binding two substrates for both dsDNA and ssDNA. The Ec MR structure and the comparison of DNA binding in the presence and absence of Rad50 NBD presented here (Figs. 2 and S1) clearly demonstrate the importance of Rad50 in DNA binding [44], as we noted a difference in cooperativity in the absence of Rad50. Thus, further solution state studies on the MRNBD complex in the presence of ATP and ADP will be necessary to understand how these various DNA-bound structures fit into the sensing and processing functions of the MR complex.
To date, there is no high-resolution structure of Mre11 bound to a ssDNA endonuclease substrate. Our DNA-induced CSPs clearly show that each substrate binds to Mre11ND differently: dsDNA predominantly affects the nuclease domain, whereas ssDNA affects the capping and nuclease domains. It was previously suggested that ssDNA approached the di-Mn2+ active site from a cleft between the capping and nuclease domains [29]; however, we do not see any significant CSPs to the methyl probe (251Val) in that cleft. Additionally, ssDNA did not greatly affect the conserved basic patch as dsDNA did. Again, further solution state studies on the MR complex bound to ssDNAs are needed to define the endonuclease competent structure, which could then be used to design the next generation of separation of function inhibitors. Nevertheless, since ssDNA substrates interact differently than dsDNA with Mre11, endonuclease activity is unaltered in Y187C and H52S. In fact, our DNA binding data, which show a 10-fold better binding affinity for ssDNA compared to dsDNA (Fig. 2 and Table 1), suggest that the Mre11 endonuclease activity may simply be more robust in the face of changes in structure and dynamics upon mutation.
Previous studies have shown that dynamics in the Mre11 dimer interface (e.g., a change in the Mre11 dimer-monomer equilibrium that favors Mre11 dissociation) are important for endonuclease activity, particularly at protein-blocked dsDNA ends [32,44,48]. For example, recent studies have reported that E. coli Mre11ND (SbcD) is monomeric in the absence of Rad50 and that the full-length MR (SbcCD) complex dissociates at the Mre11 dimer interface for proper loading onto dsDNA [44,48]. Furthermore, previous results on full-length T4 bacteriophage Mre11 have also shown that the enzyme is monomeric in the absence of Rad50 [49]. On the other hand, small angle X-ray scattering (SAXS) studies on Pf Mre11ND have indicated a dimer in solution [29]. Our solution state NMR data also does not support the idea for Pf Mre11ND dissociation. The methyl correlation spectra, which has a number of sensitive probes in the Mre11 dimer interface, and dynamics data, which is sensitive to macromolecular tumbling, support a stable Pf Mre11 dimer (Figs. 4 and S2). Upon DNA substrate binding, we did note small changes in the chemical shifts of the four helix bundle responsible for dimerization (Fig. 3b). Changes in the chemical shifts and fast timescale dynamics within this region were also observed in the apo form of the mutants (Fig. 4). Thus, small changes in structure (e.g., rigid body rotations from flexing these helices) and/or changes in the dynamics in the dimerization helices could accompany DNA binding to Pf Mre11 and be important for nuclease activity.
In conclusion, we describe the biochemical and structural characterization of two Mre11 separation of function mutants, which both lead to an exonuclease inactive/endonuclease active phenotype. In both cases, the mutants disrupt the local structure and dynamics of Mre11 resulting in a destabilized dsDNA substrate. Because of the different binding mode and higher affinity of Mre11 for endonuclease substrates, the perturbations caused by these mutations do not affect ssDNA endonuclease activity.
Materials and Methods
Protein expression and purification.
The genes for E. coli-codon optimized Pyrococcus furiosus (Pf) Mre11 were synthesized by Life Technologies, Inc. and subsequently cloned into a modified pET-29m vector (Novagen). A modified Quikchange (Stratagene) approach was used to introduce point mutations into Mre11, including a stop codon after aa342 for the truncated Pf Mre11 nuclease and capping domains construct (Mre11ND). Plasmids were transformed into E. coli BL21(DE3) C41 cells (Sigma) and grown in LB media. Unlabeled protein expression was induced with 1 mM IPTG at 37 °C for 4 h.
For Mre11 and Mre11ND protein purification, cells were lysed in 25 mM sodium phosphate, 300 mM NaCl, and 25 mM imidazole, pH 8, via sonication and then heated for 10 min at 65 °C. Clarified cell lysate was loaded onto a 5 mL HisTrap HP column (GE Healthcare) and eluted with 300 mM imidazole. The 6xHis tag was removed with TEV protease, and Mre11 was repurified, as the flow through, on the HisTrap HP column. The sample was subsequently diluted to decrease the sodium chloride concentration to ~100 mM, then loaded onto a HiTrap Q HP cation exchange column (GE Healthcare), and eluted with a linear gradient into 25 mM HEPES, 1 M sodium acetate, 0.1 mM EDTA, pH 7. The Q elution peak was concentrated using ultrafiltration (Vivaspin, Sartorius) before loading onto a HiLoad 16/60 Superdex 200 pg column (GE Healthcare) equilibrated in 25 mM HEPES, 200 mM sodium acetate, 0.1 mM EDTA, pH 7. Rad50NBD expression and purification were as described previously [43].
For Mre11ND NMR sample expression, E. coli BL21(DE3) C41 cells (Sigma) were transformed with plasmids and grown in deuterated 2x M9 minimal media [50] with 1 g/L 14NH4Cl and 3 g/L 2H,12C-glucose as the sole nitrogen and carbon sources. U-[2H], Ileδ1-[13CH3], Leuδ/Valγ-[13CH3,12CD3], Metε-[CH3] (ILVM)-labeled samples of 6xHis-tagged Mre11 were produced as previously described except 100 mg/L of each precursor was added to the bacterial culture 45 min before 1 mM IPTG induction and overexpression was for ~16 h at 37 °C [51]. The purification protocol was as described above for unlabeled protein. The final size exclusion fractions were pooled, buffer exchanged into 25 mM HEPES, 100 mM sodium chloride, 5 mM MgCl2, 0.1 mM EDTA, 1% glycerol, 1 mM TCEP, pH 7.0 in 99.9% D2O (unless otherwise indicated), and concentrated via ultrafiltration to a final volume of 500 μL for NMR samples. The overall yield of Mre11ND in deuterated 2x M9 media was ~500 μM in 0.5 mL from 1.5 L growth media, and final sample concentrations varied from 100 to 500 μM.
Mre11 exonuclease assay.
Exonuclease assays were performed with a dsDNA substrate that contained a 2-aminopurine (2-AP) at the second position from the 3’-end of one strand of the dsDNA, as previously described [29]. The two DNA strands (strand 1: 5′-GGCGTGCCTTGGGCGCGCTGCGGGCGG(2-AP)G-3′ and strand 2: 5′-CTCCGCCCGCAGCGCGCCCAAGGCACGCC-3′; IDT) were annealed at equimolar concentrations. Experiments were performed in Nuclease Buffer (50 mM Tris, 150 mM NaCl, 0.1% PEG-6000, 0.25% glycerol, 1 mM DTT, pH 7.5 at room temperature) and contained either 0.5 μM M2R NBD2 complex (MRNBD, with 1.2-fold excess Rad50NBD) or Mre11ND dimer. 1 μM 2-AP dsDNA duplex was used as the substrate without or with 1 mM MnCl2. Reactions were incubated in the dark at 60 °C for 45 min, cooled at room temperature for 2 min, and centrifuged at 21k × g for 1 min. 25 μL of each reaction was transferred to a black, flat-bottom 384-well plate, which was centrifuged at 500 × g for 1 min. 2-AP fluorescence (ex310/em375) was read in a BioTek Synergy Neo2 multimode plate reader. Reported values and errors are the average and standard deviation of n ≥ 3 experiments.
Mre11 endonuclease assays.
A 5’-end Cyanine3 (Cy3) and 3’-end Black Hole Quencher-2 (BHQ-2)-labeled 17 nucleotide ssDNA (5’-Cy3-TCTCTAGCAGTGGCGCCBHQ2–3’; IDT) was used for the quantitative endonuclease assay [52]. Experiments were performed in Nuclease Buffer using the same conditions as described for the 2-AP exonuclease assay above with the exception of 0.2 μM Cy3/BHQ-2 ssDNA as the substrate. Cy3 fluorescence (ex535/em570) was measured in a BioTek Synergy Neo2 plate reader. Reported values and errors are the average and standard deviation of n ≥ 3 experiments.
Qualitative endonuclease reactions were performed using 1 μg of ΦX174 single stranded virion circular DNA (New England Biolabs) as the substrate in 30 μL reactions. Either 2 μM MRNBD or Mre11ND dimer in 25 mM Tris, 100 mM NaCl, 2.5% glycerol, 0.1 mM EDTA, 1 mM DTT, pH 7.5 buffer (without or with 5 mM MnCl2) was incubated at 60°C. 6 μL time points were removed at 10, 20, 30, and 40 min and were stopped by adding 1% w/v SDS, 10 mM EDTA, 0.5 mg/mL Proteinase K. Samples were then centrifuged for 1 min at 21k × g and incubated at room temperature for 5 min. Endonuclease products were resolved on 1% agarose gel (in 1X TAE) run at 100 mA constant voltage for 60 min and were visualized by staining with GelRed nucleic acid stain (Biotium).
Adenosine Monophosphate (AMP) binding assay.
AMP binding was monitored via the change in fluorescence polarization of BODIPY-labeled AMP. Fluorescently labeled AMP was generated by incubating BODIPY-labeled ATP (Adenosine 5′-Triphosphate, BODIPY FL 2′-(or-3′)-O-(N-(2-Aminoethyl)-Urethane, Life Technologies) with 10 units/mL apyrase (Sigma) for 1 h at 30 °C. The apyrase was heat inactivated at 65 °C for 10 min. Thin layer chromatography (TLC) confirmed that ATP was hydrolyzed into AMP. Pf Mre11ND dimer was titrated from 0 to 100 μM in 50 mM HEPES, 100 mM NaCl, 1% glycerol, 0.1% PEG-6000, 0.1 mM EDTA, 1 mM TCEP, 0.1 mg/mL bovine serum albumin, pH 7. Each 30 μL binding reaction contained 5 nM Bodipy FL-AMP, 5 mM MgCl2, and 5 mM MnCl2. The reactions were incubated at 60 °C for 30 min. 25 μL was transferred into a black, flat bottom 384-well plate, which was centrifuged at 500 × g for 2 min. Parallel and perpendicular fluorescence intensities were measured using a FP 485/530 filter in the Synergy Neo2 plate reader. The fluorescence polarization (FP) was calculated via BioTek software. Reported points and their errors are the average and standard deviation of n ≥ 3 experiments.
DNA binding affinity.
ssDNA binding assays were performed using a FAM (carboxyfluorescein) -labeled 40-mer DNA (5′-FAMGTGTTCGGACTCTGCCTCAAGACGGTAGTCAACGTGCTTG-3’; IDT) as described [53]. Double-stranded DNA substrate (dsDNA) was made by annealing the FAM-labeled ssDNA to an unlabeled complementary strand. Binding reactions in 50 mM HEPES, 100 mM NaCl, 0.1 mM EDTA, 1% glycerol, 1 mM TCEP, 0.1 % PEG-6000, 1 mM MnCl2, pH 7 with 25 nM either ssDNA or dsDNA were titrated with increasing amounts of either MRNBD or Mre11ND dimer for a final volume of 30 μL. Binding reactions were incubated at room temperature for 15 min before reading for FAM FP in a Synergy Neo2 plate reader using the FP 485/530 filter. FP data were fit to the Hill equation for binding
where [DNA] is the concentration of DNA, KD is the dissociation constant, n is the Hill coefficient, FP0 is the FP in the absence of protein, and FPmax is FP for maximum binding. Reported points and their errors are the average and standard deviation of n ≥ 3 experiments with most error bars within the symbols.
NMR assignments.
All NMR spectra were collected at 50 °C on a 600 MHz (14.1 T) Agilent DD2 NMR spectrometer equipped with a room temperature z-axis gradient probe. Data sets were processed and analyzed with NMRPipe and associated programs as well as CCPN Analysis [54,55]. From the two-dimensional (2D) 13C,1H methyl Transverse Relaxation Optimized SpectroscopY (TROSY) Heteronuclear Multiple Quantum Coherence (HMQC) spectra [56,57], 130 13CH3 ILVM side chain methyl group probes were utilized to give insights into the Mre11ND structure and dynamics. Using the previously published crystal structure of Pf Mre11ND (PDB ID: 1II7) and nuclear Overhauser effect (NOE) experiments (250 ms mixing time), the methyl resonances in the HMQC spectra were assigned to specific ILVM residues in the structure. To aid in the assignments, an NMR sample of Mre11ND labeled with 13CH3 valines and 12CH3 leucines, which was prepared by adding unlabeled leucine (100 mg/L culture) in addition to 13CH3/12CD3 α-ketoisovaleric acid prior to protein expression, was made to differentiate between valine and leucine peaks in the 2D correlation spectra [58]. Another NMR sample was also prepared where both methyl groups of leucine and valine residues were 13CH3–labeled, using 13CH3/13CH3 α-ketoisovaleric acid during expression. Using this sample in a short mixing time (50 msec) NOE experiment revealed correlations between methyl groups in the same leucine/valine residue [59]. Stereo-specific assignments were achieved by 13CH3-labeling only the proS methyl group of leucine and valine residues using 2-(13C) methyl-4-(D3)-acetolactate prior to expression [60]. A few of the isolated ILVM methyl groups in the crystal structure could not be assigned using the above data; these residues were mutated to relatively conservative substitutions. The subsequent disappearance of a methyl resonance in the 2D 13C,1 H methyl-TROSY HMQC spectra helped to assign the respective mutant residue. Using these samples and NOEs along with the mutations, we achieved ~98% of the ILVM methyl groups assignments. Side chain methyl group chemical shifts were deposited to the Biological Magnetic Resonance Databank under accession number 28054.
Translational diffusion measurements.
Mre11 translational diffusion was measured at 50 °C on ILVM-labeled monomeric Mre11ND L97D, dimeric Mre11ND, and MRNBD complex using a 13C-edited, convection compensated sLED pulsed-field gradient NMR experiment based on the pulse sequence described by Zheng and Price [31]. Six dephasing/rephasing gradients with powers ranging from 4 – 40 G/cm were utilized with 1 ms gradient and 100 ms diffusion times. 13C decoupling was not used during acquisition to prevent heating. The total signal of the methyl group region was integrated in VnmrJ 4.2A. Translational diffusion times were inferred from plots of the natural logarithm of relative intensity versus the square of the gradient power.
Methyl chemical shift perturbation analysis.
2D 13C,1H methyl-TROSY HMQC spectra were recorded on uniformly deuterated, side chain methyl group ILVM-labeled samples of wild type Pf Mre11ND and the Y187C and H52S mutants in the absence of DNA (no DNA) or in the presence of a 1:4 molar excess of ssDNA (5’-TGTAGTGCATTGCGTTTTTGCTTCTACGTGCGTGAC-3’; Sigma) or hairpin dsDNA (5’-CACGCACGTAGAAGCTTTTGCTTCTACGTGCGTGACT-3’; Sigma). To analyze the effects that mutation or DNA binding had on each of the side chain methyl groups, a weighted methyl chemical shift perturbation (CSP) was calculated from the 13C and 1H chemical shifts (δC and δH, respectively) according to
where δi is the chemical shift value for Mre11ND wild type or apo (no DNA) and δj is the chemical shift value for mutant or DNA-bound, and wC (1.65, 1.6, 1.4, and 1.54) and wH (0.29, 0.28, 0.27, and 0.41) are the standard deviations for δC and δH from the Biological Magnetic Resonance Data Bank for Ileδ1, Leuδ, Valγ, and Metε methyl groups, respectively. CSPs for the mutants and DNA binding were plotted on the crystal structure of Pf Mre11ND (PDB ID: 1II7) [29].
For DNA-binding experiments, significant CSPs were determined by taking the absolute value of the mutant CSP from the wild type CSP (i.e., ΔCSP = |CSPmutant – CSPWT|) and identifying the methyl groups with deviations (ΔCSP) greater than the median ΔCSP plus 1.5-times the standard deviation.
Methyl group dynamics.
The build-up and decay of methyl 1H-1H ‘forbidden’ triple-quantum coherences were measured as described [34]. Pairs of forbidden (triple quantum) and allowed (single quantum) data sets at a given relaxation delay (0.002, 0.004, 0.006, 0.008, 0.01, 0.015, 0.02, 0.025 sec) were recorded in an interleaved manner. Fitting the ratios of peak intensities (Iforbid/Iallow) as a function of relaxation delay (T) gives the intra-methyl 1H-1H dipolar cross-correlated relaxation rate, η,
where Iforbid and Iallow are the intensity of the 1H triple- and single-quantum coherences, respectively, C is 0.75, and δ accounts for relaxation from external protons. The reported errors were calculated from the covariance matrix of the fit. Changes in the η relaxation rates between mutant and wild type were calculated by subtracting the wild type values from the mutant values (Δη = ηMUT – ηWT) and were highlighted on a previously published crystal structure (PDB ID: 1II7) [29].
DNA melting monitored by NMR.
NMR samples of hairpin dsDNA (same as NMR DNA above) in the absence and presence of 1:1.2 ratio of wild type Mre11ND and the Y187C, Y187F, and H52S mutants were made in 25 mM HEPES, 100 mM sodium chloride, 5 mM MgCl2, 0.1 mM EDTA, 1% glycerol, 1 mM TCEP, pH 6.6 in 10% D2O. One-dimensional (1D) 1H gradient 11-echo NMR spectra [61] of the hairpin dsDNA were collected from 30 to 70 °C (in 2.5 °C steps) for each sample. The total signal from the imino region of the 1D NMR spectra was calculated by summing the intensity of the data between 14.25 and 12.3 ppm over a noise threshold (ranging between 500 and 4000 depending on sample concentration and the number of scans). The intensities were fit to a modified version of the van’t Hoff equation
where Ff is the fraction of the hairpin dsDNA folded at a given temperature (i.e., the intensity), Fmax is the maximum intensity, T is each experimental temperature (in Kelvin), ΔH is the change in enthalpy, R is gas constant (8.314 J mol−1 K−1) and Tm is the melting temperature (in Kelvin). Errors in ΔH and Tm were obtained from the covariance of the fit.
ΔS (the change in entropy) values were obtained from the calculated ΔH using the rectangular hyperbolic expression describing enthalpy/entropy compensation found for nucleic acid melting/hybridization: , where α and To are 81 kJ mol−1 and 273 K, respectively [35]. This allowed for the determination of hairpin DNA stability according to ΔG = ΔH - TΔS.
Yeast strain preparation.
The integrated GAL10-HO cassette W303 yeast strain (MATa leu2::GAL-HO-LEU2 hmlΔ hmrΔ RAD5) was used to control DSB formation at the MAT locus (Herskowitz & Jensen, 1991) (gift from L. Symington). The mre11 gene of the parental HO yeast strain was replaced with 13x myc-tagged wild type (gift from D. Durocher) or mutated mre11 genes following established protocols [62]. The kanMX gene (cloned from the pFA6a-kanMX6 vector; Addgene plasmid # 39296 [63]) was juxtaposed to the Mre11 gene for the antibiotic selectivity, and the mre11Δ knock-out strain was prepared by replacing the mre11 open reading frame with kanMX gene. Integrated gene sequences were verified by PCR and western blot. Protein extracts for western blot analysis were prepared by TCA precipitation. Antibodies: mouse anti-c-myc (Santa Cruz, 9E10), mouse anti-PGK1 (Invitrogen, 22C5D8), goat anti-mouse-AP (Invitrogen, 31320). PGK1 was used as the loading control. Immunodetection was conducted using an alkaline phosphatase/NBT/BCIP system (Pierce).
Yeast growth curves.
To observe the growth characteristics of the mre11-myc strains, 200 μL cultures of XY + 2% glucose media were prepared in a 96-well transparent flat bottom culture plate with an initial OD600 of ~0.1. The plates were covered with a plastic film to prevent evaporation. A Synergy Neo2 multimode plate reader was used to record OD600 measurements every 10 min (with 15 s of shaking before each read) at 30 °C for 16 h. The reported data are the average of four cultures for each mre11 strain.
Yeast genotoxin sensitivity assay.
To assess survival on DNA-damage inducing drugs, wild type and mutant mre11-myc yeast cell cultures were grown in 10 mL XY media with 2% glucose overnight at 30 °C. The overnight cultures were then used to inoculate a fresh 10 mL liquid culture. Cells were collected at an OD600 of 1 and diluted to 1 ODU with water if needed. After an initial 10x dilution, five serial five-fold dilutions were prepared and 4 μL of each dilution were spotted on XY plates supplemented with either 0.03% w/v methyl methanesulfonate (MMS) or 1 μg/mL camptothecin (CPT). The plates were incubated at 30 °C for 2 to 3 days. The presented data are representative of experiments performed in triplicate.
Non-homologous end joining (NHEJ) assay.
A plasmid ligation assay was used to compare the NHEJ efficiencies of wild type and mutated mre11-myc yeast strains [39]. The empty pRS413 (ATCC plasmid # 87518 [64]) plasmid, which contains a yeast HIS3 nutritional marker, was digested overnight with EcoRI-HF (New England Biolabs). The restriction enzyme was heat inactivated at 65 °C for 10 min, and calf intestinal alkaline phosphatase (Quick CIP; New England Biolabs) was added to dephosphorylate the ends of the DNA. The digested and dephosphorylated DNA was gel purified using the NucleoSpin Gel and PCR clean up kit (Macherey-Nagel). Purified linearized pRS413 plasmid was transformed into yeast following established protocols [65]. Cells were plated on histidine drop-out XY plates and incubated at 30 °C for 2 days. The percentages of colonies that grew on the histidine drop-out plates were reported relative to the number of colonies repaired by the wild type yeast [66]. Reported values and errors are the average and standard deviation of three experiments.
End resection assay.
Genomic DNA resection assays were performed as described [41]. Yeast cells were grown at 30 °C in YP media containing 3.15% lactic acid (YPL) to repress the expression of the HO endonuclease. In log phase, cells were arrested in G2/M via a double-block of nocodazole (13.33 μg/mL for 2 h followed by an increase to 20.0 μg/mL final concentration for 1 h) before an HO-generated DNA DSB at the MAT locus was induced with the addition of 2% galactose to the media. Cells were collected at 0, 0.5, 1, 2, 4, and 6 h after galactose induction. Genomic DNA was extracted and purified from each sample using the MasterPure Yeast DNA Purification Kit (Lucigen) and quantified using a QuantiFluor dsDNA Kit (Promega). Purified genomic DNA was digested with 5 Units/μg DNA StyI-HF (New England Biolab) for 2 h at 37 °C. Mock digested DNA (i.e., no addition of StyI) was used as control. Primer sets for the MATa HO cut site, ADH1 gene, and 726-bp up-stream of the StyI cleavage site were used in qPCR reactions [41], which utilized the PowerUp SYBR Green Master Mix (Applied Biosystem). The qPCR conditions were 95 °C for 10 min and 40 cycles of 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min. QuantStudio 3 (Applied Biosystem) real-time PCR was used to calculate cycle threshold (Ct) values. The fraction of cells that experienced resection at the StyI cleavage site were calculated as described [41]. Reported values and errors are the average and standard deviation of n ≥ 3 experiments.
Supplementary Material
Highlights.
Mutation of a conserved aromatic amino acid in Mre11 causes separation of function
Separation of function mutations alter the structure and dynamics of Mre11
P. furiosus Mre11 is a stable dimer in solution
Acknowledgments
The authors thank the members of the Latham laboratory for comments, Daniel Durocher (Lunenfeld-Tanenbaum Research Institute) for S. cerevisiae mre11 and technical assistance, and Julyun Oh and Lorraine Symington (Columbia University Medical Center) for the W303 S. cerevisiae strain and technical assistance. This study was supported by Cancer Prevention and Research Institute of Texas grant RP180553 and NIGMS grant 1R35GM128906 to M.P.L.
Abbreviations used:
- DSB
double strand break
- MRN/X
Mre11-Rad50-Nbs1/Xrs2
- NHEJ
non-homologous end joining
- HR
homologous recombination
- TROSY
transverse relaxation optimized spectroscopy
- HMQC
heteronuclear multiple quantum coherence
- SAXS
small angle X-ray scattering
- CSPs
chemical shift perturbatio
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession Numbers
None.
Declaration of Interest
The authors declare that they have no conflict of interest.
References
- [1].Bunting SF, Nussenzweig A, End-joining, translocations and cancer., Nat. Rev. Cancer 13 (2013) 443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu T, Huang J, DNA End Resection: Facts and Mechanisms, Genomics, Proteomics Bioinforma. 14 (2016) 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Oh J, Symington LS, Role of the Mre11 complex in preserving genome integrity, Genes (Basel). 9 (2018) 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Syed A, Tainer JA, The MRE11–RAD50–NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair, Annu. Rev. Biochem 87 (2018) 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Paull TT, 20 Years of Mre11 Biology: No End in Sight, Mol. Cell 71 (2018) 419–427. [DOI] [PubMed] [Google Scholar]
- [6].Tainer JA, Dynamic structures in DNA damage responses & cancer, Prog. Biophys. Mol. Biol 117 (2015) 129–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stewart GS, Maser RS, Stankovic T, a Bressan D, Kaplan MI, Jaspers NG, Raams a, Byrd PJ, Petrini JHJ, Taylor a M., The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder., Cell. 99 (1999) 577–587. [DOI] [PubMed] [Google Scholar]
- [8].Carney JP, Maser RS, Olivares H, Davis EM, Le Beau MM, Yates JR, Hays L, Morgan WF, Petrini JHJ, The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response, Cell. 93 (1998) 477–486. [DOI] [PubMed] [Google Scholar]
- [9].Wang Z, Cummins JM, Shen D, Cahill DP, V Jallepalli P, Wang T-L, Parsons DW, Traverso G, Awad M, Silliman N, Ptak J, Szabo S, V Willson JK, Markowitz SD, Goldberg ML, Karess R, Kinzler KW, Vogelstein B, Velculescu VE, Lengauer C, Three classes of genes mutated in colorectal cancers with chromosomal instability., Cancer Res. 64 (2004) 2998–3001. [DOI] [PubMed] [Google Scholar]
- [10].Park YB, Chae J, Kim YC, Cho Y, Crystal structure of human Mre11: Understanding tumorigenic mutations, Structure. 19 (2011) 1591–1602. [DOI] [PubMed] [Google Scholar]
- [11].Limbo O, Moiani D, Kertokalio A, Wyman C, Tainer JA, Russell P, Mre11 ATLD17/18 mutation retains Tel1/ATM activity but blocks DNA double-strand break repair, Nucleic Acids Res. 40 (2012) 11435–11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schiller CB, Lammens K, Guerini I, Coordes B, Feldmann H, Schlauderer F, Möckel C, Schele A, Sträßer K, Jackson SP, Hopfner K-P, Structure of Mre11-Nbs1 complex yields insights into ataxia-telangiectasia-like disease mutations and DNA damage signaling., Nat. Struct. Mol. Biol 19 (2012) 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matange N, Podobnik M, Visweswariah SS, Metallophosphoesterases: structural fidelity with functional promiscuity, Biochem. J 467 (2015) 201–216. [DOI] [PubMed] [Google Scholar]
- [14].Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K, Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination, EMBO J. 17 (1998) 6412–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paull TT, Gellert M, The 3′ to 5′ Exonuclease Activity of Mre11 Facilitates Repair of DNA Double-Strand Breaks, Mol. Cell 1 (1998) 969–979. [DOI] [PubMed] [Google Scholar]
- [16].Hopfner K-P, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA, Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase., Cell. 105 (2001) 473–85. [DOI] [PubMed] [Google Scholar]
- [17].Neale MJ, Pan J, Keeney S, Endonucleolytic processing of covalent protein-linked DNA double-strand breaks, Nature. 436 (2005) 1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hartsuiker E, Neale MJ, Carr AM, Distinct Requirements for the Rad32Mre11 Nuclease and Ctp1CtIP in the Removal of Covalently Bound Topoisomerase I and II from DNA, Mol. Cell 33 (2009) 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Deshpande RA, Myler LR, Soniat MM, Makharashvili N, Lee L, Lees-Miller SP, Finkelstein IJ, Paull TT, DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP, Sci. Adv 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Myler LR, Gallardo IF, Soniat MM, Deshpande RA, Gonzalez XB, Kim Y, Paull TT, Finkelstein IJ, Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair, Mol. Cell 67 (2017) 891–898.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hopfner K-P, Tainer JA, Rad50/SMC proteins and ABC transporters: Unifying concepts from high-resolution structures, Curr. Opin. Struct. Biol 13 (2003) 249–255. [DOI] [PubMed] [Google Scholar]
- [22].Hopfner K-P, Karcher A, Shin DS, Craig L, Arthur LM, Carney JP, Tainer JA, Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily., Cell. 101 (2000) 789–800. [DOI] [PubMed] [Google Scholar]
- [23].Moreno-Herrero F, De Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C, Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA., Nature. 437 (2005) 440–3. [DOI] [PubMed] [Google Scholar]
- [24].Bhaskara V, Dupré A, Lengsfeld B, Hopkins BB, Chan A, Lee J-H, Zhang X, Gautier J, Zakian V, Paull TT, Rad50 Adenylate Kinase Activity Regulates DNA Tethering by Mre11/Rad50 Complexes, Mol. Cell 25 (2007) 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Williams RS, Dodson GE, Limbo O, Yamada Y, Williams JS, Guenther G, Classen S, Glover JNM, Iwasaki H, Russell P, Tainer JA, Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair., Cell. 139 (2009) 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lloyd J, Chapman JR, Clapperton JA, Haire LF, Hartsuiker E, Li J, Carr AM, Jackson SP, Smerdon SJ, A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage., Cell. 139 (2009) 100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hari FJ, Spycher C, Jungmichel S, Pavic L, Stucki M, A divalent FHA/BRCT-binding mechanism couples the MRE11-RAD50-NBS1 complex to damaged chromatin, EMBO Rep. 11 (2010) 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oh J, Al-Zain A, Cannavo E, Cejka P, Symington LS, Xrs2 Dependent and Independent Functions of the Mre11-Rad50 Complex, Mol. Cell 64 (2016) 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA, Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair., Cell. 135 (2008) 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois M-MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, Ismail A, Ismalaj E, Petricci E, Neale MJ, Bristow RG, Masson JY, Wyman C, Jeggo PA, Tainer JA, DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities, Mol. Cell 53 (2014) 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zheng G, Price WS, Simultaneous convection compensation and solvent suppression in biomolecular NMR diffusion experiments., J. Biomol. NMR 45 (2009) 295–9. [DOI] [PubMed] [Google Scholar]
- [32].Sung S, Li F, Park YB, Kim JS, Kim A, Song O-K, Kim J, Che J, Lee SE, Cho Y, DNA end recognition by the Mre11 nuclease dimer: insights into resection and repair of damaged DNA., EMBO J. 33 (2014) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Boswell ZK, Latham MP, Methyl-Based NMR Spectroscopy Methods for Uncovering Structural Dynamics in Large Proteins and Protein Complexes, Biochemistry. 58 (2019) 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sun H, Kay LE, Tugarinov V, An Optimized Relaxation-Based Coherence Transfer NMR Experiment for the Measurement of Side-Chain Order in Methyl-Protonated, Highly Deuterated Proteins, J. Phys. Chem. B 115 (2011) 14878–14884. [DOI] [PubMed] [Google Scholar]
- [35].Petruska J, Goodman MF, Enthalpy-entropy compensation in DNA melting thermodynamics, J. Biol. Chem 270 (1995) 746–750. [DOI] [PubMed] [Google Scholar]
- [36].Notredame C, Higgins DG, Heringa J, T-coffee: A novel method for fast and accurate multiple sequence alignment, J. Mol. Biol 302 (2000) 205–217. [DOI] [PubMed] [Google Scholar]
- [37].Seifert FU, Lammens K, Hopfner K-P, Structure of the catalytic domain of Mre11 from Chaetomium thermophilum, Acta Crystallogr. Sect. F Struct. Biol. Commun 71 (2015) 752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Boulton SJ, Jackson SP, Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing, EMBO J. 17 (1998) 1819–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boulton SJ, Jackson SP, Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways., EMBO J. 15 (1996) 5093–103. [PMC free article] [PubMed] [Google Scholar]
- [40].Moreau S, Morgan EA, Symington LS, Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism., Genetics. 159 (2001) 1423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gnügge R, Oh J, Symington LS, Processing of DNA Double-Strand Breaks in Yeast, Methods Enzymol. 600 (2018) 1–24. [DOI] [PubMed] [Google Scholar]
- [42].Llorente B, Symington LS, The Mre11 Nuclease Is Not Required for 5’ to 3’ Resection at Multiple HO-Induced Double-Strand Breaks, Mol. Cell. Biol 24 (2004) 9682–9694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Boswell ZK, Rahman S, Canny MD, Latham MP, A dynamic allosteric pathway underlies Rad50 ABC ATPase function in DNA repair, Sci. Rep 8 (2018) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Käshammer L, Saathoff J-H, Lammens K, Gut F, Bartho J, Alt A, Kessler B, Hopfner K-P, Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex., Mol. Cell 76 (2019) 382–394.e6. [DOI] [PubMed] [Google Scholar]
- [45].Zhao Q, Xue X, Longerich S, Sung P, Xiong Y, Structural insights into 5′ flap DNA unwinding and incision by the human FAN1 dimer, Nat. Commun 5 (2014) 5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA, Flipping of alkylated DNA damage bridges base and nucleotide excision repair, Nature. 459 (2009) 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tsutakawa SE, Classen S, Chapados BR, Arvai AS, Finger LD, Guenther G, Tomlinson CG, Thompson P, Sarker AH, Shen B, Cooper PK, Grasby JA, Tainer JA, Human flap endonuclease structures, DNA double-base flipping, and a unified understanding of the FEN1 superfamily, Cell. 145 (2011) 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Saathoff J-H, Käshammer L, Lammens K, Byrne RT, Hopfner K-P, The bacterial Mre11–Rad50 homolog SbcCD cleaves opposing strands of DNA by two chemically distinct nuclease reactions, Nucleic Acids Res. 46 (2018) 11303–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Albrecht DW, Herdendorf TJ, Nelson SW, Disruption of the bacteriophage T4 Mre11 dimer interface reveals a two-state mechanism for exonuclease activity, J. Biol. Chem 287 (2012) 31371–31381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Azatian SB, Kaur N, Latham MP, Increasing the buffering capacity of minimal media leads to higher protein yield, J. Biomol. NMR 73 (2019) 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tugarinov V, Kanelis V, Kay LE, Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy, Nat. Protoc 1 (2006) 749–754. [DOI] [PubMed] [Google Scholar]
- [52].Yuan S, Chu H, Singh K, Zhao H, Zhang K, Kao RYT, Chow BKC, Zhou J, Zheng BJ, A novel small-molecule inhibitor of influenza A virus acts by suppressing PA endonuclease activity of the viral polymerase, Sci. Rep 6 (2016) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Majka J, Alford B, Ausio J, Finn RM, McMurray CT, ATP hydrolysis by RAD50 protein switches MRE11 enzyme from endonuclease to exonuclease, J. Biol. Chem 287 (2012) 2328–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, A multidimensional spectral processing system based on pipes, J. Biomol. NMR 6 (1995) 277–293. [DOI] [PubMed] [Google Scholar]
- [55].Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED, The CCPN data model for NMR spectroscopy: development of a software pipeline., Proteins. 59 (2005) 687–96. [DOI] [PubMed] [Google Scholar]
- [56].Pervushin K, Riek R, Wider G, Wuthrich K, Wüthrich K, Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution, Proc. Natl. Acad. Sci. U. S. A 94 (1997) 12366–12371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE, Cross-Correlated Relaxation Enhanced 1 H− 13 C NMR Spectroscopy of Methyl Groups in Very High Molecular Weight Proteins and Protein Complexes, J. Am. Chem. Soc 125 (2003) 10420–10428. [DOI] [PubMed] [Google Scholar]
- [58].Lichtenecker RJ, Weinhäupl K, Reuther L, Schörghuber J, Schmid W, Konrat R, Independent valine and leucine isotope labeling in Escherichia coli protein overexpression systems, J. Biomol. NMR 57 (2013) 205–209. [DOI] [PubMed] [Google Scholar]
- [59].Sprangers R, Kay LE, Quantitative dynamics and binding studies of the 20S proteasome by NMR, Nature. 445 (2007) 618–622. [DOI] [PubMed] [Google Scholar]
- [60].Gans P, Hamelin O, Sounier R, Ayala I, Durá MA, Amero CD, Noirclerc-Savoye M, Franzetti B, Plevin MJ, Boisbouvier J, Stereospecific Isotopic Labeling of Methyl Groups for NMR Spectroscopic Studies of High-Molecular-Weight Proteins, Angew. Chemie Int. Ed 49 (2010) 1958–1962. [DOI] [PubMed] [Google Scholar]
- [61].Sklenář V, Bax A, Spin-echo water suppression for the generation of pure-phase two-dimensional NMR spectra, J. Magn. Reson 74 (1987) 469–479. [Google Scholar]
- [62].Gardner JM, Jaspersen SL, Manipulating the Yeast Genome: Deletion, Mutation, and Tagging by PCR, in: Methods Mol. Biol, 2014: pp. 45–78. [DOI] [PubMed] [Google Scholar]
- [63].Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, Steever AB, Wach A, Philippsen P, Pringle JR, Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe, Yeast. 14 (1998) 943–951. [DOI] [PubMed] [Google Scholar]
- [64].Sikorski RS, Hieter P, A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae., Genetics. 122 (1989) 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gietz RD, Schiestl RH, High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method, Nat. Protoc 2 (2007) 31–34. [DOI] [PubMed] [Google Scholar]
- [66].Cassani C, Vertemara J, Bassani M, Marsella A, Tisi R, Zampella G, Longhese MP, The ATP-bound conformation of the Mre11–Rad50 complex is essential for Tel1/ATM activation, Nucleic Acids Res. 47 (2019) 3550–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.