

Abstract
The detection and quantification of low-abundance molecular biomarkers in biological samples is challenging. Here, we show that a plasmonic nanoscale construct serving as an ‘add-on’ label for a broad range of bioassays improves their signal-to-noise ratio and dynamic range without altering their workflow and read-out devices. The plasmonic construct consists of a bovine-serum-albumin scaffold with approximately 210 IRDye 800CW fluorophores (with fluorescence intensity approximately 6700-fold that of a single 800CW fluorophore), a polymer-coated gold nanorod acting as a plasmonic antenna, and biotin as a high-affinity biorecognition element. Its emission wavelength can be tuned over the visible and near-infrared spectral regions by modifying its size, shape and composition. It is compatible with multiplexed bead-based immunoassays (it improves the limit of detection by up to 4,750-fold in fluorescence-linked immunosorbent assays), immuno-microarrays, flow-cytometry and immunocytochemistry methods, and it shortens overall assay times and lowers sample volumes, as shown for the detection of a pro-inflammatory cytokine in mouse interstitial fluid and of urinary biomarkers in patient samples.
Relevant concentrations of biomolecules or biomarkers related to diseases such as cancer, heart disease, inflammation, and neurological disorders can range in many orders of magnitude from μg/ml levels to sub-fg/ml, some of which possibly still remain unidentified due to the lack of sensitive bioanalytical tools.1–7 It is also highly desirable to utilize small sample volume for multiplexed detection within precious biofluids such as breath condensates, ocular fluids, cerebrospinal fluid, or serum from neonates or small animal models, which necessitates sample dilutions, further lowering the concentration. As the cornerstone of biomedical science and clinical research, fluorescence-based bioanalytical methods are widely employed in the detection, quantification and imaging of a broad range of bioanalytes.8–11 Several methods, such as enhancing antibody affinity12, reducing the background fluorescence13, promoting mass transfer14, and increasing the substrate surface area8,14, have been explored to improve the sensitivity of fluoroimmunoassays. However, weak fluorescence signal and the associated poor signal-to-noise ratio of the fluorescence label remains a persistent challenge, limiting the ultimate sensitivity of current fluorescence-based assays.10,15–17
Extensive efforts have been dedicated in creating bright fluorescence signal that involves the conversion of a single molecular event into thousands (or more) of fluorophores in a localized “enzyme-free” manner.18–24 Achieving ultrabright nanostructures by simply packing a large number of fluorophores into a nanoscale volume is extremely challenging due to the aggregation induced self-quenching of fluorophores at high local concentration, limiting the fluorophore loading and consequently their brightness.21,25 One solution to prevent the formation of non-emissive H-aggregates is to engineer the inter-fluorophore distance and orientation, for example, by modifying the fluorophore with bulky side groups26,27 or using charged dyes with hydrophobic counterions18. However, the loading is still limited to tens or hundreds of fluorophores per nanoparticle in these cases. Fluorescent nanoparticles based on Förster resonance energy transfer (FRET) represents an attractive method that can harvest incident light and transfer it to adjacent energy acceptors. However, limited by the Förster radius (typically much shorter than the radius of nanostructure, which hinders the efficient energy transfer from a whole nanoparticle to the energy acceptor) and self-quenching of donors at high local concentration, efficient FRET system (1000-fold brighter than single fluorophore) was not demonstrated until recently.23,28 Again, this method is only applicable to charged fluorophores and broad application of these nanostructures in fluorescence-based bioanalytical and bioimaging techniques has not been demonstrated.23
Here, we introduce a stable and bright fluorescent nanoconstruct, termed ‘plasmonic-fluor’, which exhibits up to 6700 (± 900)-fold brighter signal compared to the corresponding single near infrared (NIR) fluorophore (800CW, LI-COR), outperforming existing nanoengineered fluorescent structures18–24. This nanoconstruct integrates plasmon-enhanced fluorescence, a bio-linker element, and a BSA (bovine serum albumin) surface-blocking strategy, enabling it to serve as a specific and cross-assay label in various fluorescence-based bioanalytical and imaging methodologies to considerably enhance their signal-to-noise ratios. Harnessing the nanoconstruct (i.e. plasmonic-fluor), we show that plasmon-enhanced fluorescence improves the sensitivity of a variety of bioanalytical applications, such as fluorophore-linked immunosorbent assays (FLISA), multiplexed bead-based fluoroimmunoassays, high-throughput protein arrays, immunocytochemistry/immunofluorescence (ICC/IF), and flow cytometry.
Plasmonic-fluor synthesis and characterization.
“Plasmonic-fluor” is comprised of a plasmonic nanostructure (as fluorescence enhancer), a light emitter (e.g., fluorophores), spacer layer, and a universal biological recognition element (e.g. biotin) (Figure 1a). To assemble all of these functional components, we have employed bovine serum albumin (BSA) as a scaffold. As a key design element, BSA also serves as (i) a stabilizing agent, preventing the aggregation of the nanoconstructs; and (ii) a blocking agent, minimizing non-specific binding of the plasmonic-fluor to arbitrary surfaces and biomolecules, which is extremely important to achieve high signal-to-background ratios. BSA is covalently conjugated with fluorophores and biotin and subsequently coated around the plasmonic nanostructures to realize plasmonic-fluors. The synthesized plasmonic-fluor exhibited strong and specific affinity to streptavidin (a tetrameric biotin-binding protein), owing to the high affinity (Kd ≈10−14 mol/L) of biotin-streptavidin complex.29 Streptavidin conjugated with fluorophore is widely used in fluorescence-based bioanalytical techniques as a universal signal reporter. We hypothesized that following the binding of streptavidin, plasmonic-fluor can be introduced as an “add-on” step to enhance the fluorescence signal without entailing any change in the existing bioassay protocols (Figure 1b).
Fig. 1 |. Plasmonic-fluor synthesis and material characterization.
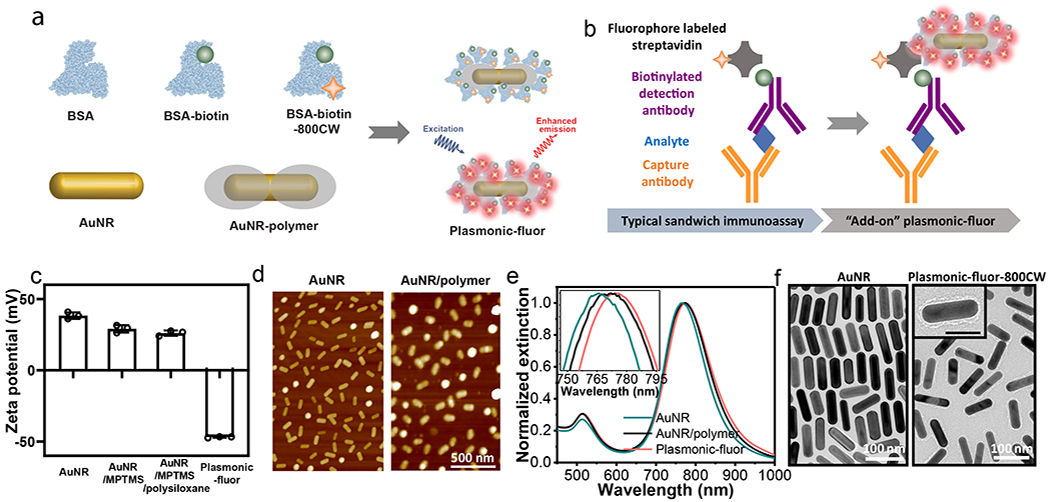
a, Schematic illustration showing the structure of “plasmonic-fluor”, which consists of a plasmonically-active core (e.g. gold nanorod (AuNR)), a polymer shell as spacer layer, light emitters, and a universal biorecognition element (biotin). Bovine serum albumin (BSA) is employed as a key design element to assemble all components into the functional nanoconstruct and to resist non-specific binding. b, Working principle of plasmonic-fluor as an “add-on” biolabel to enhance the fluorescence intensity and consequent signal-to-noise ratio of fluorescence-based assays, without changing the existing assay workflow. c, Zeta potential of AuNR, AuNR/MPTMS, AuNR/MPTMS/polysiloxane (AuNR/polymer), and the plasmonic-fluor-800CW (AuNR/polymer/BSA-biotin-800CW). Error bar, s.d. (n=3 repeated tests). d, AFM images showing the AuNR before and after coating with polymer. e, Vis-NIR extinction spectra of AuNR, AuNR/polymer, and plasmonic-fluor, showing a progressive red shift in the LSPR wavelength after each step. f, TEM images of bare AuNR and plasmonic-fluor-800CW.
Gold nanorods (AuNRs) are employed as representative plasmonic nanoantennae owing to the facile tunability of their longitudinal localized surface plasmon resonance (LSPR) wavelength with aspect ratio and large electromagnetic field enhancement at their ends (see Supporting Information, Figure S1).30–32 AuNRs (length 83.0±8.0 nm; diameter 24.3±1.8 nm) were modified with (3-mercaptopropyl)trimethoxysilane (MPTMS), which served as an interfacial layer for the copolymerization of two organosilane monomers, namely (3-aminopropyl)trimethoxysilane (APTMS) and trimethoxypropylsilane (TMPS) (Figure S2). In aqueous media, APTMS and TMPS undergo rapid hydrolysis and subsequent condensation around the MPTMS-modified AuNRs, yielding an amorphous copolymer network (Figure S2). The siloxane copolymer serves as a spacer layer between metal surface and the fluorophore to prevent fluorescence quenching (Figure 1d). This sol-gel approach enables facile control over the thickness of the spacer layer down to 1 nm, as evidenced by atomic force microscopy (AFM) (Figure S3). Modification of AuNRs with MPTMS and subsequent polymerization of APTMS/TMPS reduced the zeta potential of cetyl trimethylammonium bromide (CTAB)-capped AuNR from +38.4 ±2.3 mV to +29±2.6 mV and +25.8±1.9 mV, respectively, due to the partial replacement of the positively charged capping agent (CTAB) with less charged siloxane copolymer (Figure 1c).
Near infrared (NIR) fluorophore 800CW (LI-COR) and biotin were conjugated to BSA through carbodiimide coupling chemistry to realize conjugates with protein/biotin/fluorophore ratio of 1: 8.7: 1.2 (Supporting information, Figure S4 and S5). Subsequently, the BSA-biotin-800CW conjugates are adsorbed on polysiloxane-coated AuNR through electrostatic, hydrophobic and hydrogen bonding interactions between BSA and the functional groups (−NH3+, −CH3, −OH) of the polysiloxane layer to realize plasmonic-fluor-800CW. As formed plasmonic-fluor-800CW exhibited a negative charge (zeta potential −46.9±0.5 mV at pH=10) due to abundant carboxylic acid groups in BSA with an isoelectric point of 4.7 (Figure 1c).33 LSPR wavelength of AuNR exhibited a progressive red shift of 2.6 nm and 2.7 nm with the formation of polymer spacer layer and BSA-biotin-800CW adsorption, respectively (Figure 1e). Transmission electron microscopy (TEM) images further confirmed the presence of a thin organic layer (polymer and BSA conjugate) around AuNR with an overall thickness of ~6.3±1.4 nm (Figure 1f).
Following the structural characterization of plasmonic-fluor-800CW, we set out to determine the brightness of the fluorescent nanoconstruct. The excited state fluorescence lifetimes of free 800CW (conjugated to BSA) and plasmonic-fluor-800CW were measured to be 0.74±0.01 ns and 0.179±0.001 ns, respectively, accounting to a 7-fold increase in the quantum yield (from ~11% (800CW) to ~79% (plasmonic-fluor-800CW), see supporting information for calculation) (Figure 2a, S6, S7).34 To further understand the brightness of plasmonic-fluor-800CW, we estimated the number of fluorophores conjugated to a single AuNR. Plasmonic-fluor-800CW at concentration of 76.2 pM (extinction of ~0.63) is comprised of ~16 nM 800CW (see supporting information for detailed calculation). Therefore, we can estimate that approximately 210 fluorophores are conjugated to a single AuNR. Notably, fluorescence intensity from 76.2 pM plasmonic-fluor-800CW (containing 16 nM 800CW) was found to be equivalent to the fluorescence intensity from 544 nM 800CW (measured based on Figure 2b). Therefore, it can be concluded that each 800CW is enhanced by nearly 30-fold due to the presence of plasmonic nanoantennas. The slope of fluorescence intensity vs. molar concentration of plasmonic-fluor-800CW is 6700 (± 900)-fold steeper than that of the conventional fluorophore (800CW) (Figure 2b, S8), suggesting that each plasmonic-fluor exhibits 6700 (± 900)-fold higher brightness than the corresponding fluorophore.19,35 The observed intense emission can be attributed to the enhanced electromagnetic field (local excitation field) at the surface of the plasmonic nanostructures (Figure S1) and decrease in the fluorescence lifetime due to the coupling between excited fluorophores and surface plasmons of the nanostructures.8,10,36–43
Fig. 2 |. Plasmon-enhanced fluorescence and colloidal stability of plasmonic-fluors.
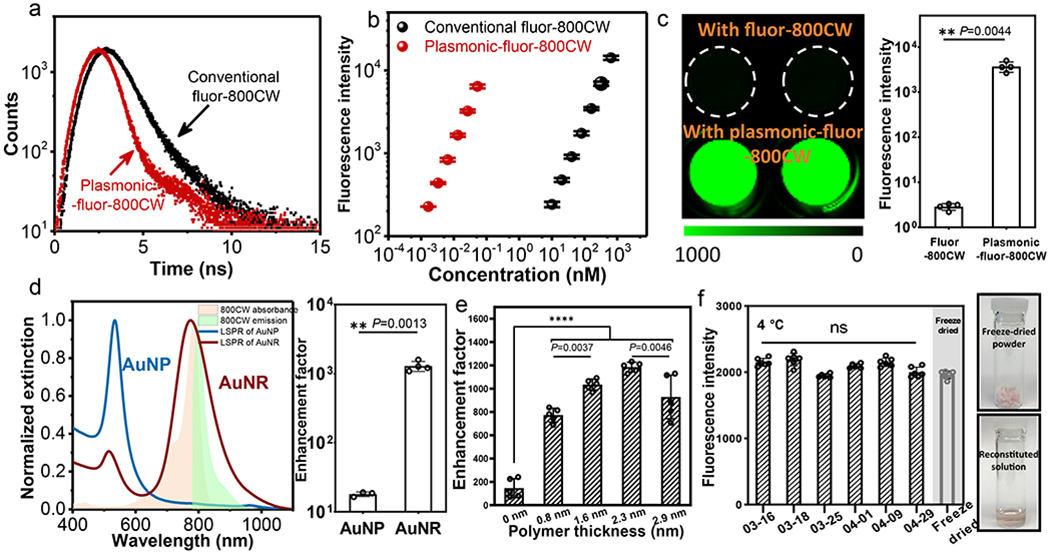
a, Excited state lifetime measurements of conventional fluor (BSA-biotin-800CW) and plasmonic-fluor-800CW (AuNR/polymer/BSA-biotin-800CW) showing a significant decrease in the lifetime of 800CW after adsorption on AuNR. b, Fluorescence intensity of conventional fluor-800CW and plasmonic-fluor-800CW at their different molar concentrations. The difference in the slopes of two curves indicates that a single plasmonic-fluor-800CW is as bright as 6700 (± 900) fluorophores. Error bar, s.d. (n=3 repeated tests). c, Fluorescence intensity of 800CW-streptavidin followed by the specific binding of plasmonic-fluor-800CW through biotin-streptavidin interaction, showing an average of 1200 (±40)-fold increase in fluorescence intensity. Error bar, s.d. (n=4 independent tests). Data statistically significant P value= 0.0044, ** P < 0.01 by two-tailed unpaired t-test with Welch’s correction. d, Left: LSPR wavelength of gold nanoparticles (AuNPs) and AuNRs with similar surface area. Plot also shows the absorption and emission spectra of 800CW. Right: Fluorescence enhancement factor upon binding of “off-resonant” AuNP-plasmonic-fluor-800CW and “on-resonant” AuNR-plasmonic-fluor-800CW to 800CW-streptavidin. Error bar, s.d. (AuNP group: n=3 independent tests. AuNR group: n=4 independent tests.). Data statistically significant P value= 0.0013, ** P < 0.01 by two-tailed unpaired t-test with Welch’s correction. e, Fluorescence enhancement factor obtained using plasmonic-fluor-800CW with different polymer spacer thickness. Error bar, s.d. (n=5 independent tests). **** P < 0.0001 and ** P < 0.01 by one-way ANOVA with Tukey’s post test. f, Left: plot showing the stability of plasmonic-fluor suspension stored at 4°C and reconstituted from lyophilized powder. Error bar, s.d. (n=6 repeated tests). NS: not significant. P value>0.9999 by one-way ANOVA with Tukey’s post test. Right: Photographs depicting the lyophilized powder of plasmonic-fluor before and after reconstitution.
We tested feasibility of using plasmonic-fluor-800CW as ultrabright fluorescent reporters by binding them to a substrate coated with streptavidin-800CW (see Methods section for detailed procedure, Figure S9). Binding of plasmonic-fluor-800CW resulted in an average of 1200 (± 40)-fold increase in the ensemble fluorescence intensity compared to streptavidin-800CW (LI-COR) (Figure 2c). The signal enhancement was achieved by using a relatively low concentration of the plasmonic-fluors (76 pM). To further validate the plasmonic enhancement of fluorescence, we employed “off-resonant” gold nanoparticle (AuNP) with similar surface area as the “on resonant” AuNR (7850 nm2/AuNP; 8064 nm2/AuNR) (Figure S10). The AuNPs exhibited LSPR wavelength around 530 nm, which was “off-resonant” with respect to the excitation laser (785 nm) and 800CW emission (800 nm) (Figure 2d). It is known that a large overlap between LSPR band of the plasmonic nanostructures and the excitation and emission bands of the fluorophores is critical for maximizing the fluorescence enhancement.44 Not surprisingly, AuNP-plasmonic-fluor-800CW resulted in only 18-fold enhancement in the fluorescence intensity, which is ~70-fold lower than that obtained with AuNR-plasmonic-fluor-800CW, confirming the plasmonically enhanced fluorescence (Figure 2d).
Our group and others have shown that an optimal distance between the metal surface and fluorophore is critical to maximize fluorescence enhancement by balancing the two opposing factors, namely, enhanced electromagnetic field and non-radiative energy transfer.45–51 Fluorescence enhancement of plasmonic-fluor-800CW with different thicknesses of the dielectric spacer (MPTMS, APTMS, and TMPS) was investigated by binding them to a substrate coated with streptavidin-800CW. The ensemble fluorescence enhancement factor (defined as the ratio of fluorescence intensities obtained after and before the addition of plasmonic-fluors on a surface coated with fluorophore-conjugated streptavidin) of the plasmonic-fluors without polymer spacer layer was found to be ~146±81. Enhancement efficiency progressively increased to ~1200(±40)-fold with the increase of the spacer thickness (Figure 2e). Notably, the colloidal solution of plasmonic-fluor exhibited stable fluorescence signal after storage in the dark at 4°C for one month (Figure 2f). For further ease of storage, transportation, and handling, the plasmonic-fluors can be lyophilized and reconstituted as needed without noticeable degradation in the fluorescence signal (Figure 2f).
Plasmonic-fluor enhanced fluorescence-linked immunosorbent assay (p-FLISA) and multiplexed bead-based assay.
Of the numerous applications of plasmonic-fluors, we first set out to demonstrate plasmon-enhanced fluorophore-linked immunosorbent assay (p-FLISA) implemented on a standard microtiter plate. Human interleukin 6 (IL-6), a pro-inflammatory cytokine, was employed as a representative protein biomarker. Conventional FLISA involves a standard sandwich format of capture antibody, analyte (IL-6), biotinylated detection antibody, followed by exposure to streptavidin-fluorophore (800CW in this study) (Figure 3a). In p-FLISA, plasmonic-fluor-800CW is introduced after the last step as the signal enhancer (Figure 3a). To determine the improvement in sensitivity and limit-of-detection ((LOD), defined as mean+3σ of the blank), serial dilutions of IL-6 of known concentration (6 ng/ml to 6 fg/ml, in 1% BSA buffered with phosphate buffered saline (PBS)) were employed as standards. Fluorescence signal obtained after applying the plasmonic-fluor-800CW revealed nearly 1440-fold enhancement in the ensemble fluorescence intensity compared to the conventional FLISA at the highest analyte concentration tested here (6 ng/ml) (Figure 3b, c, S11, and S12). The LOD of conventional FLISA was calculated to be ~95 pg/ml (Figure 3d, S11, and S12, polynomial fit). On the other hand, fluorescence signal with p-FLISA could be detected down to 20 fg/ml (~1 fM) (Figure 3e and S12, four-parameter logistic (4PL) fit), which represents a 4750-fold improvement in the LOD compared to conventional FLISA. Notably, plasmonic-fluor exhibited extremely high specificity (to streptavidin) and low non-specific binding to the interference biomolecules in the bioassays (Figure S14). We attribute this to the “BSA blocking” strategy of plasmonic-fluor, which is critical in enhancing the signal-to-background ratio. Scanning electron microscopy (SEM) images revealed an increase in the density of plasmonic-fluor-800CW at the bottom of the microtiter wells with increasing IL-6 concentration (Figure S15). Extremely low density of plasmonic-fluors was observed in the blank well, which was incubated with 1% BSA, again indicating the low non-specific binding of the plasmonic-fluors (Figure S15).
Fig. 3 |. Plasmonic-fluor enhanced fluorophore-linked immunosorbent assay (FLISA) and multiplexed bead-based immunoassay.
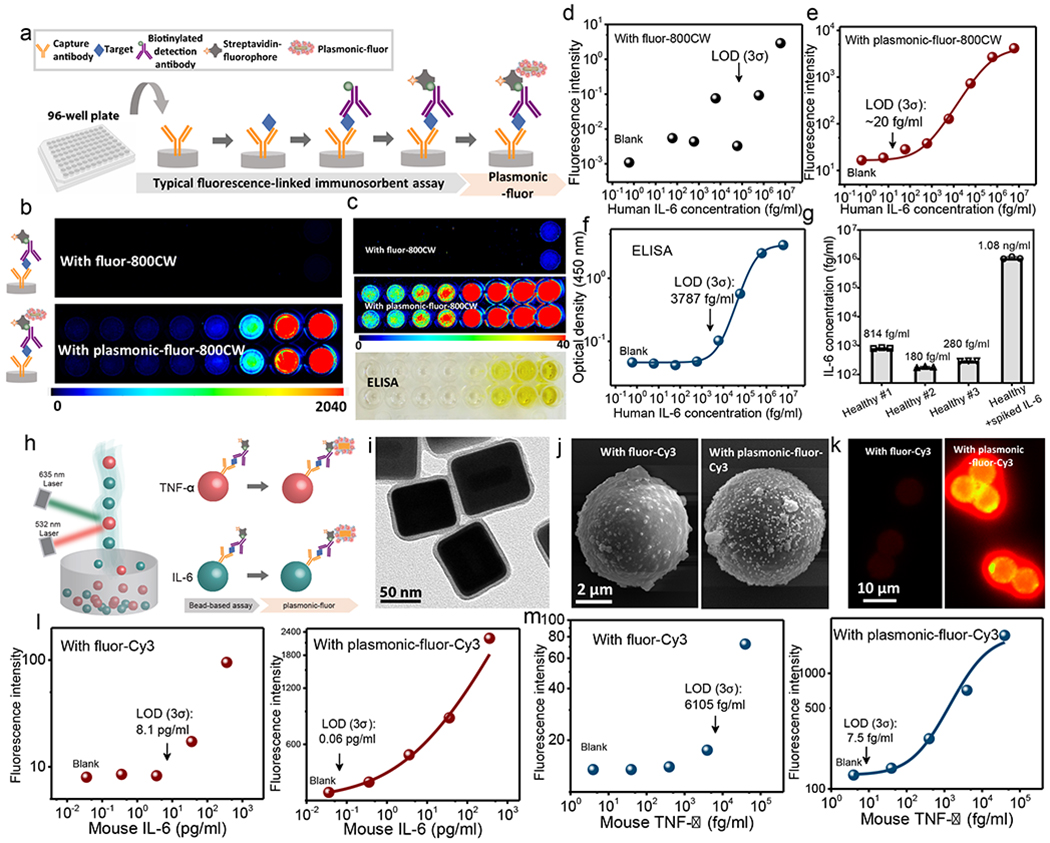
a, Schematic showing the concept of conventional FLISA (800CW) and plasmonic-fluor-800CW enhanced FLISA (p-FLISA), implemented in a standard 96-well plate. P-FLISA does not require any change in the routine workflow except adding the plasmonic-fluor as the new, last step. b, Fluorescence intensity maps of human IL-6 FLISA and p-FLISA at various analyte concentrations. c, Fluorescence intensity maps (with zoomed-in scale bar) of human IL-6 FLISA and p-FLISA and photograph of colorimetric signal of “gold standard” human IL-6 ELISA. Plots showing human IL-6 dose-dependent fluorescence intensity from (d) conventional FLISA and (e) p-FLISA (each data point represents the average value of duplicate tests, which are included in supporting information). Compared to conventional FLISA, p-FLISA exhibits 4750-fold improvement in the limit-of-detection (LOD) and more than three-order-magnitude larger dynamic range. f, Plot showing the standard curve of human IL-6 ELISA. Compared to ELISA, p-FLISA exhibited 189-fold lower LOD and more than two-order-magnitude larger dynamic range. g, IL-6 concentrations in human serum samples (diluted by 10-fold) measured using p-FLISA. Error bars, s.d. (n=3 repeated tests). h, Schematic illustration showing the concept of using plasmonic-fluor-Cy3 to enhance the sensitivity of bead-based immunoassay (Luminex assay). i, TEM image of plasmonic-fluor-Cy3 utilizing silver coated AuNR (AuNR@Ag) as the plasmonic nanoantenna. (j) SEM and (k) fluorescence images of microbead(s) before and after being probed with plasmonic-fluor-Cy3. (l) Mouse IL-6 and (m) mouse TNF-α standard curves obtained before (left) and after (right) applying plasmonic-fluor-Cy3 (each data point represents the average value of duplicate tests, which are included in supporting information). All fluorescence standard curves are performed independently on different days with different batches of plasmonic-fluors at least three times (data included in supporting information).
The LOD and lower limit of quantification ((LLOQ), defined as mean+10σ of the blank, ~82 fg/ml) of p-FLISA were found to be 189-fold and 120-fold lower than the “gold standard” enzyme-linked immunosorbent assay (ELISA), which involves enzymatic amplification of the colorimetric signal (Figure 3c, f, and S12). More importantly, p-FLISA exhibited a dynamic range (ratio between higher and lower limit of quantification) of five orders of magnitude, which is more than two-order-magnitude higher than that of ELISA. As a validation of the assay performance, we have tested healthy human serum samples and IL-6 spiked serum using p-FLISA. Serum samples were diluted by 10-fold so that only 10 μl of original sample was required for individual subjects. Concentrations of IL-6 in healthy individuals are normally in the range of 0.2-7.8 pg/ml.52 Increased level of IL-6 in serum can be indicative of systemic inflammatory, metabolic, and physiological stimuli.52 Notably, among ELISA, FLISA and p-FLISA, only the latter technique was able to determine the IL-6 concentration in healthy individuals, which were measured to be 8.1 pg/ml, 1.8 pg/ml, and 2.8 pg/ml after dilution-fold correction (Figure 3g).
Harnessing the high sensitivity and large dynamic range, we demonstrate that p-FLISA can be employed as a biomedical research tool to quantitatively analyze biomarkers in precious biofluids of extremely small volume (as low as 10 nanoliters), such as interstitial fluid (ISF). One of the existing challenges in ISF analysis is the inadequate amount of ISF that can be extracted using the exiting techniques, making downstream proteomic and metabolomic analysis challenging.53 Here, we employed p-FLISA to measure the concentrations of a proinflammation cytokine (IL-6) in skin interstitial fluid obtained from mice immune-stimulated with lipopolysaccharide (LPS) and naïve mice (as controls). It is important to note that only a small amount of ISF (less than 10 μl) could be obtained from the entire skin from the back of the mice (~6 cm2) (Figure S16a). To determine the smallest amount of sample required for p-FLISA and conventional ELISA, mouse ISF was diluted 100-fold, 1000-fold, 10,000-fold, and 100,000-fold (representing 1 μl, 0.1 μl, 0.01 μl, and 0.001 μl of ISF for each test zone, respectively) and assayed with both methods. The large dynamic range of p-FLISA enabled the detection and quantification of IL-6 concentrations in ISF samples at all the dilutions from immune-stimulated mouse (Figure S16b). Moreover, the high sensitivity of p-FLISA also enabled the detection and quantification of IL-6 concentrations in ISF from naïve mice, which was measured to be three orders of magnitude lower compared to immune-stimulated mice group (Figure S16c). Compared to p-FLISA, conventional ELISA (with ~100-fold inferior limit-of-detection of ~11.2 pg/ml) required 100-fold larger sample volume. The IL-6 in immune-stimulated mouse ISF of high dilution (> 1000-fold) and in healthy mouse ISF cannot be detected using standard ELISA (Figure S16d).
To further demonstrate the translational potential of plasmonic-fluor in clinical diagnostic settings, we show that p-FLISA can shorten the overall sample-to-answer time, making them attractive for point-of-care applications. In many pathological conditions such as acute kidney injury, myocardial infarction, and sepsis, shortening the time-to-treatment is critical to improve the clinical outcomes. The large enhancement in the fluorescence intensity and signal-to-noise ratio of p-FLISA allows us to shorten the overall assay time to 20 minutes. The ultrafast p-FLISA (20-minute) exhibited the same sensitivity as the conventional ELISA (280-minute) (Figure S17a, b), in measuring the concentrations of urinary neutrophil gelatinase-associated lipocalin (NGAL), which can be elevated above normal level in patients with renal masses and kidney diseases such as acute kidney injury (AKI) and chronic kidney disease (CKD)54,55. The ultrafast p-FLISA was able to accurately quantify the urinary NGAL concentrations from 10 kidney patients with imaged renal masses and 10 self-described healthy individuals and the assay revealed that NGAL concentrations in patients to be higher (by more than 10-fold) compared to that of the healthy individuals (Figure S17c, d). Moreover, NGAL concentrations determined using 20-minute p-FLISA showed a good correlation (linear regression with R2=0.96) with those acquired from the standard 280-minute ELISA, proving that the accuracy of the ultrafast assay is not compromised (Figure S17e). ELISA, when shortened to 20 minutes, showed deteriorated performance, and cannot detect urinary NGAL concentrations in several patients or any of the healthy volunteers (Figure S17f).
In addition to the microtiter plate format, we have also investigated the application of plasmonic-fluors as ultrabright reporters in micro bead-based multiplexed fluoroimmunoassays, which utilize a non-planar sampling surface. Luminex assay was employed as an example, which utilizes magnetic microbeads embedded with ratio-set fluorophores as barcode for each unique analyte (Figure 3h). The antibody conjugated microbead captures and facilitates the detection of the analyte in a typical sandwich format and is subsequently probed by streptavidin conjugated with phycoerythrin (PE), a bright fluorescent protein isolated from red algae or cyanobacteria. However, PE employed in Luminex assays is structurally unstable and prone to photobleaching.56 Here, Cy3 (Abcam), a highly stable fluorophore with absorption and emission at 554 nm and 568 nm respectively, similar to PE, was employed as a substitute. As discussed above, it is extremely important to choose plasmonic nanostructure with LSPR wavelength matching the excitation/emission of the fluorophore.44,57 To this end, we have employed AuNR@Ag (silver (Ag) coated AuNR) nanocuboids with LSPR wavelength of 520 nm to fabricate plasmonic-fluor-Cy3 (Figure 3i, S18).58,59 Notably, as synthesized plasmonic-fluor-Cy3 exhibited high brightness and individual nanoconstructs can be easily identified under a common epifluorescence microscope (Figure S19).
We customized the Luminex assay to simultaneously detect mouse IL-6 and mouse tumor necrosis factor-α (TNF-α), which are important pro-inflammatory cytokines involved in cell signaling and immune modulation. The microbeads were incubated with a mixture of serial dilutions of TNF-α and IL-6, followed by the detection antibody cocktail, streptavidin-Cy3, and plasmonic-fluor-Cy3 (Figure 3H). The beads are subsequently read using a dual laser flow-based instrument (Luminex 200), with the classification laser (635 nm) deciphering the barcode of each bead and the reporter laser (532 nm) determining the intensity of the Cy3 fluorescence, which is in direct proportion to the amount of analyte bound (Figure 3h). SEM image of the microbead shows uniform binding of plasmonic-fluor-Cy3 with no sign of aggregation (Figure 3j). The binding of plasmonic-fluor-Cy3 did not alter the size and shape of the bead (Figure S20) or the optical barcode signal (Figure S21). A significant increase in the microbead fluorescence intensity was observed after the binding of plasmonic-fluor-Cy3 (Figure 3k). The LODs of plasmon-enhanced mouse IL-6 and TNF-α assays were determined to be 56.6 fg/ml (2.7 fM) and 7.5 fg/ml (0.3 fM), respectively (Figure 3l, m, and S23). Compared to unenhanced counterpart (Figure 3l, m, S22, and S23), the plasmon-enhanced assay exhibited 143-fold and 814-fold lower LOD for mouse IL-6 and mouse TNF-α, respectively. Notably, the vendor-specified LOD (using PE-streptavidin) for mouse IL-6 (2.3 pg/ml) and mouse TNF-α (1.47 pg/ml) were noted to be 41-fold and 196-fold inferior to the plasmon-enhanced Luminex assay. In essence, plasmonic-fluors serve as a powerful technology to enhance the bioanalytical parameters (LOD, LLOQ, dynamic range) of various existing immunoassays without requiring tedious steps or any specialized instruments.
Plasmonic-fluor enhanced high throughput multiplexed proteomic array.
Biomolecular (micro-)arrays based on fluorescence read-out is an important clinical and research tool, especially for simple, high-throughput and rapid proteomic and genetic analysis, allowing miniaturization of thousands of assays on one small piece of analytical substrate.60 Despite advantages such as high multiplexity, rapid screening, and low sample volume, this methodology suffers from low sensitivity (even inferior to ELISA), which hinders its widespread application.
We have investigated the applicability of plasmonic-fluors for enhancing the sensitivity of immuno-arrays. An array of antibodies to biomarkers of human kidney disease was employed as a representative example (Figure 4a). This array is comprised of 38 capture antibodies corresponding to human kidney disease protein biomarkers, printed in duplicates on a microporous nitrocellulose membrane (Figure S26). Biotinylated IgGs and PBS were printed as reference positive control and negative control, respectively (Figure S26). A human urine sample from a patient with kidney disease was diluted 10-fold using blocking buffer, mixed with biotinylated detection antibody cocktail, and added onto the nitrocellulose membrane. After incubation, the membrane was exposed to streptavidin-800CW. Finally, plasmonic-fluor-800CW suspension is added on the array, incubated, and thoroughly rinsed to remove the unbound nanoconstructs (Figure 4a).
Fig. 4 |. Plasmonic-fluor enhanced high-throughput proteome profiler array.
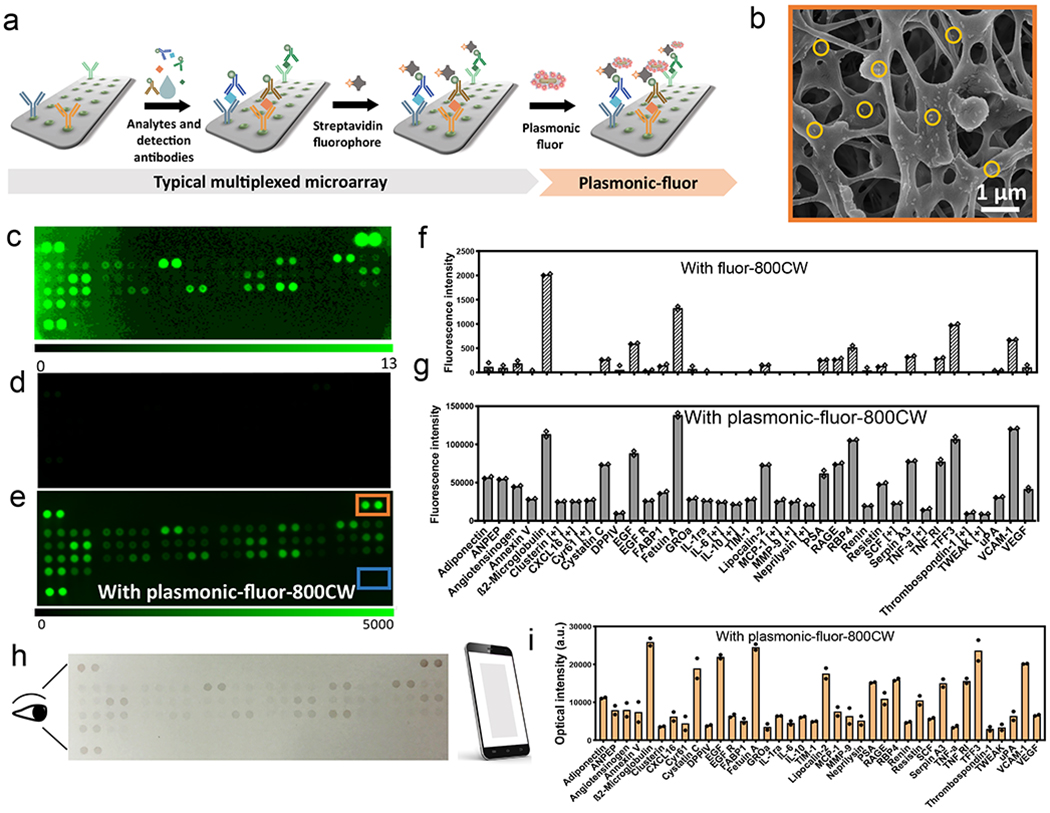
a, Illustration showing the application of plasmonic-fluor-800CW to enhance the bioanalytical parameters of multiplexed proteome profiler for human kidney disease biomarkers implemented on a nitrocellulose membrane. b, SEM image showing the uniform distribution of plasmonic-fluor-800CW (a few highlighted by the yellow circles) on and in subsurface regions of the nitrocellulose membrane. Fluorescence intensity map representing kidney disease protein biomarker profile of a kidney disease patient obtained (c, d) using conventional fluorophores (streptavidin-800CW) and (e) after the addition of plasmonic-fluor-800CW (note the difference in fluorescence intensity scale bar). Fluorescence intensity corresponding to the concentrations of various urinary biomarkers (f) before (typical assay using conventional fluorophore) and (g) after the addition of plasmonic-fluor-800CW. [+] indicates biomarkers detected only with plasmonic-fluor-800CW. Experiment was repeated three times independently on different days with different batches of plasmonic-fluors and the data is shown in supporting information. h, Photograph (acquired by mobile phone) showing the colour change of the nitrocellulose membrane with urine sample from kidney disease patient after the addition of plasmonic-fluor-800CW. i, Histogram showing the optical intensity of the mobile phone acquired photograph corresponding to each analyte in the patient urine sample.
SEM images from the positive control region revealed a uniform distribution of plasmonic-fluors on membrane (including porous subsurface regions) (Figure 4b). Concurrently, no signal was detected from the negative control (Figure 4e: blue box) and plasmonic-fluors were not observed in the SEM images from these locations, indicating their minimal non-specific binding (Figure S27). Using conventional fluorophores, out of the 38 target protein biomarkers, only 26 were detectable, most of them exhibiting weak intensity (Figure 4c, d, f and S28). After addition of the plasmonic-fluor-800CW, the fluorescence signal intensity from each spot of the protein array increased significantly (Figure 4e, g, and S28), enabling the detection and relative quantification of all of the other targets that could not be detected by the conventional fluors (Figure 4g, [+] mark indicating biomarker detected only with plasmonic-fluors). Additionally, we have employed a commercially available 40-plex cytokine microarray as another validation for plasmonic-fluor, where significant improvement in the microarray sensitivity was observed as well (Figure S31).
It is known that the plasmonic nanostructures at the LSPR wavelength exhibit large extinction cross-section, which can be up to 5–6 orders of magnitude larger than light absorption of most organic dyes.61 This unique property of plasmonic nanostructures renders the possibility of utilizing plasmonic-fluors as multimodal bio-label.62 Indeed, the binding of plasmonic-fluor to the sensing domains resulted in analyte concentration-dependent colour spots, which can be directly visualized by the naked eye (Figure 4h). The colour intensity of each spot in a digital photograph, acquired using a smartphone camera under ambient light condition, was analyzed and compared to the corresponding fluorescence intensity (Figure 4i). We observed a good correlation between the two acquisition modes (R2=0.86, Figure S32), which indicates the potential applicability of this nanoconstruct as a “visible label” in resource-limited settings to alleviate the reliance on a dedicated and expensive readout instrument.
Plasmonic-fluor enhanced immunocytochemistry/immunofluorescence (ICC/IF).
Immunocytochemistry based on immunofluorescence is a well-developed semi-quantitative method for analyzing the relative abundance, conformation, and subcellular localization of target antigens in cells. Again, this method lacks the sensitivity to distinguish low abundant biomolecules from the noise level due to the feeble fluorescence signal of conventional fluorophores. Autofluorescence, the natural emission of light by biological structures, further contributes to the overall low signal-to-noise ratio.
To test the applicability of plasmonic-fluor in ICC/IF, we employed ErbB2 (human epidermal growth factor receptor 2)-positive epithelial breast cancer cells (SK-BR-3) as a model cell line. The surface receptor ErbB2 was immuno-stained using standard approach (biotinylated ErbB2 primary antibody and streptavidin-800CW), followed by the addition of plasmonic-fluor-800CW (Figure 5a). ErbB2 primary antibody (1 mg/ml) was diluted to different concentrations before incubation with cells. SEM images revealed the uniform distribution of plasmonic-fluors on the cell membrane (Figure S33). Confocal laser scanning microscopy (CLSM) images of the cells revealed up-to 100-fold higher fluorescence signal (background subtracted) after the addition of plasmonic-fluors (20 pM) (Figure 5a, b, S34 and S35), and the expression of ErBb2 receptors could be imaged even at 100,000-fold dilution of the primary antibody (10 ng/ml) (Figure 5a, S34). In stark contrast, the fluorescence signal could only be imaged at a 100-fold (typical dilution; 10 μg/ml) dilution of primary antibody using conventional fluorophores (Figure 5a). These results demonstrate not only the applicability of plasmonic-fluor in significantly reducing the amount of antibody (and consequent cost) required in ICC/IF but also the ability to image low-abundance biomarkers on the cell surface using plasmonic-fluors.
Fig. 5 |. Plasmonic-fluor enhanced immunocytochemistry and flow cytometry.
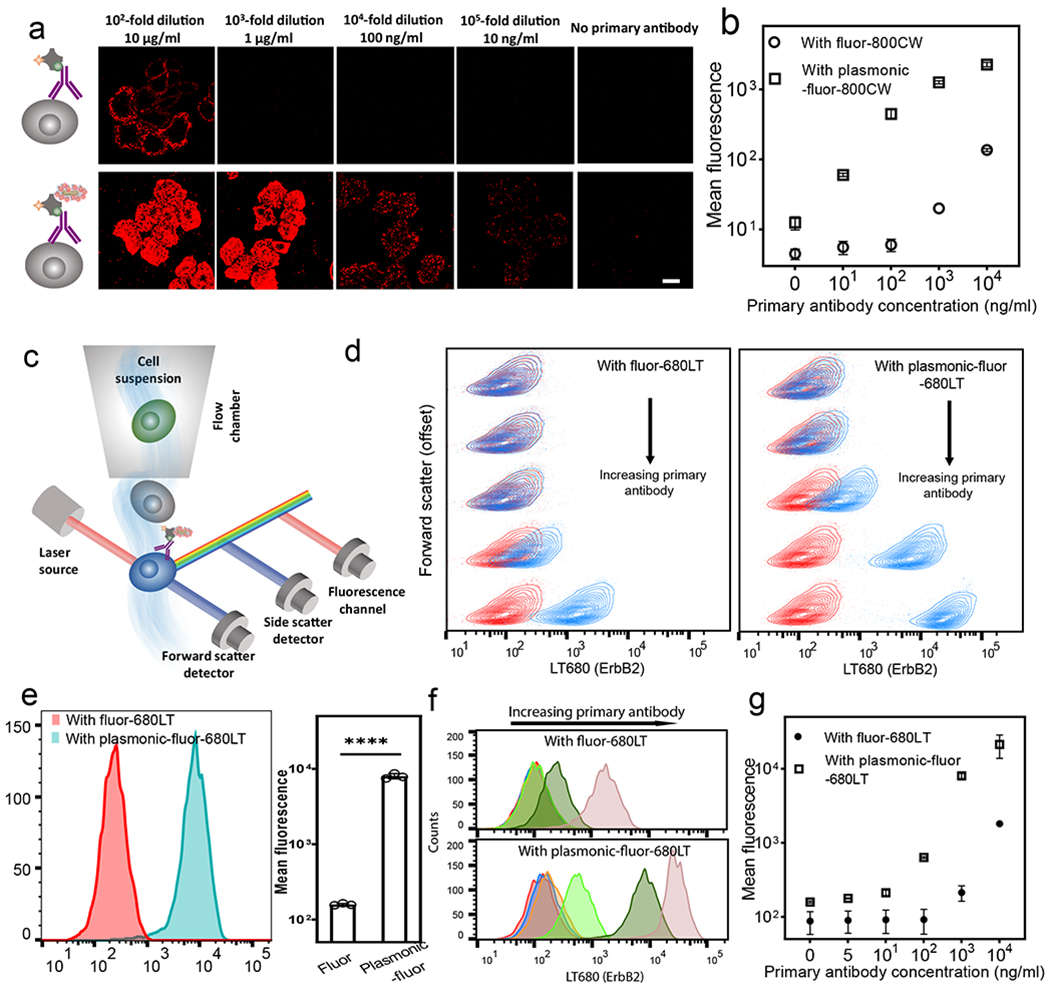
a, Confocal laser scanning microscopy (CLSM) images of breast cancer cells (SK-BR-3) probed with conventional fluor (800CW, top row) and plasmonic-fluor-800CW (bottom row) at different concentrations of ErbB2 primary antibody. Scale bar, 10 μm. b, Plot showing the fluorescence intensity of SK-BR-3 cells stained with conventional fluor and plasmonic-fluor-800CW. Error bars, s.d. (over three different locations). c, Schematic showing flow cytometry of ErbB2-stained SK-BR-3 cells probed by conventional fluor (680LT) followed with plasmonic-fluor-680LT. d, Flow contour plot (with outliers) of fluorescence vs. forward scatter (vertically offset for clarity) of SK-BR-3 cells probed using different concentrations of ErbB2 primary antibody (Red: control group without adding primary antibody. Blue: cells treated with different dilutions of primary antibody). Cells are stained with conventional fluor (680LT, left plot) followed by the addition of plasmonic-fluor-680LT (right plot). e, Fluorescence histogram of SK-BR-3 cells probed using conventional fluor (680LT) followed by the addition of plasmonic-fluor-680LT (at 103-fold dilution of primary antibody). Error bars, s.d. (n=3 independent tests). ****p < 0.0001 by two-tailed unpaired t-test with Welch’s correction. f, Histogram showing fluorescence for SK-BR-3 cells before (top) and after (bottom) the addition of plasmonic-fluor-680LT. Red: no primary antibody; blue: 2×105-fold dilution; orange:105-fold dilution; light green: 104-fold dilution; green: 103-fold dilution; rose: 102-fold dilution of the stock solution provided by the vendor. g, Plot showing the mean fluorescence intensity obtained from flow cytometry at different primary antibody concentrations. Error bars, s.d. (n=3 independent tests). All experiments were repeated three times independently on different days with different batches of plasmonic-fluors and the data is shown in supporting information.
Plasmonic-fluor enhanced flow cytometry measurement.
Flow cytometry is extensively employed in cell analysis to measure the expression and relative abundance of specific analytes on or within the cells at rates of thousand or more cells per second (Figure 5c). However, flow cytometry also suffers from significant challenges in terms of fluorescence signal-to-noise ratio due to the high speed of the target species as they cross the laser focus, limiting the time for fluorescence readout.63 Again, background fluorescence (autofluorescence) from cells poses difficulty in delineating small changes in the expression levels of intra- and extracellular targets.
To test the ability of plasmonic-fluors to enhance the signal-to-noise ratio in flow cytometry-based cell analysis (Figure 5c), SK-BR-3 cell suspensions were incubated with ErbB2 primary antibody, streptavidin-680LT (LI-COR), followed by the addition of plasmonic-fluor-680LT. Subsequently, the labeled cells were collected by mild centrifugation (1000 rpm) with concomitant removal of unbound plasmonic-fluors. To match the excitation laser and fluorophore emission, we have employed AuNRs with LSPR wavelength around 647 nm as the nanoantennae to create plasmonic-fluor-680LT (Figure S38). Specific binding of the plasmonic-fluor-680LT caused a change in the colour of the cell pellet (Figure S39). The presence of plasmonic-fluors-680LT on the cell surface did not change the forward scatter or side scatter intensity (Figure S40), indicating that the cell size and granularity/complexity remained virtually unaltered after binding of the plasmonic-fluor-680LT. Flow cytogram of fluorescence vs. forward scatter (vertically offset for clarity) of SK-BR-3 cells revealed a more obvious separation of cell populations stained with plasmonic-fluor-680LT compared to that obtained with conventional fluorophores (Figure 5d). Histograms of cell fluorescence signals revealed up-to 60-fold higher intensity (background subtracted) using plasmonic-fluor-680LT compared to its conventional counterpart (Figure 5e). Fluorescence histogram revealed that the expression of ErbB2 on the cell surface can be detected even at 200,000-fold dilution of primary antibody (5 ng/ml) using plasmonic-fluor-680LT labeling (Figure 5f, g). On the other hand, conventional labeling required the antibody to be diluted less than 1000-fold (i.e. concentration > 0.5 μg/ml) to ensure a detectable increase in fluorescence signal compared to the background (blank) (Figure 5f, g).
To further validate the performance of plasmonic-fluors in delineating cell populations with small differences in surface receptor expression levels, we employed bone marrow-derived dendritic cells (BMDCs) as a model system in which the surface expression of receptors can be modulated using immunogenic stimulus. Dendritic cells after exposure to an immunogenic stimulus undergo activation and maturation, which leads to cytokine secretion and upregulation of maturation markers such as cluster of differentiation 40 (CD40), CD80, CD86, and major histocompatibility complex (MHC I and MHC II). Here, BMDCs were isolated from 6–8 weeks old C57BL/6 mice and lipopolysaccharide (LPS) was employed as immunogenic stimulus to trigger the upregulation of CD80 and cytokine release in a dose-dependent manner. Subsequently, the cells were fixed and treated with biotinylated CD80 antibody. Finally, BMDCs were probed by conventional fluorophore (680LT, LI-COR) followed by plasmonic-fluor-680LT, and the fluorescence levels were compared using flow cytometer (Figure 6a).
Fig. 6 |. Flow cytometry measurement of BMDC maturation maker probed by conventional fluor (680LT) and plasmonic-fluor-680LT.
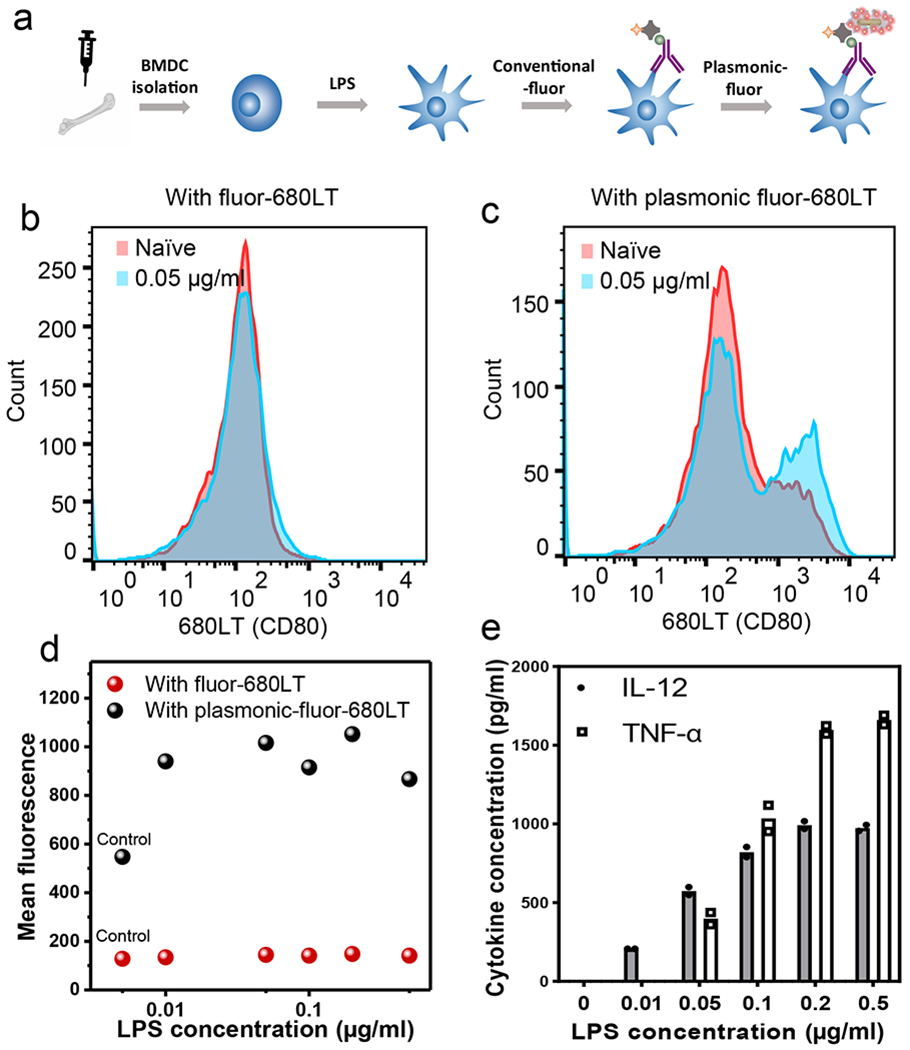
a, Schematic illustration showing bone marrow derived dendritic cells (BMDCs) treated with the immuno-stimulant (lipopolysaccharide (LPS)). The small changes of maturation markers (CD80) expression after stimulation are detected by immunofluorescence staining followed by addition of plasmonic-fluor-680LT. Fluorescence intensity distribution corresponding to naïve (control) and LPS-stimulated BMDCs obtained using (b) conventional fluors (680LT) and (c) plasmonic-fluor-680LT. d, Plot showing mean fluorescence intensity of BMDCs (corresponding to the expression level of CD80) after stimulation with different amounts of LPS. Experiment was repeated three times independently on different days with different batches of plasmonic-fluors and the data is shown in supporting information. e, Secretion levels of pro-inflammatory cytokines (TNF-α and IL-12), which confirmed the dose-dependent activation and maturation of BMDCs. each data point represents the average value of duplicate tests, which are included in supporting information.
Figure 6b, c show the fluorescence histograms corresponding to naïve and LPS (0.05 μg/ml)-stimulated BMDCs obtained using conventional fluors (680LT, Figure 6b) and plasmonic-fluor-680LT (Figure 6c). Clearly, plasmonic-fluor stained BMDCs exhibited a significant fluorescence difference between activated (blue) and naïve (red) cell populations (Figure 6b, c, S43). LPS dose-dependent (0 to 0.05 μg/ml) stimulation of BMDCs was further investigated, where a steep increase in the mean fluorescence intensity was observed using plasmonic-fluor-680LT followed by plateau at higher LPS dose (Figure 6d, S44), indicating an increase in the expression of CD80. BMDCs stained with conventional fluorophore, however, exhibited a shallow fluorescence increase with LPS dose, which was obscured by the high fluorescence background (Figure 6d, and S44). Moreover, the secretion levels of pro-inflammatory cytokines (TNF-α and interleukin 12 (IL-12)) exhibited an increasing trend with the increase of LPS concentration (Figure 6e, S45). This further confirmed the dose-dependent activation and maturation of BMDCs as well as the specificity and accuracy of plasmonic-fluor in differentiating the minute changes in the cell surface maturation markers.
Harnessing plasmon-enhanced fluorescence, we have designed and synthesized ultrabright nanoconstructs as fluorescence reporter. In addition to the improved bioanalytical parameters, plasmonic-fluors can potentially simplify the readout instrumentation, decrease the required sample volume, shorten the overall assay time, and enable bioassays to be implemented with minimal effort and reduced cost. Through a series of experiments, we have demonstrated that plasmonic-fluors are highly customizable in terms of the excitation/emission wavelength over the entire visible and near infrared range and can be applied in microtiter plates, on porous membranes, on microbeads, and cells. The cross-assay signal amplification approach introduced here is a disease-, biomarker-, and application-agnostic ubiquitously-applicable fundamental and enabling biomedical technology to immediately improve the sensitivity of existing bioanalytical methodologies in an easy-to-implement and cost-effective manner.
Methods
Synthesis of AuNR.
AuNR-760 (LSPR wavelength ~760 nm) was prepared by a seed-mediated method.64,65 Au seed was synthesized by adding 0.6 ml of ice-cold NaBH4 solution (10 mM) (Sigma-Aldrich, 71321) into a solution containing 0.25 ml HAuCl4 (10 mM) (Sigma-Aldrich, 520918) and 9.75 ml CTAB (0.1 M) (Sigma-Aldrich, H5882) under vigorous stirring at room temperature for 10 min. The colour of the solution changed from yellow to brown indicating the formation of Au seed. For the synthesis of AuNR, the growth solution was prepared by the sequential addition of aqueous HAuCl4 (0.01 M, 2 ml), CTAB (0.1 M, 38 ml), AgNO3 (0.01 M, 0.5 ml, Sigma-Aldrich, 204390), HCI (1M, 0.8 ml, Sigma-Aldrich, H9892) and ascorbic acid (0.1 M, 0.22ml, Sigma-Aldrich, A92902) followed by gentle inversion to homogenize the solution. The AgNO3 and HCI volume ratio may vary to obtain the right wavelength. Subsequently, 5 μl of the seed solution was added into the growth solution and left undisturbed in the dark for 24 hours. AuNR solution was centrifuged at 7000 rpm for 40 minutes to remove the supernatant and the AuNR was re-dispersed into nanopure water to achieve a final peak extinction ~2.0. For AuNR-647 (LSPR wavelength ~647 nm), the growth solution contained HAuCl4 (0.01 M, 2 ml), CTAB (0.1M, 38 ml), AgNO3 (0.01 M, 0.2 ml, this value may vary), and ascorbic acid (0.1 M, 0.32ml).
Synthesis of AuNR@Ag.
AuNR with LSPR wavelength around 711 nm was employed as the core for the synthesis of AuNR@Ag nanostructures. Specifically, 3 ml of 711 nm AuNR (peak extinction~4) was incubated with 8 ml of CTAC (20 mM) at 60 °C for 20 minutes under stirring. Then, 8 ml of AgNO3 (4 mM), 4 ml of CTAC (20 mM), and 0.8 ml of ascorbic acid (0.1M) were added sequentially and the mixture was incubated at 60 °C for 4 h under magnetic stirring to form AuNR@Ag nanocuboids. Finally, AuNR@Ag nanocuboids solution was centrifuged at 6000 rpm and the nanocuboids were redispersed in nanopure water.
Conjugation of biotin and 800CW on BSA.
Biotin and 800CW were sequentially conjugated to BSA through EDC/NHS chemistry. In pH 7-9 buffers, NHS esters react efficiently with primary amino groups (-NH2) by nucleophilic attack, forming an amide bond and releasing the NHS. Specifically, 2 mg of NHS activated biotin (NHS-PEG4-Biotin, Thermo Scientific, Prod #: 21329) was added to 2.2 ml of BSA (Sigma-Aldrich, A7030) solution (5 mg/ml in 1X PBS). The mixture was incubated at room temperate (~22°C) for 1 hour to complete the reaction. Excess NHS-PEG4-Biotin was removed from the solution using a desalting column (5mL, 7000 MWCO, Thermo Scientific, Prod #: 21329) pre-equilibrated with 1X PBS. Next, 800CW was conjugated to BSA-biotin. 0.1 ml of 1M potassium phosphate buffer (K2HPO4, pH=9) was added into 1ml of purified BSA-biotin solution to raise the pH. Next, 25 μl of 4 mg/ml NHS-800CW (LI-COR, 929-70020) was added to the mixture and the solution was incubated at 23°C for 2.5 hours. Free NHS-800CW was then separated from the conjugate using a Zeba desalting column pre-equilibrated with nanopure water. BSA-biotin-680LT and BSA-biotin-Cy3 were prepared using a similar method, except for changing the fluorophore to NHS-680LT (LI-COR, 929-70010) and NHS-Cy3 (ab146452).
Synthesis of plasmonic-fluor.
To fabricate plasmonic-fluor with high fluorescence enhancement efficacy, it is extremely important to choose an “on-resonant” plasmonic nanostructure for a given fluorophore. For 800CW, AuNR-760 (length and diameter of 83 and 24 nm, respectively) was employed as the nanoantenna. 1 μl of MPTMS (Sigma Aldrich, 175617) was added to 1 ml AuNR with extinction ~2.0 and the mixtures was shaken for 1 hour allowing the formation of an interfacial layer on the AuNR. MPTMS-modified AuNR was further mixed with different volumes of APTMS (Sigma Aldrich, 281778) and TMPS (Sigma Aldrich, 662275) (from 0.5 μl to 2 μl) to form the polymer spacer layer on AuNR. Finally, AuNR/polymer solution was centrifuged twice each at 6000 rpm for 10 minutes to remove the free monomer. After second centrifugation, AuNR/polymer was concentrated into a final volume of 10 μl.
Next, BSA-biotin-800CW conjugate was coated around AuNR/polymer. We have adopted previously reported method to coat BSA around metal nanostructures with a few modifications.66 Specifically, 1 μl of 20 mg/ml citric acid (Alfa Aesar, 36664) was added into 100 μl of BSA-biotin-800CW (~4 mg/ml) to lower the pH. Concentrated AuNR/polymer solution was subsequently added into this mixture and sonicated for 20 minutes under dark condition. The nanostructures were then collected using mild centrifugation (5000 rpm for 3 minutes). Subsequently, the AuNRs were incubated with 0.5 ml BSA-biotin-800CW (~0.4 mg/ml, pH=10) for 3 days under dark condition in 4°C. Finally, the nanostructures were washed four times using nanopure water (pH=10) by centrifugation at 6000 rpm. After the last washing step, the particles were re-dispersed into 1% BSA (buffered with 1X PBS).
Material characterization.
Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100F field emission (FE) instrument. A drop of aqueous solution was dried on a carbon-coated grid, which had been made hydrophilic by glow discharge. SEM images were obtained using a FEI Nova 2300 field‐emission scanning electron microscope at an acceleration voltage of 10 kV. AFM imaging was performed on a Dimension 3000 using silicon cantilevers with a nominal spring constant of 40 N/m in light tapping mode. The extinction spectra of plasmonic nanostructures were obtained using a Shimadzu UV-1800 spectrophotometer. Fluorescence lifetime was measured using time correlated single photon counting (TCSPC implemented in Fluorolog-3, Horiba Jobin Yvon) with a 740 nm excitation source NanoLed® (impulse repetition rate 1 MHz) at 90º to the PMT R928P detector (Hamamatsu Photonics, Japan). Most of the fluorescence mappings were recorded using LI-COR Odyssey CLx imaging system. Luminex 200 system was employed to read the fluorescence signal from the microbeads. Cell imaging was performed using Olympus FV1000 LSM confocal laser scanning microscopy (785 nm excitation laser) under 40X water-immersion objective. Guava easyCyte was employed to acquire the flow cytometry data.
Fluorescence enhancement of 800CW-streptavidin using AuNR-plasmonic-fluor-800CW and AuNP-plasmonic-fluor-800CW.
Experimental procedure employed for this test is illustrated in Figure S9 and the data in shown in figure 2c, d. Specifically, BSA-biotin was first immobilized on the bottom of plastic 96-well plate by incubating the well with 50 ng/ml BSA-biotin (in 1X PBS) at room temperature for 15 minutes. The plate was washed three times using PBST (0.05% Tween 20 in 1X PBS) and then blocked using Odyssey® Blocking Buffer (PBS) (LI-COR, P/N 927-40100). Wells coated with BSA-biotin were subsequently incubated with 1 μg/ml streptavidin-800CW (in Odyssey® Blocking Buffer) for 10 minutes to allow specific binding of streptavidin to biotin. Next, the plate was washed three times using PBST and then incubated with ~76 pM plasmonic-fluor-800CW (in 1% BSA). The plate was washed three more times using PBST to remove free plasmonic-fluor. Finally, 200 μl of PBST was added into each well and the fluorescence signal before and after the addition of plasmonic-fluor was recorded using the LI-COR CLx fluorescence imager with the following scanning parameters: laser power~L2; resolution~169 μm; channel: 800; height: 4 mm. The experiment was repeated four times independently and the fluorescence intensities before and after adding plasmonic-fluor-800CW were compared. The data is statistically significant, and the P value was calculated to be 0.0044, ** P < 0.01 by two-tailed unpaired t-test with Welch’s correction.
To compare the fluorescence enhancement efficiency of plasmonic-fluor-800CW comprised of AuNR and AuNP, similar experiment was performed except using AuNP-plasmonic-fluor-800CW instead of AuNR-plasmonic-fluor-800CW after the binding step of streptavidin-800CW. The molar concentration of AuNP-plasmonic-fluor-800CW was maintained to be similar as that of AuNR. The experiment was repeated three times independently. Fluorescence enhancement factor obtained from AuNP-plasmonic-fluor-800CW was compared with that obtained from AuNR-plasmonic-fluor-800CW. Data is statistically significant with P value= 0.0013, ** P < 0.01 by two-tailed unpaired t-test with Welch’s correction.
Human IL-6 ELISA.
Human IL-6 DuoSet ELISA kit (R&D, catalog# DY206, lot# P173353) was employed in the study. Specifically, 96-well plates were first coated with capture antibodies (2 μg/ml in PBS, PART# 840113) through overnight incubation at room temperature, followed by blocking with 300 μl reagent diluent (1X PBS containing 3% BSA, 0.2 μm filtered). After three times washing with PBST, 100 μl of serial diluted standard samples (PART# 840115) as well as patients’ serum samples (10-fold dilution using reagent diluent) were added into different wells and the plate was incubated at room temperature for 2 hours. The plate was washed subsequently and incubated with biotinylated detection antibodies (PART# 840114, 50 ng/ml in reagent diluent) for 2 hours, washed again with PBST, and incubated with HRP-labeled streptavidin (PART# 893975, 200-fold dilution using reagent diluent) for 20 mins. 100 μl of substrate solution (1:1 mixture of Colour Reagent A (H2O2) and Colour Reagent B (tetramethylbenzidine) (R&D Systems, Catalog # DY999)) was added to each well and the reaction was stopped by adding 50 μl of H2SO4 (2 N) (R&D Systems, Catalog # DY994) after 20 mins. Optical density of each well was determined immediately using a microplate reader set to 450 nm.
Human IL-6 FLISA and p-FLISA.
Human IL-6 FLISA was implemented adopting the similar approach as the ELISA described above, expect that HRP-labeled streptavidin was replaced by 800CW-labeled streptavidin (LI-COR P/N 926–32230, 50 ng/ml for 20 minutes). The plate was washed three times using PBST followed by washing with nanopure water. In p-FLISA, plasmonic-fluor-800CW was added subsequently (extinction ~1), incubated for 1 hour, and the plate was washed 3 times each with reagent diluent followed by PBST. The plate was imaged using LI-COR CLx fluorescence imager with the following scanning parameters: laser power~L2; resolution~169 μm; channel: 800; height: 4 mm. The results from independent experiment are shown in the main manuscript (Figure 3d,e) as well as in Figure S13.
Human NAGL ELISA (280 minutes) and p-FLISA (20 minutes).
Human NGAL DuoSet ELISA kit (R&D, catalog# DY1757, lot# P195696) was employed in the study. Specifically, 100 μl of serial diluted standard samples (PART#842273) as well as urinary samples from patients and self-described healthy volunteers (10-fold dilution using reagent diluent) were added into different wells and the plate was incubated at room temperature for 2 hours. The plate was washed subsequently and incubated with biotinylated detection antibodies (PART#844865, 25 ng/ml in reagent diluent) for 2 hours, washed again with PBST, and incubated with HRP-labeled streptavidin (PART#893975, 40-fold dilution using reagent diluent) for 20 mins. Human NGAL p-FLISA was performed by adopting a similar procedure as the ELISA with significantly shortened assay time. Specifically, the incubation time for standards/samples, biotinylated detection antibodies, 800CW-labeled streptavidin (LI-COR P/N 926-32230, 50 ng/ml), and plasmonic-fluor-800CW (extinction~1) was set to 5 minutes. The study was approved by Washington University IRB 201601082 “Nanotech Biomarkers for Renal Cancer Intervention: Clinical Validation and Utility” and IRB 201202051 “Urine Proteome of Surgical Patients and Healthy Volunteers”. Informed consent was obtained from the participants.
Measurement of mouse IL-6 concentration in skin interstitial fluid (ISF).
C57BL/6J female mice (9 to 10 weeks of age) were intraperitoneally injected with LPS (1 μg/g) to induce acute phase response and systemic inflammation. Mouse ISF was collected 4 hours after LPS injection and the concentrations of pro-inflammation cytokine (IL-6) in ISF was analyzed by ELISA and p-FLISA. Mouse ISF was extracted according to previously reported centrifugation method.67 Briefly, the skin on the back of the mice was harvested and placed on a basket of nylon mesh with pore size ∼15-20 μm, with the subcutis facing the mesh. The skin sample was subsequently transferred to a centrifuge tube and was immediately spun at 424g at 4°C to collect the ISF. ISF samples from naïve mice without any immune stimulation were collected using the same method.
Mouse IL-6 DuoSet ELISA kit (R&D, catalog# DY406-05, lot# P195781) was employed to measure the IL-6 concentration in mouse ISF. Specifically, 100 μl of serial diluted standards (PART#840173) as well as diluted ISF samples were added into different wells and the plate was incubated at room temperature for 3 hours. The plate was washed subsequently and incubated with biotinylated detection antibodies (PART#840172, 75 ng/ml in reagent diluent) for 2 hours, washed again with PBST, and incubated with HRP-labeled streptavidin (PART#893975, 40-fold dilution using reagent diluent) for 20 mins. Mouse IL-6 p-FLISA was performed by adopting a similar approach as the ELISA described above, expect that HRP-labeled streptavidin was replaced by 800CW-labeled streptavidin (LI-COR P/N 926-32230, 20 ng/ml for 20 minutes). Finally, plasmonic-fluor-800CW (extinction~0.5) was added, incubated for 30 minutes, and washed by PBST.
Plasmonic-fluor enhanced Luminex bead-based assay.
Mouse magnetic Luminex assay was purchased from R&D systems (catalog number: LXSAMSM-03, lot# L126064), which was customized to simultaneously detect mouse TNF-α and mouse IL-6. To begin with, 50 μl of standards that contain different concentrations of TNF-α and IL-6 (PART# 984658) were mixed with 50 μl of diluted microbead cocktail (PART# 894724) in 96-well plate. The mixture was homogenized by shaking horizontally using a microplate orbital shaker (0.12” orbit) set at 800 rpm for 2 hours. Microbeads were subsequently collected using a magnetic device (Millipore Sigma 40-285) designed to accommodate the microplate and were washed by removing the liquid and filling with wash buffer (PART# 895003). The washing step was repeated by three times. Next, 50 μl of diluted biotin-antibody cocktail (PART# 894666) was introduced to each well and incubated for 1 hour on the shaker at 800 rpm. The microbeads were washed three times again and then incubated with 100 ng/ml Cy3-streptavidin (in 3% BSA buffered with 1X PBS) for 30 minutes at 800 rpm. After three-time washing, the microbeads were incubated with 50 μl plasmonic-fluor-Cy3 (extinction ~5) for 1 hour at 800 rpm and washed 3 times each with 3% BSA and washing buffer. Finally, the microbeads were resuspended in 100 μl washing buffer and incubated for 2 minutes at 800 rpm prior to reading. Luminex 200 instrument was employed for fluorescence readout. Dual mode fluorescence of the microbead was observed on a Confocor II LSM system (Carl Zeiss-Evotec, Jena, Germany) using a ×40 water-immersion objective. The results from independent experiment are shown in the main manuscript (Figure 3l, m) as well as in Figure S24, 25.
Plasmonic-fluor enhanced human kidney biomarker array.
Human kidney biomarker array kit was purchased from R&D system (catalog# ARY019, lot# 1311110). Urine sample from kidney disease patient (ID #25, age 61, male) was employed for this study. The study was approved by Washington University IRB 201601082 “Nanotech Biomarkers for Renal Cancer Intervention: Clinical Validation and Utility”. Informed consent was obtained from the participants. The nitrocellulose membrane (PART# 893967) was blocked by incubation with 2 ml of blocking buffer (PART# 893573) in the 4-well multi-dish for 1.5 hour under gentle rocking. During blocking process, kidney disease patient (ID #25) urine sample (150 μl) was diluted with 500 μl of blocking buffer and 850 μl of array buffer (PART# 895876), resulting in a total 10-fold dilution. The diluted urine sample was mixed with 15 μl of reconstituted detection antibody cocktail (PART# 893966) and the mixture was incubated at room temperature for 1 hour. The nitrocellulose membrane was taken out from the blocking solution and incubated with the mixture of urinary sample and biotinylated detection antibodies for overnight at 4°C. The membrane was subsequently washed with 20 ml of 1X washing buffer (PART# 895003) for 10 minutes under gentle rocking, and the washing process was repeated for two more times. Next, the membrane was incubated with 800CW-streptavidin (50 ng/ml in 1% BSA) for 30 minutes under gentle rocking, washed three times, and incubated with plasmonic-fluor-800CW (extinction~0.5) for one more hour. Finally, the membrane was imaged using the LI-COR CLx imager at L2 laser power, with focusing height at 0.5 mm and resolution of 169 μm. The photograph of the protein array was acquired using the iPhone6 camera and the image was analyzed using Image Studio Lite software to measure the median intensity of each spot (background subtracted). The results from independent experiment are shown in the main manuscript (Figure 4f, g) as well as in Figure S29, 30.
Plasmonic-fluor enhanced human cytokine microarray.
Forty-plex human cytokine microarray (RayBiotech, catalog #: QAH-CYT-4) was employed to further test the efficacy of plasmonic-fluor. To begin with, the glass substrate of the microarray was blocked with 100 μl sample diluent (catalog #: QA-SDB) followed by incubation with sample standard (catalog #: QAH-CYT-4-STD) at room temperature for 2 hours with gentle rocking. The microarray was washed by five times using 1X wash buffer I (catalog #: AA-WB1-30ML) followed by twice washing with 1X wash buffer II (catalog #: AA-WB2-30ML). Next, 80 μl of reconstituted detection antibody cocktail was added into each well and incubated for another 2 hours under gentle rocking. Following the incubation, washing process was repeated again as described above. Subsequently, 80 μl of 800CW-streptavidin (50 ng/ml in 1% BSA) was added to the array slide and incubated for 20 minutes, washed, and immersed with plasmonic-fluor-800CW (extinction~1) for one hour. The slide was scanned using LI-COR CLx scanner with the following parameters: laser power~3.5; resolution~21 μm; channel: 800; height: 1.8 mm.
Plasmonic-fluor enhanced immunocytochemsitry/immunofluorescence (ICC/IF).
Human epithelial breast cancer cells SK-BR-3 [SKBR3] (ATCC® HTB30™) were purchased from ATCC (Manassas, VA) and sub-cultured in Mc.Coy’s 5A medium with 10% fetal bovine serum (FBS) and antibiotics (100 μg/ml penicillin and 100 μg/ml streptomycin) (Sigma, St. Louis, MO). Cells were grown in water jacketed incubator at 37°C with 5% CO2-humidified atmosphere in T-25 tissue culture flasks. Once the cells reached to 90% confluence, they were washed with PBS and detached from the flask bottom using a scraper. After centrifugation, cells were re-dispersed in culture medium and seeded on 6-well plate for overnight to allow attachment to the plate bottom. Cells were subsequently fixed using 3.7% formaldehyde (in 1X PBS) for 30 minutes, washed three times with 1X PBS, and blocked with 3% BSA for 1 hour. Next, ErbB2 primary antibody (anti-human HER-2/biotin, eBioscience, clone 2G11, REF # BMS120BT, lot # 186281000) was diluted using 1% BSA and incubated with SK-BR-3 cells for 1.5 hours. The cells were subsequently washed three times, incubated with 800CW-streptavidin (1 μg/ml in 1% BSA) for 30 minutes, washed for another three times, and probed with plasmonic-fluor-800CW (extinction~0.3). The cells were finally imaged using Olympus FV1000 LSM confocal laser scanning microscopy (785 nm excitation laser) under 40X water-immersion objective. The results from independent experiment are shown in the main manuscript (Figure 5a) as well as in Figure S36, 37.
SK-BR-3 flow cytometry measurements.
SK-BR-3 cells were grown and harvested using the method described above. The cells were centrifuged at 1000 rpm for 10 minutes to remove the culture medium and were subsequently fixed using 3.7% formaldehyde in 1X PBS for 30 minutes. The cell suspension was centrifuged again to remove the free formaldehyde and the cells were subsequently blocked with 3% BSA for overnight. Next, different amounts of ErbB2 primary antibody was added into the cell suspension and the mixture was incubated for 1 hour under gentle shaking. The cells were centrifuged at 1000 rpm and washed once with 1X PBS to remove the free antibody, incubated with streptavidin-680LT (LI-COR: P/N 926-6803; 1 μg/ml in 1% BSA) for 1 hour, washed two more times, and incubated with plasmonic-fluor-680LT (extinction~2.0) for 1 hour. Finally, 5000 cells were analyzed by Guava easyCyte to acquire the fluorescence signal (RED-R channel (excitation laser: 642nm; filter: 662/15nm)) in combination with forward scatter (FSC) and side scatter (SSC). The results from independent experiment are shown in the main manuscript (Figure 5f, g) as well as in Figure S41, 42.
BMDC isolation and flow cytometry measurement.
Female C57BL/6 (H-2b) mice that were 5 to 6 weeks of age were purchased from Jackson Labs (Bar Harbor, ME, USA). The mice were maintained under pathogen-free conditions. All experiments employing mice were performed in accordance with laboratory animal protocol approved by the School of Medicine Animal Studies Committee of Washington University in St. Louis. Mouse were euthanized using CO2 asphyxiation and cervical dislocation. The euthanized mouse was kept in 70% (v/v) ethanol for 1 min. Both the femurs and tibiae were isolated, and the muscle attachments were carefully removed using gauze pads. Both ends of the bones were cut with scissors and the marrow was centrifuged in an adapted centrifuge tube (0.6 ml tube with a hole inserted in 1.5 ml tube) at 1000 rpm for 10 seconds. The pellet was resuspended by vigorous pipetting in RPMI 1640 media. The cells were passed through a 70 μm cell strainer to prepare a single cell suspension. After one washing (1200 rpm, 5 min), red blood cells were depleted with RBC lysis buffer (Sigma-Aldrich). The bone marrow cells were collected and cultured in 100-mm Petri dishes containing 10 mL RPMI medium supplemented with 10% heat-inactivated FBS, 50 IU mL−1 penicillin, 50 μg mL−1 streptomycin, and 20 ng mL-1 mouse recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF, R&D Systems, MN, USA). 1 x106 BMDCs were cultured in 6 well plates and were stimulated by adding 1 ml of different concentrations of LPS (0.5 μg/ml, 0.2 μg/ml, 0.1 μg/ml, 0.05 μg/ml, 0.01 μg/ml, and 0 μg/ml) for 24 hours. Cells were harvested using a cell scraper for further staining and flow cytometry analysis.
CD80 overexpressed on the cell surface was probed using conventional fluorophore followed with plasmonic-fluor-680LT. Specifically, stimulated BMDCs were washed once with 1X PBS to remove the culture medium (centrifugation at 2000 rpms for 5mins) and fixed using 10% neutral buffered formalin for 20 minutes. The cells were then washed (2000 rpms for 5mins) and blocked with 3% BSA for overnight at 4 °C. Next, biotinylated CD80 primary antibody (anti-Mo CD80/biotin (Invitrogen, REF# 13-0801-82, Clone 16-10A1, lot# 1934784)) was added into the BMDC suspension to achieve a final antibody concentration of 100 ng/ml and the mixture was incubated for 1 hour. The BMDCs were washed once (2000 rpms for 5mins) and were subsequently incubated with 1 μg/ml streptavidin-680LT (in 1% BSA) for 40 minutes. Finally, the cells were washed two more times and incubated with plasmonic-fluor-680LT (extinction~2) for 1 hour, followed by once more washing to remove unbound plasmonic-fluor-680LT. 10, 000 cells were analyzed by Guava easyCyte to acquire the fluorescence signal (RED-R channel (excitation laser: 642nm; filter: 662/15nm)) in combination with forward scatter (FSC) and side scatter (SSC). The results from independent experiment are shown in the main manuscript (Figure 6) as well as in Figure S46, 47.
Statistics.
For analyzing the statistical difference between two groups, unpaired two-tailed t-test with Welch’s correction was used. For analyzing the statistical difference between more than two groups, one-way ANOVA with post-hoc Tukey’s honest significance test was used. Statistical significance of the data was calculated at 95% (p < 0.05) CIs. All values are expressed as mean ± standard deviation. GraphPad Prism 6 (San Diego, CA, USA) was used for all statistical analysis. We employed four-parameter logistic (4PL) or polynomial fit to calculate the limit-of-detection in the standard curves of bioassays. The limit-of-detection is defined as the analyte concentration corresponding to the mean fluorescence intensity of blank plus three times of its standard deviation (mean+3σ). Origin 2016 (Northampton, MA, USA) was employed for calculating the limit-of-detection.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We acknowledge support from National Science Foundation (CBET-1512043 and CBET-1900277), National Institutes of Health (R01DE027098 and R01CA141521) and a grant from the Barnes-Jewish Hospital Research Foundation (3706). The authors thank Prof. Kristen Naegle for providing access to a LI-COR Odyssey CLx scanner, Prof. Lori Setton for flow cytometer, Prof. Jai Rudra and Dr. Pietka for Luminex readers, and the Nano Research Facility (NRF) and Institute of Materials Science and Engineering (IMSE) at Washington University for providing access to electron microscopy facilities. We also thank Prof. Guy Genin for inspiring discussions and suggestions.
Footnotes
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Raw imaging data ((collected and analysed via the software indicated in the reporting summary) are available from figshare with the identifier https://doi.org/10.6084/m9.figshare.11888055.
Competing interests
J.L., J.J.M., and S.S. are inventors on a provisional patent related to this technology. The technology has been licensed by the Office of Technology Management at Washington University in St. Louis to Auragent Bioscience LLC, which is developing plasmonic-fluor products. J.L., J.J.M., and S.S. are co-founders/shareholders of Auragent Bioscience LLC. These potential conflicts of interest have been disclosed and are being managed by Washington University in St. Louis. The remaining authors declare no competing interests.
References
- 1.Cohen L & Walt DR Highly Sensitive and Multiplexed Protein Measurements. Chemical reviews (2018). [DOI] [PubMed] [Google Scholar]
- 2.Hanash SM, Pitteri SJ & Faca VM Mining the plasma proteome for cancer biomarkers. Nature 452, 571 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Shaw LM, Korecka M, Clark CM, Lee VM-Y & Trojanowski JQ Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nature reviews Drug discovery 6, nrd2176 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Blennow K & Zetterberg H Understanding biomarkers of neurodegeneration: ultrasensitive detection techniques pave the way for mechanistic understanding. Nature medicine 21, 217 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Savage MJ et al. A sensitive aβ oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. Journal of Neuroscience 34, 2884–2897 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westermann D, Neumann JT, Sörensen NA & Blankenberg S High-sensitivity assays for troponin in patients with cardiac disease. Nature Reviews Cardiology 14, 472 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Rissin DM et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nature biotechnology 28, 595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabakman SM et al. Plasmonic substrates for multiplexed protein microarrays with femtomolar sensitivity and broad dynamic range. Nature communications 2, 466 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang B et al. Diagnosis of Zika virus infection on a nanotechnology platform. Nature Medicine 23, 548–550 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zhang B, Kumar RB, Dai H & Feldman BJ A plasmonic chip for biomarker discovery and diagnosis of type 1 diabetes. Nature medicine 20, 948–953 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang B et al. Plasmonic micro-beads for fluorescence enhanced, multiplexed protein detection with flow cytometry. Chemical Science 5, 4070–4075 (2014). [Google Scholar]
- 12.Steward MW & Lew AM The importance of antibody affinity in the performance of immunoassays for antibody. Journal of immunological methods 78, 173–190 (1985). [DOI] [PubMed] [Google Scholar]
- 13.Roth S et al. Photobleaching: Improving the Sensitivity of Fluorescence-Based Immunoassays by Photobleaching the Autofluorescence of Magnetic Beads (Small 3/2019). Small 15, 1970016 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Zhang P et al. Ultrasensitive detection of circulating exosomes with a 3D-nanopatterned microfluidic chip. Nature Biomedical Engineering, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Espina V et al. Protein microarray detection strategies: focus on direct detection technologies. Journal of immunological methods 290, 121–133 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Chen Z et al. Protein microarrays with carbon nanotubes as multicolor Raman labels. Nature biotechnology 26, 1285 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Luan J et al. Add-on plasmonic patch as a universal fluorescence enhancer. Light: Science & Applications 7, 29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reisch A et al. Collective fluorescence switching of counterion-assembled dyes in polymer nanoparticles. Nature Communications 5, 4089 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Hu J et al. Sensitive and quantitative detection of C-reaction protein based on immunofluorescent nanospheres coupled with lateral flow test strip. Analytical chemistry 88, 6577–6584 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Huang L et al. Brilliant Pitaya-Type Silica Colloids with Central–Radial and High-Density Quantum Dots Incorporation for Ultrasensitive Fluorescence Immunoassays. Advanced Functional Materials 28, 1705380 (2018). [Google Scholar]
- 21.Reisch A & Klymchenko AS Fluorescent polymer nanoparticles based on dyes: seeking brighter tools for bioimaging. Small 12, 1968–1992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shulov I et al. Fluorinated counterion-enhanced emission of rhodamine aggregates: ultrabright nanoparticles for bioimaging and light-harvesting. Nanoscale 7, 18198–18210 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Melnychuk N & Klymchenko AS DNA-functionalized dye-loaded polymeric nanoparticles: ultrabright FRET platform for amplified detection of nucleic acids. Journal of the American Chemical Society 140, 10856–10865 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Xu Z et al. Broad-Spectrum Tunable Photoluminescent Nanomaterials Constructed from a Modular Light-Harvesting Platform Based on Macrocyclic Amphiphiles. Advanced Materials 28, 7666–7671 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Kasha M, Rawls H & El-Bayoumi MA The exciton model in molecular spectroscopy. Pure and applied Chemistry 11, 371–392 (1965). [Google Scholar]
- 26.Méallet-Renault R et al. Fluorescent nanoparticles as selective Cu (II) sensors. Photochemical & Photobiological Sciences 5, 300–310 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Trofymchuk K, Reisch A, Shulov I, Mély Y & Klymchenko AS Tuning the color and photostability of perylene diimides inside polymer nanoparticles: towards biodegradable substitutes of quantum dots. Nanoscale 6, 12934–12942 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Trofymchuk K et al. Giant light-harvesting nanoantenna for single-molecule detection in ambient light. Nature photonics 11, 657 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmberg A et al. The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 26, 501–510 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Chen H, Shao L, Li Q & Wang J Gold nanorods and their plasmonic properties. Chemical Society Reviews 42, 2679–2724, doi: 10.1039/C2CS35367A (2013). [DOI] [PubMed] [Google Scholar]
- 31.Huang X, Neretina S & El-Sayed MA Gold nanorods: from synthesis and properties to biological and biomedical applications. Advanced Materials 21, 4880–4910 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Dreaden EC, Alkilany AM, Huang X, Murphy CJ & El-Sayed MA The golden age: gold nanoparticles for biomedicine. Chemical Society Reviews 41, 2740–2779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge S, Kojio K, Takahara A & Kajiyama T Bovine serum albumin adsorption onto immobilized organotrichlorosilane surface: influence of the phase separation on protein adsorption patterns. Journal of Biomaterials Science, Polymer Edition 9, 131–150 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Ashitate Y, Tanaka E, Stockdale A, Choi HS & Frangioni JV Near-infrared fluorescence imaging of thoracic duct anatomy and function in open surgery and video-assisted thoracic surgery. The Journal of thoracic and cardiovascular surgery 142, 31–38. e32 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu C et al. Fluorescence-converging carbon nanodots-hybridized silica nanosphere. Small 12, 4702–4706 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Flauraud V et al. In-plane plasmonic antenna arrays with surface nanogaps for giant fluorescence enhancement. Nano Letters 17, 1703–1710 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Tam F, Goodrich GP, Johnson BR & Halas NJ Plasmonic enhancement of molecular fluorescence. Nano letters 7, 496–501 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Kinkhabwala A et al. Large single-molecule fluorescence enhancements produced by a bowtie nanoantenna. Nature Photonics 3, 654–657 (2009). [Google Scholar]
- 39.Cai Y et al. Photoluminescence of gold nanorods: Purcell effect enhanced emission from hot carriers. ACS nano (2017). [DOI] [PubMed] [Google Scholar]
- 40.Liu B et al. High Performance, Multiplexed Lung Cancer Biomarker Detection on a Plasmonic Gold Chip. Advanced Functional Materials 26, 7994–8002 (2016). [Google Scholar]
- 41.Mayer KM & Hafner JH Localized surface plasmon resonance sensors. Chemical reviews 111, 3828–3857 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Haes AJ, Chang L, Klein WL & Van Duyne RP Detection of a biomarker for Alzheimer’s disease from synthetic and clinical samples using a nanoscale optical biosensor. Journal of the American Chemical Society 127, 2264–2271 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Bardhan R, Grady NK, Cole JR, Joshi A & Halas NJ Fluorescence enhancement by Au nanostructures: nanoshells and nanorods. Acs Nano 3, 744–752 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Khatua S et al. Resonant plasmonic enhancement of single-molecule fluorescence by individual gold nanorods. ACS nano 8, 4440–4449 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Anger P, Bharadwaj P & Novotny L Enhancement and quenching of single-molecule fluorescence. Physical review letters 96, 113002 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Tian L et al. Gold nanocages with built-in artificial antibodies for label-free plasmonic biosensing. Journal of Materials Chemistry B 2, 167–170 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Abadeer NS, Brennan MR, Wilson WL & Murphy CJ Distance and plasmon wavelength dependent fluorescence of molecules bound to silica-coated gold nanorods. ACS nano 8, 8392–8406 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Mishra H, Mali BL, Karolin J, Dragan AI & Geddes CD Experimental and theoretical study of the distance dependence of metal-enhanced fluorescence, phosphorescence and delayed fluorescence in a single system. Physical Chemistry Chemical Physics 15, 19538–19544 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Yan B et al. Engineered SERS substrates with multiscale signal enhancement: nanoparticle cluster arrays. Acs Nano 3, 1190–1202 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Pierre MCS & Haes AJ Purification implications on SERS activity of silica coated gold nanospheres. Analytical chemistry 84, 7906–7911 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Bardhan R, Grady NK & Halas NJ Nanoscale Control of Near-Infrared Fluorescence Enhancement Using Au Nanoshells. Small 4, 1716–1722 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Thompson DK, Huffman KM, Kraus WE & Kraus VB Critical appraisal of four IL-6 immunoassays. PloS one 7, e30659 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samant PP & Prausnitz MR Mechanisms of sampling interstitial fluid from skin using a microneedle patch. Proceedings of the National Academy of Sciences 115, 4583–4588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mishra J et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. The Lancet 365, 1231–1238 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Han W et al. Urinary biomarkers in the early diagnosis of acute kidney injury. Kidney international 73, 863–869 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munier M et al. Physicochemical factors affecting the stability of two pigments: R-phycoerythrin of Grateloupia turuturu and B-phycoerythrin of Porphyridium cruentum. Food chemistry 150, 400–407 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Yuan H, Khatua S, Zijlstra P, Yorulmaz M & Orrit M Thousand-fold Enhancement of Single-Molecule Fluorescence Near a Single Gold Nanorod. Angewandte Chemie International Edition 52, 1217–1221 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Ma Y et al. Au@ Ag core− shell nanocubes with finely tuned and well-controlled sizes, shell thicknesses, and optical properties. ACS nano 4, 6725–6734 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu K-K, Tadepalli S, Tian L & Singamaneni S Size-dependent surface enhanced Raman scattering activity of plasmonic nanorattles. Chemistry of Materials 27, 5261–5270 (2015). [Google Scholar]
- 60.Dixit CK & Aguirre GR Protein microarrays with novel microfluidic methods: current advances. Microarrays 3, 180–202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jain PK, Lee KS, El-Sayed IH & El-Sayed MA Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine. The journal of physical chemistry B 110, 7238–7248 (2006). [DOI] [PubMed] [Google Scholar]
- 62.De La Rica R & Stevens MM Plasmonic ELISA for the ultrasensitive detection of disease biomarkers with the naked eye. Nature nanotechnology 7, 821 (2012). [DOI] [PubMed] [Google Scholar]
- 63.Deng W, Xie F, Baltar HT & Goldys EM Metal-enhanced fluorescence in the life sciences: here, now and beyond. Physical Chemistry Chemical Physics 15, 15695–15708 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Lee K-S & El-Sayed MA Dependence of the enhanced optical scattering efficiency relative to that of absorption for gold metal nanorods on aspect ratio, size, end-cap shape, and medium refractive index. The Journal of Physical Chemistry B 109, 20331–20338 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Gole A & Murphy CJ Azide-derivatized gold nanorods: functional materials for “click” chemistry. Langmuir 24, 266–272 (2008). [DOI] [PubMed] [Google Scholar]
- 66.Tebbe M, Kuttner C, nnel M, Fery A & Chanana M Colloidally stable and surfactant-free protein-coated gold nanorods in biological media. ACS applied materials & interfaces 7, 5984–5991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nedrebø T, Reed RK, Jonsson R, Berg A & Wiig H Differential cytokine response in interstitial fluid in skin and serum during experimental inflammation in rats. The Journal of physiology 556, 193–202 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.