

Abstract
Multiple myeloma (MM) is an incurable refractory hematological malignancy arising from plasma cells in the bone marrow. Here we investigated miR-26a function in MM and tested single-wall carbon nanotube delivery of miR-26a in vitro and in vivo. miR-26a was downregulated in patient MM cells compared with plasma cells from healthy donors. miR-26a overexpression inhibited proliferation and migration and induced apoptosis in MM cell lines. To identify the targets of miR-26a, RPMI8226-V-miR-26-GFP and RPMI8226-V-GFP cells were cultured using Stable isotope labeling by amino acids in cell culture (SILAC) medium followed by mass spectrometry analysis. In MM cells overexpressing miR-26a, CD38 protein was downregulated and subsequently confirmed to be a direct target of miR-26a. Depletion of CD38 in MM cells duplicated the MM inhibition observed with exogenous expression of miR-26a, whereas restoration of CD38 overcame the inhibition of miR-26a in MM cells. In a human MM xenograft mouse model, overexpression of miR-26a inhibited CD38 expression, provoked cell apoptosis, and inhibited cell proliferation. Daratumumab is the first CD38 antibody drug for monotherapy and combination therapy for MM patients, but eventually resistance develops. In MM cells, CD38 remained at low level during daratumumab treatment, but a high quality response is sustained. In daratumumab-resistant MM cells, CD38 expression was completely restored but failed to correlate with daratumummab-induced cell death. Therefore, a therapeutic strategy to confer selection pressure to maintain low CD38 expression in MM cells may have clinical benefit.
Keywords: miR-26a, multiple myeloma, CD38
Introduction
Multiple myeloma (MM), a cancer of terminally differentiated post-germinal center B cells or plasma cells, is the second most frequently diagnosed hematologic cancer in the US (1). Despite recent advances in treatment, including proteasome inhibitors, immunomodulatory drugs, and CD38 monoclonal antibody therapies, patient survival has increased by only about 1.5 years, from 3.5 years to 5.5 years; further, it remains incurable due to frequent relapse and emergence of drug resistance. Fully understanding the mechanisms of MM development, progression, and drug resistance are essential to developing new treatments that will improve patient outcome.
The microRNAs (miRNAs) are short non-coding RNAs that suppress the expression of protein-coding genes by partial complementary binding, particularly to the 3’-untranslated regions of messenger RNAs (mRNAs). Disturbance of miRNA expression is involved in the tumorigenesis and metastasis of human cancers(2,3), including MM (4,5). Studies of the MM patient miRNA signature have consistently shown that several miRNAs are commonly upregulated in MM, such as miR-21, miR-221/222 and miR-181a/b, but that others are downregulated, such as miR-30s and miR-15/16 (6–8). Functionally, miR-21 has been reported to promote proliferation by inhibiting PTEN (9), and miR-30s represses MM growth via inhibiting Wnt/BCL9 (10). In addition, miRNAs can be therapeutic targets for MM — for instance, inhibitors of miR-34a and miR-21 reduced MM proliferation in vitro and in vivo (10–12).
The human miR-26 family is composed of 3 members, miR-26a-1, miR-26a-2 and miR-26b, which are located on chromosomes 3, 12, and 2, respectively. miR-26a-1 and miR-26a-2 have identical sequences, which differ by two nucleotides from miR-26b. miR-26a is usually dysregulated in cancer: it induces cell cycle arrest through downregulating CCND2 and CCNE2 in hepatocellular cancer (13); functions as a tumor suppressor in breast carcinogenesis by repressing MTDH and EZH2 (14); and in leukemia, inhibits proliferation, migration, invasion, angiogenesis, and metabolism through targeting EZH2, CDK6, and Mcl1(15,16). miR-26a delivered via adeno-associated virus suppresses proliferation and promotes apoptosis in xenograft mouse models, suggesting its potential clinical use (13).
In this study, we first investigated the function of miR-26a in cell proliferation and apoptosis in MM and identified CD38 as its direct target in vitro and in vivo. Furthermore by knocking down and then restoring CD38 expresssion, we verified that miR-26a performed its function directly by targeting CD38 and observed a synergistic effect between miR-26a and bortezomib or melphalan treatments, providing proof-of-concept support for systemic delivery of miR-26a as a specific anti-MM treatment.
Materials and Methods
miR-26 expression profiling data analysis
We downloaded the GSE16558 dataset(17), and normalized expression levels of miR-26a and miR-26b to the mean Ct value of RNU44 and RNU48, which are consistently expressed across the dataset. Relative quantification of miR-26 expression was calculated with the 2−ΔΔCt method, where ΔCt = Ct(miR-26) − Ct((RNU44+RNU48)/2) and ΔΔCt = ΔCt(MM) − ΔCt(average normal PC)(18).
Patient tissue preparation and cell line culture
Bone marrow, lymph node, and peripheral blood specimens were obtained from healthy donors and from MM and CLL patients in accordance with Cleveland Clinic Foundation Institutional Review Board approval, and written informed consent was obtained in compliance with the Declaration of Helsinki. Normal plasma cells were purified using CD138 magnetic beads (Miltenyi Biotec) as previously described (19). The MM cell lines H929, MM.1S, RPMI8226, U266, and the 293T cell line were purchased from ATCC. OPM1, OPM2, and KMS26 were kindly provided by Dr. Teru Hideshima at the Dana-Farber Cancer Institute. All cell lines were routinely genotyped using the Human Cell Line Genotyping System (Promega) and mycoplasma tested. Myeloma cells were cultured in RPMI-1640 medium, and the 293T cells were cultured in DMEM medium, supplemented with 10% fetal bovine serum in 5% CO2 in a humidified incubator at 37°C.
Primary human B cell purification and in vitro differentiation assay.
Peripheral blood mononuclear cells were isolated from healthy volunteers, and the naïve B cells were purified by negative selection using magnetic cell separation by using the Naive B Cell Isolation Kit II (Miltenyi Biotec) with anti-CD10 antibodies (Miltenyi Biotec). The purity of the isolated CD19+ CD27− naïve B cell population was routinely >95%. The cells were labeled with 1 μM CFSE (Invitrogen) in serum-free medium at 37 °C for 10 min and washed in complete medium so as to monitor cellular division. Purified naive B cells were cultured at 7.5 × 105 cells/mL in 24-well plates and stimulated during 4 days with 2.6 μg/ml F(ab’)2 fragment goat anti-human IgA+IgG+IgM (H+L) (Jackson ImmunoResearch Laboratories), 100 ng/ml recombinant human soluble CD40L (Millipore), 1.0 mg/ml CpG oligodeoxynucleotide 2006 (Invivogen), and 50 U/ml recombinant IL-2 (R&D Systems). Day 4-activated B cells were washed and cultured at 4 × 105 cells/ml for up to 3 days with 50 U/ml IL-2, 50 ng/ml IL-6, 50 ng/ml IL-10, and 2 ng/ml IL-12 (R&D Systems). At day 7 of culture, cells were washed and cultured with 50 ng/ml IL-6, 10 ng/ml IL-15 and 500U/ml IFN-α for 3 days. The cells were subjected to flow cytometry to analyze cell surface CD38, CD138. QRT-PCR was used to examine the expression of BLIMP1 mRNA and miR-26a.
Lentivirus packaging, infection, and transient transfection
Lentivirus packaging and infection of MM cells were performed using pPACKH1 HIV Lentivector Packaging kit according to the manufacturer’s protocol (System Biosciences). MiR-26a over-expression or control plasmid (System Biosciences) was transfected with packaging plasmids into 293T cells using Lipofectamine 2000 (Thermo Fisher Scientific), which were labeled as V-miR-26a-GFP and V-GFP, respectively. Lentiviral particles were then collected from the culture supernatant at 48 h intervals and filtered with 0.22 μm filters. To obtain stable cell lines overexpressing miR-26a, MM cells were infected with recombinant lentivirus in the presence of polybrene overnight; GFP-positive cells were sorted by flow cytometry 4 days after infection. The expression levels of miR-26a in stable clones and transiently transduced cells were verified with real-time reverse transcriptase quantitative PCR (qRT-PCR). CD38 was knocked down using specific siRNA (Santa Cruz Biotechnology), and protein levels were detected by western blot.
To obtain CD38 overexpression vector, full length of CD38 ORF was cloned into pCDH-MSCV-MCS-EF1-copGFP-T2A-Puro plasmid (System Biosciences) between Xba I and Not I sites. Primer sequences were:
CD38-Xba-F: TTATCTAGATGGCCAACTGCGAGTTCAGCC
CD38-Not-R: ATAAGAATGCGGCCGCTCAGATCTCAGATGTGCAAGATGAATCCTC
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
qRT-PCR analysis was used to determine the relative expression levels of miR-26a and its downstream mRNAs. Total RNA was extracted from cultured cells or healthy donor samples using TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions. For miRNA analysis, cDNA was synthesized using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific) and then amplified with TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) together with miR-26a-5p primer (Thermo Fisher Scientific). RNU44 was used as internal control for miR-26a expression. For CD38 and Blimp1 mRNA quantification, cDNA was synthesized using the High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) and then amplified using SYBR Green PCR Master Mix (Thermo Fisher Scientific) with gene-specific primers. GAPDH was used as the internal control. Gene relative expression was calculated with the 2−ΔΔCt method. Primer sequences were:
CD38-F, CTCAATGGATCCCGCAGTAAA
CD38-R, ATGTATCACCCAGGCCTCTA
Blimp1-F, TGTGGTATTGTCGGGACTTTG
Blimp1-R, TCAGTGCTCGGTTGCTTTAG
GAPDH-F, GGTGTGAACCATGAGAAGTATGA
GAPDH-R, GAGTCCTTCCACGATACCAAAG
In situ hybridization
Slides were deparaffinized and rehydrated through immersion in xylene and an ethanol gradient and then digested with 20 μg/mL proteinase K in pre-warmed 50 mM Tris for 20 min at 37°C. After fixation in 4% paraformaldehyde for 5 min at room temperature, slides were dehydrated by immersion in an ethanol gradient and air drying; slides were pre-hybridized using DIG Easy Hyb (Roche, Mannheim, Germany) at 50℃ for 1h. The 10 pmol digoxin-labeled miR-26a locked nucleic acid (LNA) probe (5’-AGCCTATCCTGGATTACTTGAA-3’) was denatured in hybridization buffer at 95°C for 2 min and then chilled on ice. The probe was diluted in 250 μL pre-warmed hybridization buffer. Each sample was covered with 50 to 100 μL diluted probe and incubated in a humidified hybridization chamber at 50°C overnight. Slides were washed twice in 50% formamide in 4×SSC at 37°C for 30 min, and then washed three times in 2×SSC at 37°C for 15 min. After washing twice with maleic acid buffer containing Tween-20 (MABT), slides were blocked using blocking buffer (Roche) at room temperature for 30 min, the blocking buffer was drained off, and the samples were incubated with 1:250 diluted anti-digoxigenin-AP fab fragments (Roche) at 37°C for 1h. After washing twice in MABT and once in detection solution (0.1M Tris-HCl, 0.1M NaCl, pH9.5), the slides were stained with freshly diluted NBT/BCIP detection solution (Roche) and incubated at 37°C for 30 min. Slides were washed in PBS twice, air dried for 30 min, and then mounted with Eukitt quick-hardening mounting medium (Sigma Aldrich, St. Louis, MO, USA). Images were obtained under phase-contrast microscopy (Leica DM2000 LED) and a digital camera (Leica DMC 2900).
Immunohistochemistry
Formalin-fixed paraffin-embedded sections were deparaffinized and then incubated with rabbit anti-CD38 polyclonal antibody (Cell Signaling Technology, #14637S) or rabbit anti-Ki-67 polyclonal antibody (Cell Signaling Technology, #9027S) or rabbit anti-cleaved caspase-3 monoblonal antibody (Cell Signaling Technology, #9664S) at 4℃ overnight. After incubation with HRP-conjugated goat anti-rabbit secondary antibody, signal was detected using a DAB Substrate kit (Abcam, ab64238) following the manufacturer’s instructions. Images were obtained using phase contrast microscopy (Leica DM2000 LED) and a digital camera (Leica DMC 2900).
Cell viability assay
To determine the effect of miR-26a on MM proliferation, 5×103 V-miR-26a-GFP/V-GFP-infected RPMI8226, MM.1S, and H929 cells were seeded in 96-well plates and incubated with bortezomib in DMSO (0, 4, or 8 nM for RPMI8226; and 0, 2, or 4 nM for MM.1S and H929), or melphalan in DMSO (0, 10 or 20 μM). Cell viability was evaluated at 24 h intervals using the CellTiter-GloLuminescent Cell Viability Assay Kit (Promega) according to the manufacturer’s protocol. All treatments and measurements were performed in 3 independent experiments with 4 replicates of each cell line.
Cell apoptosis analysis
RPMI8226, MM.1S, and H929 cells infected with V-miR-26a-GFP/ V-GFP cells were cultured in 6-well plates (5×105 cells/well); in experiments to test for synergy, cells were also exposed to bortezomib or melphalan for 48 h. To quantify apoptosis, cells were washed with PBS and stained with annexin V–APC and propidium iodide (Biolegend). Stained cells were analyzed by flow cytometry (BD FACSCalibur), 10,000 cells were recorded, and the data were statistically evaluated using FlowJo software OSX 10.6 (Tree Star, Inc.). Some cells were harvested for Western blot analysis to detect expression of cleaved PARP1 and caspase3 (c-PARP1 and c-caspase3).
For CD38 knockdown experiments, RPMI8226, MM.1S, and H929 cells were transfected with 50 nM CD38 siRNA or control siRNA (Santa Cruz) for 5 days. Cells were stained with annexin V–FITC and propidium iodide (Biolegend) and then subjected to flow cytometry (BD FACSCalibur) to analyze cell apoptosis.
Transwell cell migration assay
For migration studies, a transwell assay was conducted in 24-well plates with 8-μm pore inserts (Corning, Inc.). The bottom chambers were filled with culture medium containing 10% serum. Then 3×104 RMPI8226-V-miR-26a-GFP/V-GFP, MM.1S-V-miR-26a-GFP/V-GFP, or H929-V-miR-26a-GFP/V-GFP cells were plated onto the upper chambers with serum-free medium. Monitoring of migration was initiated immediately using an IncuCyte ZOOM system (Essen Bioscience); the bottom transwell chambers were imaged every 2 h for a total of 9 h. The scanned images were analyzed using Essenbio software (version 2016B).
Western Blot
Total protein samples were isolated with RIPA buffer, separated on 4% to 12% Bis-Tris polyacrylamide gels (Thermo Fisher Scientific), and electroblotted onto nitrocellulose membranes. Membranes were blocked with 5% non-fat milk and incubated overnight with rabbit anti-PARP1 antibody (9532S, Cell Signaling), rabbit anti-caspase3 antibody (9662, Cell Signaling), rabbit anti-CD38 antibody (14637, Cell Signaling), or actin-HRP (sc-1615, Santa Cruz Biotechnology), as appropriate. Then, membranes were washed and incubated with HRP-linked anti-rabbit IgG secondary antibody (7074, Cell Signaling) or HRP-linked anti-mouse IgG secondary antibody (7076S, Cell Signaling).
Stable isotope labeling with amino acids in cell culture (SILAC)
SILAC experiments were performed using the Pierce SILAC Protein Quantitation Kit (Thermo Fisher Scientific). Briefly, RPMI8226-V-miR-26a-GFP cells were cultured in heavy SILAC medium (100 mg/L 13C6 L-lysine-HCl and 100 mg/L L-arginine-HCl), whereas the RPMI8226-V-GFP cells were cultured in light medium (100 mg/L L-lysine-HCl and 100 mg/L L-arginine-HCl). After 6 doublings, cells were lysed with RIPA buffer. After incorporation efficiency was confirmed by mass spectrometry, 20 μg protein from each cell lysate was mixed in a 1:1 (heavy:light) ratio, electrophoresed on 4% to 12% Bis-Tris polyacrylamide gels, and stained with Coomassie Brilliant Blue for band visualization. Protein bands were cut out and digested with trypsin followed by liquid chromatography–mass spectrometry (LC-MS) analysis performed in the Proteomics and Metabolomics Core at the Cleveland Clinic Lerner Research Institute. Proteins were identified if their detection probability was greater than 99.9% and at least 2 specific peptides for 1 protein could be identified.
3′-UTR luciferase reporter assays
To construct the CD38 reporter vector, a 25-bp segment of the CD38 mRNA 3’UTR that contained the predicted miR-26a binding site was cloned into pmirGLO vector after the firefly luciferase coding sequence. For the dual luciferase assay, the CD38 reporter was transiently transfected into HEK293T cells with miR-26a overexpression (pCDH-CMV-MCS-EF1-copGFP-miR26a) or knockdown vector (pmiRZIP-26a). The cells were lysed, and luciferase activity was detected 48 h after transfection. Results are presented as mean ± SD of 3 independent experiments.
Multiple myeloma mouse xenograft model
A total of 5×106 RMPI8226-V-26a-GFP/V-GFP or MM.1S-V-26a-GFP/V-GFP cells in 100 μL PBS together with an equal volume of Matrigel basement membrane matrix (BD Bioscience) was subcutaneously injected into the shoulder of Fox Chase SCID beige mice (strain code: 250, Charles River) to establish a human MM xenograft model. The two orthogonal diameters (x and y) of tumors were monitored once a week, and xenograft volume was calculated as V=xy2/2. Mice were sacrificed at 28 days post-injection. RNA was isolated from xenograft determine miR-26a level, protein was extracted for CD38 expression, and remaining tissue was paraffin-embedded for immunohistochemistry analysis (Ki-67 and c-caspase3).
Functionalization of purified single-wall carbon nanotubes
(6,5)-enriched single-wall carbon nanotubes (SWCNTs) were purified by a polymer aqueous two-phase separation method using the DNA sequence: T(TA)2T4(AT)2T(20). (6,5) SWCNTs were mixed with 10 mg PL-PEG2000-NH2 (Avanti Polar Lipids, 880128P) in 5 ml double-distilled water in a glass scintillation vial. The vial was sonicated in a bath sonicator (97043–992, VWR) for 1 h at room temperature with water changes every 20 min to avoid overheating. The SWCNT suspension was centrifuged at 24,000g for 6 h at room temperature, and the supernatant was collected. The SWCNT supernatant, 1 mL, was washed 5 times, by adding 1 mL SWCNT supernatant to a 4 mL centrifugal filter (Amicon; MilliporeSigma, UFC910008) and 33 mL double-distilled water, and centrifuging for 10 min, 4,000g, room temperature. After the final wash, the SWCNT concentration was measured using a UV/VIS spectrometer (Thermo Fisher Scientific, accuSkan GO UV/Vis Microplate Spectrophotometer) with an extinction coefficient of 0.0465L/mg/cm at 808 nm. The SWCNT concentration was adjusted to ~ 50mg/L by adding the required amount of double distilled water.
Conjugation of miR-26a to SWCNTs through cleavable disulfide bond
Functionalized SWCNTs, 500 μL, were mixed with 0.5 mg of Sulfo-LC-SPDP (c1118, ProteoChem). 50 μL of 10X PBS was added and incubated for 2 hours at room temperature. After incubation, the SWCNT solution was washed 5–6 times using a centrifugal filter (Amicon) by adding 3 to 4 mL DNase/RNase-free water and centrifuging for 6 to 8 min at 10,000g each time. 15 μL miR-26a (100 μM) was mixed with 1.5 μL DTT solution (Sigma, #43815), incubated for 1.5 hours at room temperature, and then DTT-treated miR-26a was purified using a NAP-5 column (GE Healthcare, 17-0853-01) following the manufacturer’s protocol. 500 μL miR-26a was eluted and collected from the column with DNase/RNase free 1X PBS. The activated SWCNTs were suspended with the 500 μL purified miR-26a solution, and the conjugation was allowed to proceed for 24 h at 4°C.
Delivery of SWCNT-miR-26a to disseminated MM mouse model
A murine disseminated model of human MM cells were established in 8-week-old female NOD.CB17-Prkdcscid/J mice (Charles River). All mice were irradiated and then intravenously injected with 5×106 MM.1S-Luc-GFP cells and were randomized to separate to control and treatment groups. Mice were subsequently injected with 100 uL (40mg/mL) SWCNT-miR-26a or SWCNT-ctrl, or bortezomib (0.5mg/kg) plus SWCNT-ctrl, or bortezomib (0.5 mg/kg) plus SWCNT-miR-26a once a week through the tail veins in a masked fashion, then observed daily and sacrificed once mice developed hind limb paralysis (endpoint). Images were acquired using an in vivo imaging system (IVIS) (PerkinElmer). Hind limb paralysis was used as the end point in this disseminated disease model. All experiments involving animals were pre-approved by the Cleveland Clinic IACUC(Institutional Animal Care and Use Committtee).
Statistical analyses
Statistical analysis was performed using SPSS (version 17.0). Comparisons between two independent groups were performed using a two-tailed Student’s t-test. In our mouse model, time to our endpoint of hind limb paralysis was measured using the Kaplan–Meier method, with Cox proportional hazard regression analysis for group comparisons. P ≤ 0.05 was considered as statistically significant. Correlation analysis was perfomred using the Pearson correlation test; R2 > 0.3 was considered as positive. Isobologram analysis was performed using the CompuSyn software program (ComboSyn, Inc. Paramus, NJ, USA). A combination index (CI) less than 1.0 indicates synergism, and a CI of 1 indicates additive activity (21,22).
Results
MiR-26a inhibited cell proliferation and migration and induced apoptosis in MM
Analysis of the GSE16558 dataset (60 MM patients and 5 healthy donors) (17) revealed that miR-26a, but not miR-26b, expression was significantly down-regulated in MM patients compared with healthy donors (Fig. 1A). We confirmed this result in CD138+ plasma cells from the healthy donors and MM cell lines (Fig. 1B). As post-transcriptional regulators, miRNAs may inhibit protein expression without influencing mRNA level (23,24). Thus, to identify the downstream targets of miR-26a in MM, we performed SILAC combined with LC-MS instead of mRNA microarray analysis to uncover all proteins regulated by miR-26a. RPMI8226-V-miR-26a-GFP and RPMI8226-V-GFP cells were cultured in heavy or light medium separately using SILAC followed by protein separation and MS-LC. A total of 2,724 unique proteins were recognized, of which 180 were up-regulated (68 proteins) or down-regulated (112 proteins) (Table S1). Because miRNAs are negative regulators of gene expression, we further screened the 112 down-regulated proteins using web-based query tools (TargetScan Release 7.1 and miRBase), and identified CD38 (H/L = 0.49, P = 0.02) as potential target of miR-26a (Fig. 1C).
Figure 1. MiR-26a was down-regulated in MM, and targeted CD38 in MM.
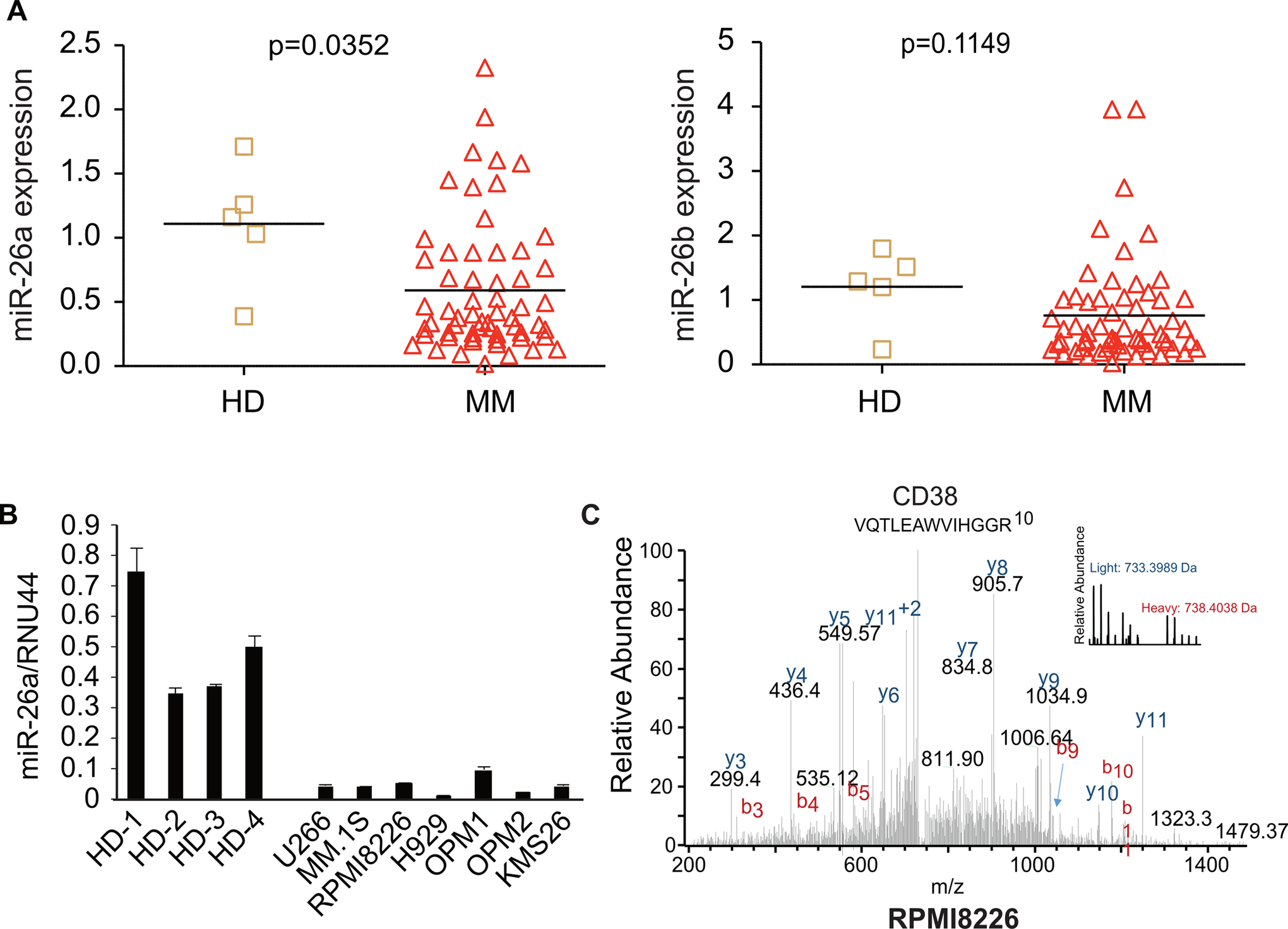
A. Analysis of the GSE16558 dataset showed that miR-26a was down-regulated in MM patients (n = 60) compared with healthy donors (HD, n = 5) (left panel, p = 0.04), whereas miR-26b was not (right panel, p = 0.12). B. Expression of miR-26a in 7 MM cell lines was determined by qRT-PCR; plasma cells from 4 healthy donors were used as control. C. Histogram analysis of protein expression ratios for all 2724 proteins identified and quantified after RPMI8226 cells were transduced with V-miR-26a-GFP/V-GFP, cultured using SILAC, and analyzed by LC-MS. Proteins were binned into groups based on ln value (heavy/light ratio).
To investigate the function of miR-26a in MM, we overexpressed miR-26a in RPMI8226 (Fig. 2A), MM.1S (Fig. 2B), and H929 (Fig. 2C) cells by infecting them with V-miR-26a-GFP/V-GFP. The overexpression of miR-26a was confirmed by qRT-PCR. To determine the influence of miR-26a on cell growth, proliferation was assessed in these stable cell lines. Results showed that overexpressed miR-26a slowed proliferation significantly. To assess the effect of miR-26a on migration, a transwell migration assay was performed using the IncuCyte live cell analysis system, which showed that MM migration was inhibited by overexpressed miR-26a in both RPMI8226 (Fig. 2A), MM.1S (Fig. 2B), and H929 (Fig. 2C) lines. To uncover the apoptotic status of cells with miR-26a overexpression, we performed western blots for PARP1/c-PARP1 and caspase3/c-caspase3, two proteins that reflect apoptotic status, and found miR-26a induced greater expression of c-PARP1 and c-caspase3 in RPMI8226, MM.1S, and H929 cells (Fig. 2A–C). Furthermore, a reporter assay demonstrated that miR-26a directly regulated expression of CD38 mRNA through binding to its 3’UTR (Fig. 2D). In addition, overexpressed miR-26a inhibited CD38 protein expression in RPMI8226, MM.1S, and H929 cells (Fig. 2D).
Figure 2. Overexpressed miR-26a suppressed proliferation and migration and induced apoptosis in multiple myeloma cell lines.
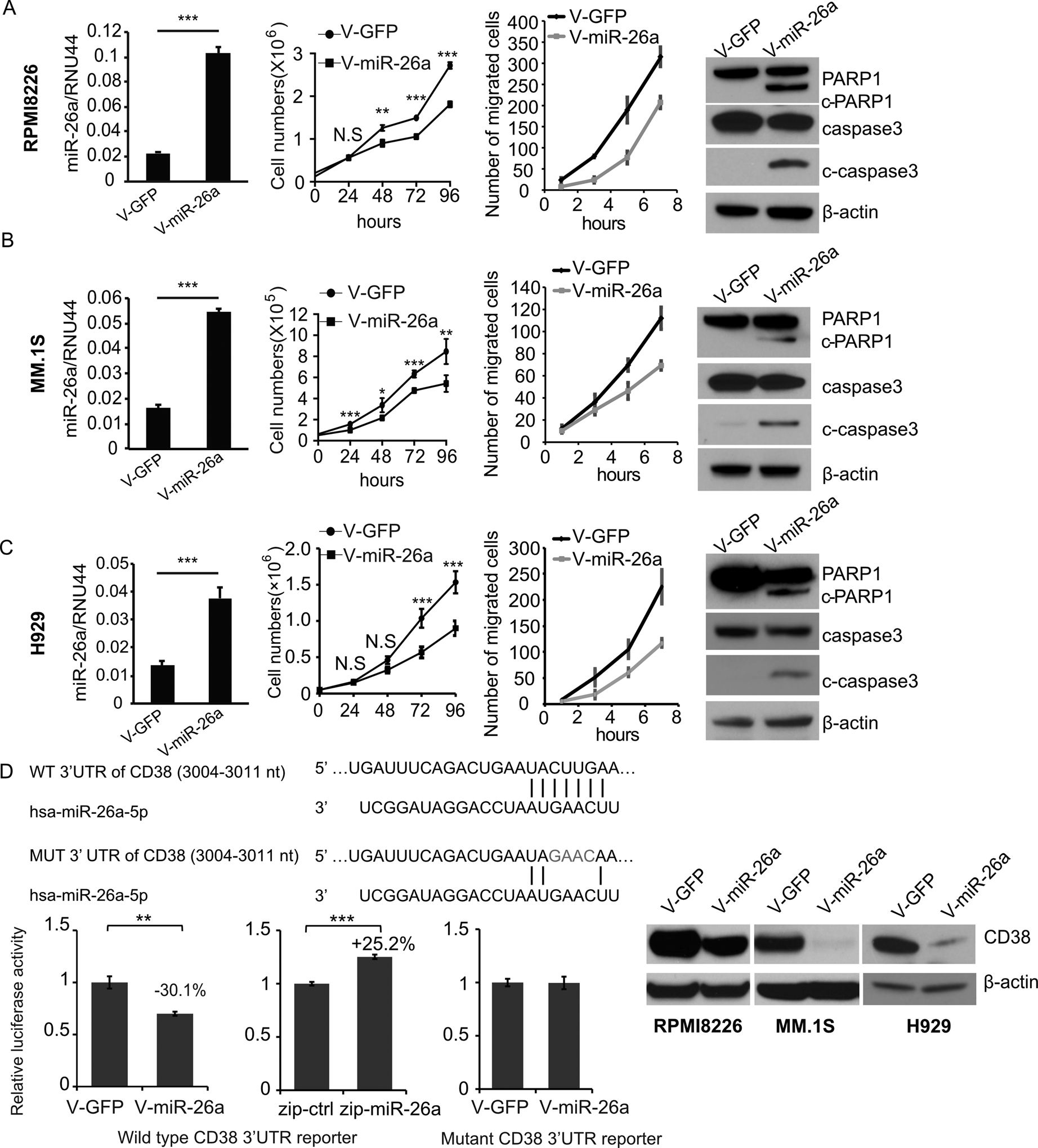
The expression of miR-26a in RPMI8226 (A), MM.1S (B), and H929 (C) cells stably transduced with V-miR-26a-GFP or V-GFP was determined by qRT-PCR; RNU44 was the internal control. The effect of miR-26a on MM cell proliferation and migration was investigated. Expression of PARP1, c-PARP1, caspase3 and c-caspase3 in RPMI8226 and MM.1S cells transduced with V-miR-26a-GFP or V-GFP were determined by western blot. D. Predicted miR-26a target sequence on the 3’UTR of CD38. To interrupt the binding between the 3’UTR of CD38 and seed sequence of miR-26a, 4 nucleotides on the predicted region of the CD38 3’UTR were changed to the complementary sequence. A luciferase reporter assay was performed in HEK293T cells transduced with V-miR-26a-GFP or V-GFP. Expression of CD38 in RPMI8226, MM.1S, and H929 cells infected with V-miR-26a-GFP or V-GFP as determined by western blot. (*p<0.05, **p<0.01, ***p<0.001)
MiR-26a directly inhibited CD38 translation in MM cells
We did not find a direct association between levels of miR-26a and CD38 mRNA in our analysis of the GSE16558 dataset (Fig. S1A, R2 = 0.126, and p = 0.0037). However, our qRT-PCR results revealed that the CD38 mRNA level increased after miR-26a overexpression in RPMI8226, but decreased in MM.1S cells, which indicated that the inhibition of CD38 protein expression caused by miR-26a is independent of its mRNA level (Fig. S1B).
To further verify whether miR-26a repressed MM cell growth through CD38 inhibition, we knocked down CD38 expression by siRNA in RPMI8226, MM.1S, and H929 cells. Five days after CD38 siRNA transfection, apoptosis was assayed (Fig. 3A) and found to be increased significantly by about 3–5 fold in CD38-knockdown cells compared to cells transfected with control siRNA, indicating that CD38 knockdown directly increased apoptosis in MM. Protein expression also decreased after CD38 knock-down (Fig. 3A). In addition, we restored CD38 expression by infecting miR-26a mimic-treated MM.1S cells with V-CD38; V-GFP was the control (Fig. 3B). Restoring CD38 expression diminished the apoptosis induced by miR-26a overexpression, indicating the suppression of cell growth and accumulation of apoptosis caused by miR-26a were mediated by CD38 inhibition.
Figure 3. CD38 knockdown induced apoptosis and reduced cell viability, and replenishing CD38 overrode the inhibition of miR-26a in MM cells.
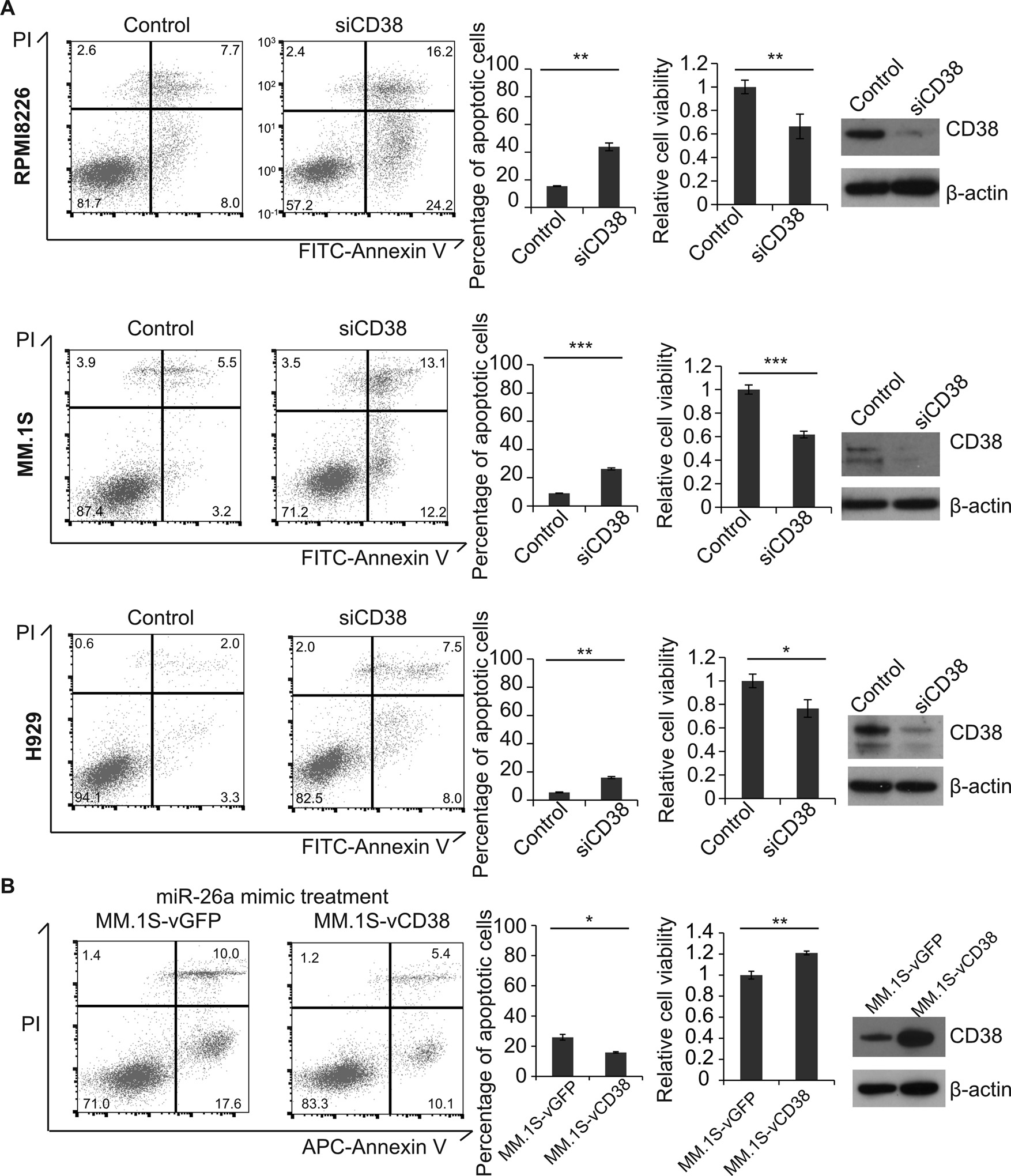
A. CD38 siRNA and control siRNA were transfected into RPMI8226, MM.1S, and H929 cells. The expression of CD38 was verified by Western blot, the population of apoptotic cells was determined by flow cytometry, and cell viability was measured by using CellTiter-GloLuminescent Cell Viability Assay Kit. B. MM.1S cells treated with miR-26a mimic were infected with V-CD38-GFP and V-GFP; cell apoptosis and viability were determined. (*p<0.05, **p<0.01)
MiR-26a synergistically enhanced the effects of bortezomib or melphalan in MM
Our results indicate that miR-26a inhibits MM growth through directly inhibiting CD38. To investigate whether miR-26a improved the therapeutic effect of bortezomib or melphalan in MM cells, RPMI8226, MM.1S, and H929 cells stably transduced with V-miR-26a-GFP and V-GFP were treated with various doses of bortezomib in DMSO or melphalan in DMSO. Cells were collected for apoptosis determination by flow cytometry and Western blot, and viability was evaluated using a CellTiter-Glo Luminescent kit. Exogenous expression of miR‑26a markedly increased bortezomib- or melphalan-induced apoptosis as measured by the percentage of annexin V-APC-positive cells (Fig. 4A, Fig. S2A) and protein levels of c-PARP1 and c-caspase3 (Fig. 4B, Fig. S2B). As shown in Fig. 4C and Fig. S2C, enforced expression of miR-26a strongly increased growth inhibition induced by bortezomib or melphalan at 24 h after treatment, and the inhibition became more pronounced over time. These findings indicate that overexpression of miR-26a in MM cells act synergistically improve the curative effect of bortezomib or melphalan (Fig. 4D, Fig. S2D).
Figure 4. MiR-26a enhanced the therapeutic effect of bortezomib in MM.
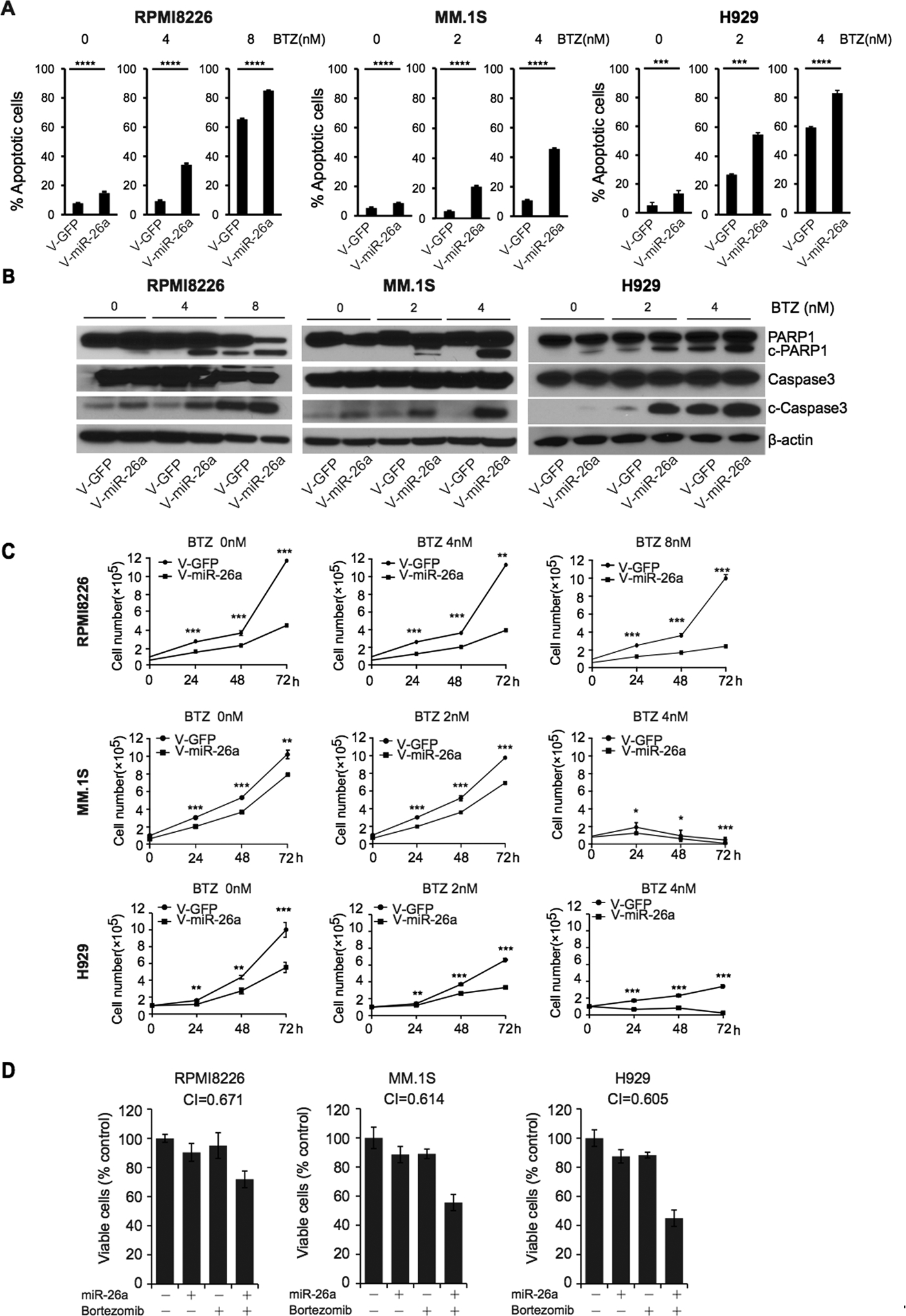
RPMI8226, MM.1S, and H929 cells transduced with V-miR-26a-GFP or V-GFP were treated with bortezomib at different doses. A. Apoptosis was analyzed by flow cytometry with annexin V B. protein levels of PARP1, c-PARP1, caspase3 and c-caspase3 were detected by western blot. Β-actin was the internal control. C. Cell viability assay showing the effect of miR-26a enforced expression on RPMI8226, MM.1S, and H929 cells with bortezomib (BTZ) or DMSO treatment. miR-26a enhanced the effect of bortezomib in a dose-dependent fashion. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001) D. RPMI8226, MM.1S, and H929 cells were transfected with 500 nM miR-26a or treated with 2 nM bortezomib or combined with miR-26a plus bortezomib and then assessed for viability using cell viability assay. Isobologram analysis was performed using the CompuSyn software program. A combination index (CI) of less than 1.0 indicates synergism.
MiR-26a decelerated MM growth in xenograft murine models
To validate our in vitro findings that a miR-26a mimic can inhibit MM, we examined its ability to suppress tumor growth in an in vivo xenograft murine model. RPMI8226 or MM.1S cells infected with V-miR-26a-GFP/V-GFP were injected subcutaneously into SCID mice. Xenograft growth of both cell lines was strikingly inhibited in the V-miR-26a mice compared with V-GFP control mice (Fig. 5A and 5B). V-miR-26a-transduced xenografts weighed significantly less than controls when they were sacrificed on post-injection day 28. RNA was extracted for miR-26a expression, tissues were fixed and embedded for immunohistochemistry for Ki-67 and c-caspase3, and protein was extracted to determine expression of CD38. miR-26a overexpression not only inhibited CD38 expression and reduced proliferation as indicated by Ki-67 staining, but also induced c-caspase3 in vivo (Fig. 5A and 5B), as we had observed in vitro, indicating that miR-26a played a tumor suppressor role in vivo through inhibiting the function of CD38, suppressing proliferation, and provoking cell apoptosis in MM.
Figure 5. MiR-26a inhibited MM growth in a xenograft mouse model.
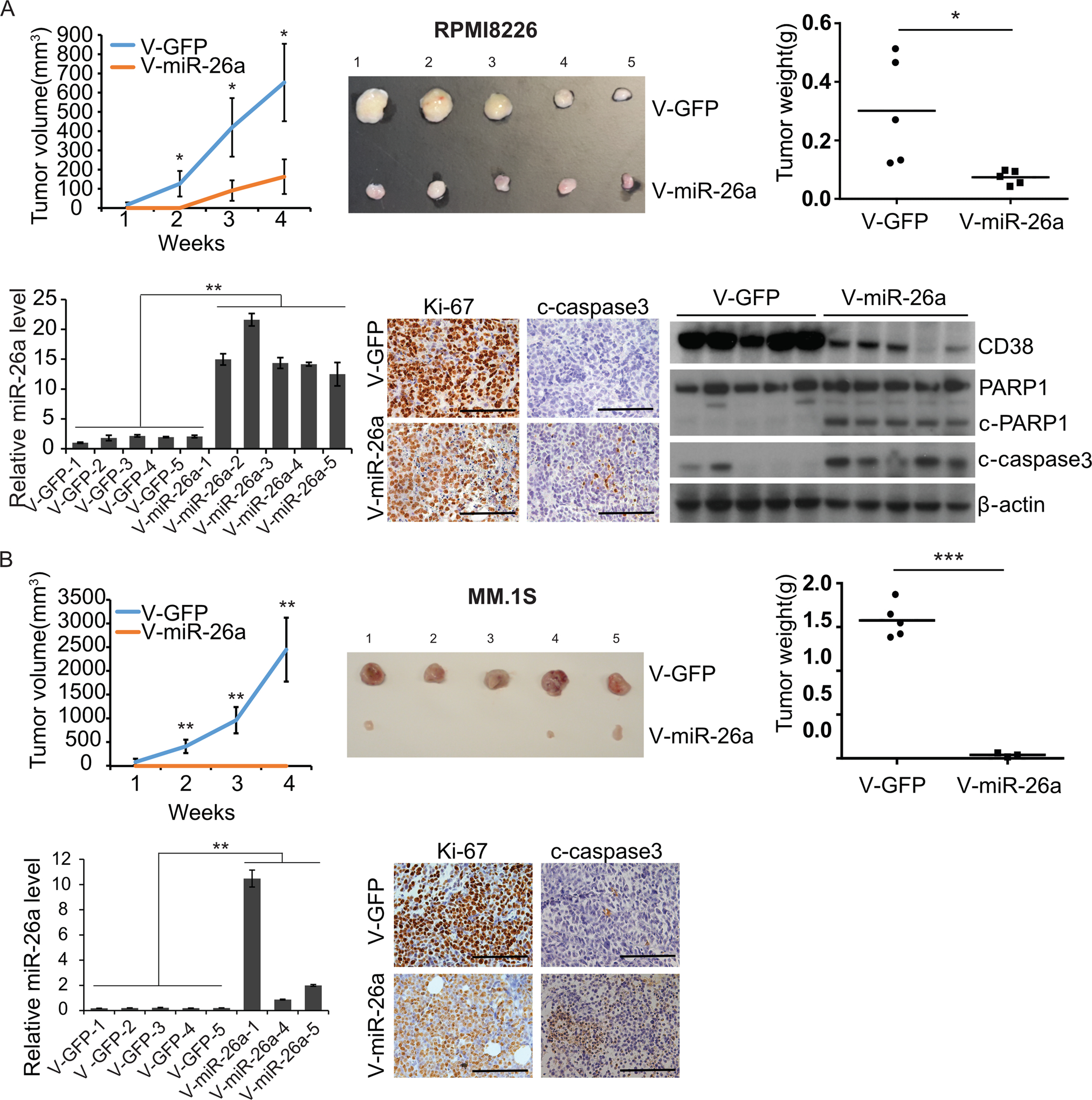
RPMI8226 (A) and MM.1S cells (B) transduced with V-miR-26a-GFP or V-GFP were injected subcutaneously in shoulders of SCID mice (5 mice/group). Xenograft growth was monitored for 4 weeks. Tumor diameter was measured once a week, and tumor volume was calculated. Mice were sacrificed on day 28 post-injection; xenografts were isolated and weighed. Expression of miR-26a in xenografts was determined by qRT-PCR; RNU44 was used as an internal control. Expression of Ki-67 and c-caspase3 was detected by immunohistochemistry. Protein levels of CD38 in RPMI8226 and MM.1S xenografts were measured by western blot; β-actin was used as control. (*p<0.05, **p<0.01, ***p<0.001)
SWCNT-miR-26a delivery prolongs the lifespan in a disseminated human MM cell mouse model
Our data indicated that miR-26a was a robust tool to provoke cell apoptosis in MM by targeting CD38. In our next step, we delivered miR-26a in vivo to mimic drug delivery in the clinical setting. We had used SWCNT to deliver antisense nucleic-acid drugs effectively and efficiently with good tolerability and minimal toxicity in vitro and in vivo (25). To test SWCNT delivery of microRNA, we conjugated SWCNTs with miR-26a (SWCNT-miR-26a)(Fig. 6A and Fig. S3)(26,27), then added the conjugated SWCNT to culture medium of H929, RPMI8226, and MM.1S cells to validate delivery efficiency. As shown in Fig. 6B, SWCNT-miR-26a was delivered into the MM cells efficiently and led to substantial overexpression of the endogenous miR-26a in RPMI8226, MM.1S, and H929 cells.
Figure 6. SWCNT-miR-26a treatment repressed myeloma growth disseminated murine models.
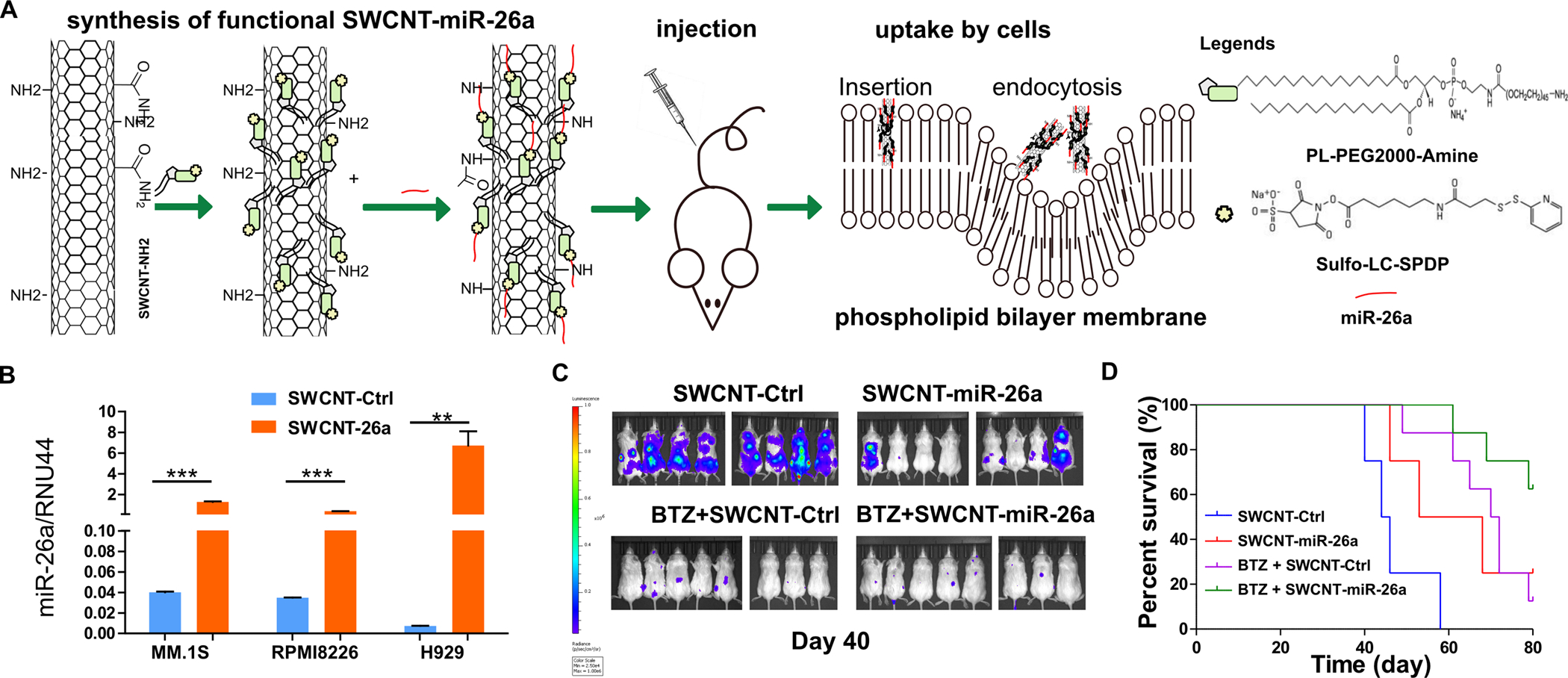
(A) The schematic diagram of SWCNT functioanlization, miR-26a conjugation, mouse model construction and miR-26a uptake processes. (B) RPMI8226, MM.1S, and H929 cells were cultured with SWCNT-ctrl or SWCNT-mir-26a for 48 hours and then were subjected to RNA extraction. The miR-26a level was determined by qRT-PCR with RUN44 as loading control. (C) SCID mice (8 mice each group) were intravenously injected with 1×106 MM.1S-Luc-GFP cells, then injected with SWCNT-miR-26a or SWCNT-ctrl or BTZ (0.5 mg/kg) plus SWCNT-ctrl, or SWCNT-miR-26a combined with BTZ (0.5 mg/kg) once a week through the tail vein. Images were acquired using an in vivo imaging system (IVIS) (PerkinElmer). Hind limb paralysis was used as the end point. (D) Time to the endpoint of hind limb paralysis was measured using the Kaplan–Meier method, with Cox proportional hazard regression analysis for group comparison. SWCNT-Ctrl vs. SWCNT-miR-26a P = 0.02; SWCNT-Ctrl vs. bortezomib, P = 0.0003; SWCNT-Ctrl vs. BTZ + SWCNT-miR-26a, P < 0.0001; BTZ + SWCNT-Ctrl vs. BTZ + SWCNT-miR-26a, P = 0.026; BTZ + SWCNT-Ctrl vs SWCNT-miR-26a, P = 0.75.
To further estimate the treatment potential of SWCNT-miR-26a in vivo, we IV injected MM.1S-Luc-GFP cells through the tail vein of SCID mice to establish a human MM cells dissemination murine model (Fig. 6A). At day 7 after tumor cell injection, SWCNT-miR-26a or SWCNT-ctrl oligos or bortezomib (0.5mg/kg) plus SWCNT-ctrl, or SWCNT-miR-26a combined with bortezomib (0.5 mg/kg) were IV injected once a week. We observed tumor burden with IVIS after luciferin injection, and found the miR-26a level was dramatically high in tumor cells isolated from SWCNT-miR-26a-treated mice compared with the cells iolated from SWCNT-ctrl-treated mice (Fig. 6B). The luciferin signal was significantly lower in SWCNT-miR-26a-treated mice compared with the SWCNT-ctrl-treated group and even further lower when combined with bortezomib treatment (Fig. 6C). We found SWCNT-miR-26a treatment extended time to hind limb paralysis significantly compared with SWCNT-ctrl treated group. We also found that SWCNT-miR-26a and bortezomib combination therapy, compared with SWCNT-miR-26a or bortezomib single treatment, dramatically prolongs the lifespan of the treated MM mice (Fig. 6D).
miR-26a regulates CD38 expression during human B cells development to plasma cells
To understand whether the control exerted by miR-26a on CD38 expression is involved in normal B-cell differentiation toward plasma cells, naïve B cells were isolated from peripheral blood obtained from healthy donors. The purified naïve B cells were differentiated to plasma cells in vitro, and then subjected to qRT-PCR to examine miR-26a expression. As shown in Fig. 7A and B, naïve B cells were successfully isolated and differentiated to CD38-positive and CD138-positive plasma cells. The miR-26a level was markedly reduced in the differentiated plasma cells (Fig. 7C). We also determined the CD38 and miR-26a levels in serial histological sections of normal human tonsil tissues, finding the miR-26a level was low in germinal center B cells and plasma cells whereas the CD38 signal was extremely high (Fig. 7D).
Figure 7. miR-26a negatively regulated CD38 during B cells differentiation to plasma cells and daratumumab resistant MM patient.
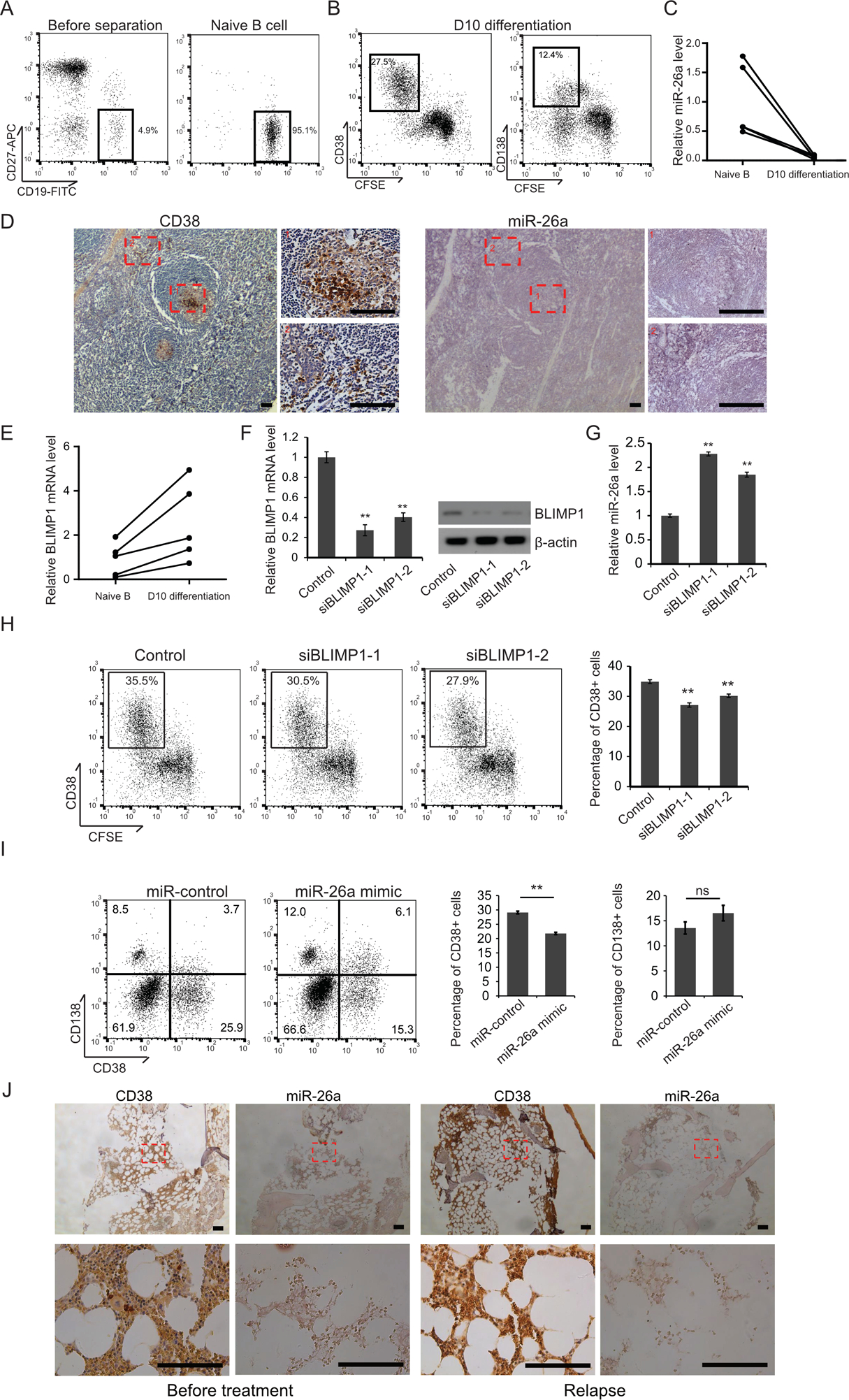
(A) Human naïve B cells were isolated from mormal donor peripheral blood, and the purity was deteremined by flowcytometry. (B) The naïve B cells were labeled by CFSE, cultured in 24-well plates and stimulated for 3 days with 2.6 μg/ml F(ab’)2 fragment goat anti-human IgA+IgG+IgM (H+L), 100 ng/ml recombinant human soluble CD40L, 1.0 mg/ml CpG oligodeoxynucleotide 2006, and 50 U/ml recombinant IL-2. Day 3 activated B cells were washed and cultured up to 4 days with 50 U/ml IL-2, 50ng/ml IL-6, 50 ng/ml IL-10, and 2 ng/ml IL-12 and then followed by a 3 days culture with 50ng/ml IL-6, 10ng/ml IL-15 and 500U/ml IFN-α. Cell surface CD38 and CD138 expression was determined by flowcytometry. (C) qRT-PCR was used to determine the expression of miR-26a. (D) The CD38 signal in human tonsil tissue samples was detected by immunohistochemistry, and the miR-26a signal was examined by in situ hybridization. Area 1: Germinal center B cells. Area 2: plasma cells. Scale bar = 100 μM. (E) BLIMP1 mRNA level in naïve B cells and day 10 differentiated B cells was examined by qRT-PCR. Naïve B cells were activated for 3 days and transfected with one of the BLIMP1 siRNAs, with seqeunce scrambled siRNA as control. The cells were differentiated for another 7 days and the cell surface CD38 expression was determined by flowcytometry (F) BLIMP1 expression was determined by qRT-PCR and immunoblotting (G) and the miR-26a level was determined by qRT-PCR (H). Naïve B cells were activated for 3 days and then transfected with miR-control or miR-26a mimic. The cells were cultured with IL-2, IL-6, IL-12 and IL-10, allowed to differnentiate for 4 days and then followed by a 3 days culture with IL-6, IL-15 and IFN-α. Cell surface CD38 and CD138 expression was determined by flowcytometry (I). (J) Bone marrow biopsies were obtained from the same MM patient before daratumumab treatment and after became daratumumab resistant. The CD38 expression was examined by IHC and miR-26a level was examined by ISH. Scale bar = 100μM.
PR domain zinc finger protein 1, also known as BLIMP1, is a key transcription factor that controls the differentiation of activated B cells into plasma cells by silencing several important B cells genes, including Pax5 (28), Myc (29) and Bcl6 (30). We examined BLIMP1 expression in day-10 differentiated plasma cells and found the mRNA level of BLIMP1 was increased compared with naïve B cells (Fig. 7E). To investigate whether BLIMP1controls the expression of miR-26a, we knocked down the BLIMP1 level with siRNA in day-3 activated B cells. After another 7 days’ differentiation, the cells were subjected to flow cytometry, qRT-PCR, and immunoblotting. As shown in Fig. 7F, BLIMP1 mRNA and protein levels were both reduced in the siRNA-treated cells compared with control siRNA-treated cells. The miR-26a level was significantly increased in BLIMP1 knockdown cells (Fig. 7G), indicating that BLIMP1 negatively regulated miR-26a expression. At the same time, the number of CD38-positive cells was significantly reduced in BLIMP1 knockdown groups (Fig. 7H), confirming that BLIMP1 potentially regulates miR-26a and CD38 during B cell differetiation. To understand whether miR-26a was functionally associated with plasma cell differentiation in human B cells, we ectopically overexpressed miR-26a in B cells that had differentiated into plasma cells. Surprisingly, we found that even though CD38 expression was inhibited in miR-26a-transfected diffentiated plasma cells, the total CD138+ plasma cell number was not reduced (Fig. 7I).
MiR-26a has been reported recently to be downregulated in patients with CLL (31) and can predict prognosis after adjustment for covariate confounders (e.g., FISH analyses, IGHV mutational status, and ZAP-70 or CD38 expression) (32). Whether miR-26a level and CD38 expression correlate is unknown. The topographic distribution of CD38 expression in CLL, which is often characterized by the formation of CD38-high hot spots within a CD38 negative milieu offer an optimal ground to investigate topographic distribution of the miR-26a expression. We first analyzed the GSE51529 microarray dataset (210 CLL patients)reported by Maura F. et al (33), and found a significant inverse correlation between miR-26a level and CD38 mRNA expression level (p=0.0004) (Fig. S4A). In situ hybridization and IHC studies formiR-26a ISH and CD38 in consecutive histological sections from 25 CLL patients lymph node revealed 5 cases with CD38 patchy positive staining and 2 cases with strong CD38-positve cell clusters. miR-26a level and CD38 expression inversely correlated in some CD38 positive cells spots on serial histological sections (Fig. S4B). The data also supported the rational of previous miR-26a delivery studies using CD38 conjugated nanoparticles in CLL (34).
Daratumumab-resistant patient MM cells express high level of CD38 and low level of miR-26a
In patients, surface CD38 level was decreased 90% in nondepleted myeloma cells after the first daratumumab infusion by selective killing, CD38 microvesicle release, and internalization of CD38-daratumumab (35). CD38 returned to basal level after about 6 months treatment indicating the development of daratumumab resistance (35). To further study CD38 and miR-26a expression after disease becomes daratumumab resistant, we examined all the multiple myeloma patients treated and followed up at the Cleveland Clinic Tausig Cancer Center from 2015–2019 and who donated tissue to our MM tissue bank. Of the 20 patients identified, 2 have experienced relapse and bone marrow from 1 of these patients was biopsied successfully. IHC revealed that the MM cell surface CD38 level was even higher in the relapsed bone marrow biopsy sample compared with pre-daratumumab treatment. At the same time, miR-26a levels were undetectable in both pre- and post- daratumumab treatment MM cells (Fig. 7J). Consistent with previous studies (35), greater CD38 expression that daratumumab no longer killed these MM cells, even when combined with other drug treatment including dexamethasone, pomalidomide, ixazomib, cafizomib, and panoblinostat.
Discussion
MM is a refractory plasma cell malignancy characterized by accumulation of antibody-secreting malignant plasma cells and osteolytic lesions in the bone marrow. The genetic heterogeneity of MM presents obstacles to treatment. New treatments, such as proteasome inhibitors and immunomodulatory agents, have increased patient survival to a limited extent, from 3.5 years to 5.5 years (36–38), and patients with relapse have few treatment choices. MM is a disease with complex and variable genomic changes. Consequently, effective treatments thus far developed, including bortezomib, lenalidomide and dexamethasone, are multiple target drugs. Therefore, we decided to focus on miRNAs, which are natural small RNAs with multiple targets. By analyzing established data sets, we found that miR-26a was markedly downregulated in MM cells. miR-26a has been verified in patient tissues as a tumor suppressor in multiple cancers, including osteosarcoma (39), lymphoma (40), colorectal cancer, (41), and others. Given our database findings, we believed miR-26a also plays a role in MM.
A type II transmembrane glycoprotein, CD38 is uniformly overexpressed on MM cells and regulates signal transduction of specific cell surface proteins (42). The CD38-specific monocolonal antibody, daratumumab (43), is FDA-approved for refractory MM and has produced impressive therapeutic effects in clinic. Daratumumab induces MM cell death mainly via antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis, but it also can induce apoptosis through acting as an ectoenzyme (44–46). We showed that knocking down CD38 induces apoptosis in MM cells; furthermore, restoring CD38 expression in MM cells overexpressing miR-26a suppressed apoptosis significantly. These results indicate that insufficient CD38 expression will cause MM cell death without the need for Fc-mediated immunoreactions. Some studies have reported that in vitro, antibody-dependent cell-mediated and complement-dependent cytotoxicity of IgG1-based mAbs may translate into only limited clinical anti-MM activity when used alone because MM patients are usually immunocompromised, especially when their disease is resistant to current treatments (47,48). However, another CD38 monoclonal antibody, isatuximab, has showed shown efficacy and tolerability as a mono- and combination therapy in Phase 1 and Phase 2 studies in patients with relapse or refractory MM; it is in Phase III clinical trials now. Isatuximab has strong proapoptotic activity via inhibiting CD38 ectoenzyme function, which is independent of CD38 inter-crosslinking (49–51).
miRNA regulates protein expression at the post-transcriptional level and so can induce cleavage of mRNA or inhibit translation without reducing mRNA level (23,24). For this reason we used SILAC to screen for downstream target screening. We are the first to report that miR-26a inhibits MM cell proliferation and migration and promotes cell apoptosis by targeting CD38 in MM. As we were preparing this manuscript, a report was published in Cancer Research that uncovered a novel CD38 function in which CD38 drives mitochondrial trafficking to promote bioenergetic plasticity in MM (52). Consistent with what these investigators found, we found that siRNA mediated knockdown of CD38 improved animal survival. Thus we believe miR-26a mimic oligonucleotide treatment will have more consistent effects in MM patients.
We also showed that miR-26a enhanced bortezomib and melphalan treatment effects in vitro and in vivo. The FDA has approved several antisense oligonucleotide drugs, including nusinersen for spinal muscular atrophy (53), mipomersen for homozygous familial hypercholesterolemia (54), fomivirsen for cytomegalovirus retinitis (55), and eteplirsen for Duchenne muscular dystrophy (56). As a natural oligonucleotide, a miR-26a mimic will have advantages because it won’t induce an immune response or deposit in organs due to inappropriate metabolism. In our mouse xenograft experiments, miR-26a inhibited growth of RPMI8226 and MM.1S xenografts as compared with V-GFP infected xenografts. In addition, CD38 protein level was inhibited significantly in miR-26a-transduced xenografts, and Ki-67 was suppressed whereas c-caspase3 was intensely induced, all of which are consistent with our in vitro results. These results indicated that the miR-26a treatment alone effectively inhibits growth of MM cells with (RPMI8226) or without (MM.1S and H929) p53 mutation (57). Thus miR-26a is potentially an effective in MM as a single treatment or in combination with standard chemotherapeutics.
All-trans retinoic acid (58), the histone deacetylase inhibitor panobinostat (59), and recently DNA methyltransferase inhibitors (60) are being used as a combination therapy to overexpression CD38 to increase the myeloma cell sensitivity to daratumumab. However, this strategy may not work after once daratumumab resistance develops because CD38 expression level is high in myeloma cells from patients with daratumumab resistance. Loss of cell surface CD38 expression on MM cells during long-term daratumumab treatment does not underly the resistance mechanism that develops. Although CD38 expression remained low during daratumumab treatment, the response was high as others have reported (35). A strategy that uses miR-26a to MM cells under pressure to maintain CD38 expression at a certain level may resensitize resistant cells to daratumumab treatment and have clinical benefit.
Supplementary Material
Significance.
These results highlight the tumor suppressor function of miR-26a via its targeting of CD38 and suggest the therapeutic potential of miR-26a in MM patients.
Acknowledgements:
The authors thank the Lerner Research Institute proteomic, genomic and imaging cores for their assistance and support and Dr. Cassandra Talerico provided editorial assistance and helpful comments. Funding: This work was financially supported by grant from: NCI R00 CA172292 (to J.Z) and start-up funds (to J.Z) and two Core Utilization Pilot Grant (to J.Z) from the Clinical and Translational Science Collaborative of Cleveland, V Foundation Scholar Award (to J.Z), National Institutes of Health training grant T32 CA094186, Training in Computational Genomic Epidemiology of Cancer (CoGEC) career development program (to J.L), 5UL1TR002548 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH roadmap for Medical Research. The Orbitrap Elite instrument was purchased via an NIH shared instrument grant, 1S10RR031537-01.
Footnotes
Competing interests: The authors report no potential conflicts of interest.
References
- 1.Kazandjian D Multiple myeloma epidemiology and survival: A unique malignancy. Semin Oncol 2016;43:676–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005;122:6–7 [DOI] [PubMed] [Google Scholar]
- 3.Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A 2004;101:11755–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Li C, Zhang R, Gao X, Qu X, Zhao M, et al. miR-17–92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett 2011;309:62–70 [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Zhou L, Shi M, Kuang Y, Fang L. Downregulation of miRNA-15a and miRNA-16 promote tumor proliferation in multiple myeloma by increasing CABIN1 expression. Oncol Lett 2018;15:1287–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Chen L, Barlogie B, Stephens O, Wu X, Williams DR, et al. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. Proc Natl Acad Sci U S A 2010;107:7904–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmad N, Haider S, Jagannathan S, Anaissie E, Driscoll JJ. MicroRNA theragnostics for the clinical management of multiple myeloma. Leukemia 2014;28:732–8 [DOI] [PubMed] [Google Scholar]
- 8.Zhao JJ, Carrasco RD. Crosstalk between microRNA30a/b/c/d/e-5p and the canonical Wnt pathway: implications for multiple myeloma therapy. Cancer Res 2014;74:5351–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lou Y, Yang X, Wang F, Cui Z, Huang Y. MicroRNA-21 promotes the cell proliferation, invasion and migration abilities in ovarian epithelial carcinomas through inhibiting the expression of PTEN protein. Int J Mol Med 2010;26:819–27 [DOI] [PubMed] [Google Scholar]
- 10.Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gulla A, et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin Cancer Res 2013;19:2096–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res 2012;18:6260–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarone MR, Misso G, Grimaldi A, Zappavigna S, Russo M, Amler E, et al. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci Rep 2017;7:17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kota J, Chivukula RR, O’Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 2009;137:1005–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B, Liu XX, He JR, Zhou CX, Guo M, He M, et al. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis 2011;32:2–9 [DOI] [PubMed] [Google Scholar]
- 15.Salvatori B, Iosue I, Djodji Damas N, Mangiavacchi A, Chiaretti S, Messina M, et al. Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer 2011;2:585–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Abundo L, Callegari E, Bresin A, Chillemi A, Elamin BK, Guerriero P, et al. Anti-leukemic activity of microRNA-26a in a chronic lymphocytic leukemia mouse model. Oncogene 2017 [DOI] [PubMed] [Google Scholar]
- 17.Gutierrez NC, Sarasquete ME, Misiewicz-Krzeminska I, Delgado M, De Las Rivas J, Ticona FV, et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010;24:629–37 [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25:402–8 [DOI] [PubMed] [Google Scholar]
- 19.Lin J, Zhang W, Zhao JJ, Kwart AH, Yang C, Ma D, et al. A clinically relevant in vivo zebrafish model of human multiple myeloma to study preclinical therapeutic efficacy. Blood 2016;128:249–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ao G, Streit JK, Fagan JA, Zheng M. Differentiating Left- and Right-Handed Carbon Nanotubes by DNA. J Am Chem Soc 2016;138:16677–85 [DOI] [PubMed] [Google Scholar]
- 21.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews 2006;58:621–81 [DOI] [PubMed] [Google Scholar]
- 22.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer research 2010;70:440–6 [DOI] [PubMed] [Google Scholar]
- 23.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 2008;9:102–14 [DOI] [PubMed] [Google Scholar]
- 24.Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 2007;8:9–22 [DOI] [PubMed] [Google Scholar]
- 25.Perrot A, Lauwers-Cances V, Corre J, Robillard N, Hulin C, Chretien ML, et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018;132:2456–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo JH, Ren B, Keryanov S, Tseng GC, Rao UN, Monga SP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology 2006;44:1012–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Lin J, Fang H, Fang J, Li C, Chen W, et al. Targeting the MALAT1/PARP1/LIG3 complex induces DNA damage and apoptosis in multiple myeloma. Leukemia 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Molecular and cellular biology 2002;22:4771–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Y, Wong K, Calame K. Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 1997;276:596–9 [DOI] [PubMed] [Google Scholar]
- 30.Shaffer AL, Lin KI, Kuo TC, Yu X, Hurt EM, Rosenwald A, et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002;17:51–62 [DOI] [PubMed] [Google Scholar]
- 31.Van Roosbroeck K, Bayraktar R, Calin S, Bloehdorn J, Dragomir MP, Okubo K, et al. The involvement of microRNA in the pathogenesis of Richter syndrome. Haematologica 2019;104:1004–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Negrini M, Cutrona G, Bassi C, Fabris S, Zagatti B, Colombo M, et al. microRNAome expression in chronic lymphocytic leukemia: comparison with normal B-cell subsets and correlations with prognostic and clinical parameters. Clin Cancer Res 2014;20:4141–53 [DOI] [PubMed] [Google Scholar]
- 33.Maura F, Cutrona G, Mosca L, Matis S, Lionetti M, Fabris S, et al. Association between gene and miRNA expression profiles and stereotyped subset #4 B-cell receptor in chronic lymphocytic leukemia. Leuk Lymphoma 2015;56:3150–8 [DOI] [PubMed] [Google Scholar]
- 34.D’Abundo L, Callegari E, Bresin A, Chillemi A, Elamin BK, Guerriero P, et al. Anti-leukemic activity of microRNA-26a in a chronic lymphocytic leukemia mouse model. Oncogene 2017;36:6617–26 [DOI] [PubMed] [Google Scholar]
- 35.van de Donk N, Usmani SZ. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front Immunol 2018;9:2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906–17 [DOI] [PubMed] [Google Scholar]
- 37.Benboubker L, Dimopoulos MA, Dispenzieri A, Catalano J, Belch AR, Cavo M, et al. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med 2014;371:906–17 [DOI] [PubMed] [Google Scholar]
- 38.Mateos MV, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N Engl J Med 2018;378:518–28 [DOI] [PubMed] [Google Scholar]
- 39.Tan X, Fan S, Wu W, Zhang Y. MicroRNA-26a inhibits osteosarcoma cell proliferation by targeting IGF-1. Bone Res 2015;3:15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao X, Lwin T, Zhang X, Huang A, Wang J, Marquez VE, et al. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013;27:2341–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez-Urrutia E, Coronel-Hernandez J, Garcia-Castillo V, Contreras-Romero C, Martinez-Gutierrez A, Estrada-Galicia D, et al. MiR-26a downregulates retinoblastoma in colorectal cancer. Tumour Biol 2017;39:1010428317695945. [DOI] [PubMed] [Google Scholar]
- 42.Zhao YJ, Lam CM, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal 2012;5:ra67. [DOI] [PubMed] [Google Scholar]
- 43.Bhatnagar V, Gormley NJ, Luo L, Shen YL, Sridhara R, Subramaniam S, et al. FDA Approval Summary: Daratumumab for Treatment of Multiple Myeloma After One Prior Therapy. Oncologist 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanchez L, Wang Y, Siegel DS, Wang ML. Daratumumab: a first-in-class CD38 monoclonal antibody for the treatment of multiple myeloma. J Hematol Oncol 2016;9:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nijhof IS, Lammerts van Bueren JJ, van Kessel B, Andre P, Morel Y, Lokhorst HM, et al. Daratumumab-mediated lysis of primary multiple myeloma cells is enhanced in combination with the human anti-KIR antibody IPH2102 and lenalidomide. Haematologica 2015;100:263–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood 2018;131:13–29 [DOI] [PubMed] [Google Scholar]
- 47.Zonder JA, Mohrbacher AF, Singhal S, van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012;120:552–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jurisic V, Srdic T, Konjevic G, Markovic O, Colovic M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med Oncol 2007;24:312–7 [DOI] [PubMed] [Google Scholar]
- 49.Richardson PG, Attal M, Campana F, Le-Guennec S, Hui AM, Risse ML, et al. Isatuximab plus pomalidomide/dexamethasone versus pomalidomide/dexamethasone in relapsed/refractory multiple myeloma: ICARIA Phase III study design. Future Oncol 2017 [DOI] [PubMed] [Google Scholar]
- 50.Jiang H, Acharya C, An G, Zhong M, Feng X, Wang L, et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016;30:399–408 [DOI] [PubMed] [Google Scholar]
- 51.Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin Cancer Res 2017;23:4290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, et al. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res 2019 [DOI] [PubMed] [Google Scholar]
- 53.Aartsma-Rus A FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther 2017;27:67–9 [DOI] [PubMed] [Google Scholar]
- 54.Smith RJ, Hiatt WR. Two new drugs for homozygous familial hypercholesterolemia: managing benefits and risks in a rare disorder. JAMA Intern Med 2013;173:1491–2 [DOI] [PubMed] [Google Scholar]
- 55.Highleyman L FDA approves fomivirsen, famciclovir, and Thalidomide. Food and Drug Administration. BETA 1998:5 [PubMed] [Google Scholar]
- 56.Nelson SF, Miceli MC. FDA Approval of Eteplirsen for Muscular Dystrophy. JAMA 2017;317:1480. [DOI] [PubMed] [Google Scholar]
- 57.Pichiorri F, Suh SS, Rocci A, De Luca L, Taccioli C, Santhanam R, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010;18:367–81 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Nijhof IS, Groen RW, Lokhorst HM, van Kessel B, Bloem AC, van Velzen J, et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015;29:2039–49 [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Guerrero E, Gogishvili T, Danhof S, Schreder M, Pallaud C, Perez-Simon JA, et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017;129:3386–8 [DOI] [PubMed] [Google Scholar]
- 60.Choudhry P, Mariano MC, Geng H, Martin TG 3rd, Wolf JL, Wong SW, et al. DNA methyltransferase inhibitors upregulate CD38 protein expression and enhance daratumumab efficacy in multiple myeloma. Leukemia 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.