

Abstract
Watermelon mosaic virus (WMV) is an important virus causing adverse effects on cucurbits throughout the world. In this study, we recorded WMV infection in the watermelon (Citrullus lanatus)-growing area of Alwar and Sikar in districts of Rajasthan, India. The RT-PCR-based detection was performed to confirm the presence of WMV, by using potyvirus-degenerated coat protein primers. Further, the complete genome sequences of two WMV isolates were compared with previously reported genome sequences. The complete genome of each isolate was 10,030 nt long, excluding the poly-A tails. Phylogeny relationships of the WMV isolates in the present study revealed the presence of uneven evolutionary pressure among the different WMV viral genomic segments. The analysis revealed that all the WMV isolates were divided into three clusters and the Indian WMV isolates cluster together with the French isolate. Recombination analysis of WMV exhibited significant recombination hotspots in the P1, NIa-Pro and Nib-CP regions. Our finding highlights the importance of genetic variability and recombination analysis to provide a better understanding of WMV molecular diversity.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02248-8) contains supplementary material, which is available to authorized users.
Keywords: Potyvirus, WMV, Phylogeny, Sequence variability, Recombination
Introduction
Watermelon, which originated in the tropical regions of the African subcontinent, is nowadays widely cultivated in hina, Iran, Turkey, Brazil and the USA. In India, the fruit constitutes an important part of the cuisine, and the area under watermelon cultivation was recorded to be 95,520 hectares with an annual production of 2,362,160 tons in the year 2016–2017 (Kumar and Kulkarni 2018). It is a major riverbed crop chiefly grown in the states of West Bengal, Uttar Pradesh, Karnataka, Orissa, Andhra Pradesh, Tamil Nadu, Rajasthan, Gujarat, Punjab, Haryana, Assam and Himachal Pradesh (Kumar and Kulkarni 2018). Watermelon is a rich source of natural antioxidants, such as lycopene, ascorbic acid and citrulline, which reduce the risk of various diseases, including cardiovascular disorders, asthma, atherosclerosis, cancer insurgence, diabetes, colon cancer and arthritis (Ellis et al. 2019; Naz et al. 2014; Zhang and Hamauzu 2004).
Watermelon mosaic virus (WMV) (family: Potyviridae), a single-stranded positive-sense RNA virus transmitted by aphids in a non-persistent manner, is prevalent in the temperate and Mediterranean regions of the globe (Ouibrahim et al. 2014; Sharifi et al. 2008). Its genome has terminal untranslated (UTR) regions flanked by a single large open reading frame (~ 10 kb) cleaved into ten fragments by three virus-encoded proteases (P1, HC-Pro and NIa-Pro) with motifs conserved among the homologous proteins of other potyviruses (Revers and García 2015; Desbiez and Lecoq 2004). Recently, an 11th protein, P3N-PIPO, was identified containing the N-terminal region of P3 and PIPO generated by the transcriptional slippage mechanism (Cheng et al. 2017). WMV exhibits high similarity with the soybean mosaic virus (SMV) and might have emerged by inter-specific recombination between the bean common mosaic virus (BCMV) and SMV-like potyvirus in the P1 protein-coding region (Desbiez and Lecoq 2004). Although the complete genomic sequences of WMV have been studied in many parts of the world, the virus not yet been characterized extensively in India at the molecular level.
Watermelon, an important cash crop of India, is prone to infection by several pathogens, including microbes and insects. Among them, potyviruses, particularly WMV, cause severe losses every year by limiting the quality and quantity of the crop (Santosa et al. 2018). Experimentally, WMV has shown to be transmitted in more than 170 plant species belonging to 27 families (Ali et al. 2006) and cause symptoms such as mosaic, leaf distortion, stunting and mottling (Desbiez et al. 2011). WMV incidence in cucurbits, peas, orchids, weeds and carrots has also been reported worldwide (Aragonés et al. 2019; Desbiez et al. 2007). In this study, we made an attempt to identify and characterize the WMV isolates from two locations of Rajasthan and analyzed the genetic variability in different cistronic regions of WMV genomes and their recombination breakpoints along with phylogenetic relationships.
Materials and methods
Sample collection and nucleic acid extraction
A survey was conducted during the year 2013–2014, to investigate the incidence of WMV in Alwar and Sikar districts of Rajasthan, India. Twenty watermelon (C. lanatus L) leaf samples, showing severe mosaic and mottling symptoms, and four asymptomatic leaf samples were collected from the two districts (Fig. 1a). Total RNA from the diseased and healthy samples was extracted by using the Trizol method (Invitrogen, Carlsbad, CA, USA). The RNA aliquots were tested for the existence of WMV by RT-PCR using potyvirus-degenerated coat protein (CP) primer. To further verify that the sample was infected with WMV, PCR-based amplification of entire genomes of WMV was performed using seven sets of previously reported oligo primers (Desbiez and Lecoq 2004; Ali et al. 2006). The PCR products were resolved on agarose gel, eluted by using a gel extraction kit (GeNei TM Gel Extraction Kit, GeNei, India) and subsequently ligated into the PCR cloning vector pGEM-T (Promega, Madison, WI) as per the manufacturer’s instructions. The authenticity of the WMV clones was confirmed through PCR amplification, followed by restriction digestion and sequencing in both directions with a Sanger sequencer (Eurofins Genomics India Pvt. Ltd, India). The sequence data were further assembled and combined to obtain the full-length genome sequences of WMV by employing the Clustal W alignment tool in DAMBE software (Xia, 2000).
Fig. 1.

a WMV-infected watermelon (Citrullus lanatus L.) plant exhibiting mottling and mosaic symptoms. b Detection of Watermelon mosaic virus (WMV) by RT-PCR. Amplification using coat protein (CP)-degenerated primer pair from infected samples. Lane M: 1 kb ladder, lane 3: healthy and lane 1–2 and 4–9: amplified product ~ 800 bp of coat protein (CP) region of WMV
Alignment analysis
The nucleotide sequences of WMV isolates (RKG1 and RKG2) were aligned with those available in NCBI GenBank database by using BLASTn (https://blast.ncbi.nlm.nih.gov) program (Altschul et al. 1990). A total of 86 full-length sequences of WMV originating from different countries and exhibiting high similarity with the sequences were selected and retrieved from the GenBank database. DnaSP 6(Rozas et al. 2017) was used to estimate genetic variability such as nucleotide diversity (π), the total number of mutations (Ƞ), Watterson's diversity of segregating sites (θ–w), the average of nucleotide differences (k) and total number of segregating sites (s).
Phylogeny and recombination analysis
For phylogenetic and recombination analysis of WMV, complete genome sequences of WMV isolates were retrieved from the NCBI and aligned, followed by phylogenetic analysis using CLUSTALW incorporated in MEGA X software (Kumar et al. 2018). The model selection tool implemented in TOPALi v2.5 software was used to predict the best substitution matrix for the genome sequences. The tree was visualized as a cladogram using FigTree 1.4.0 (https://tree.bio.ed.ac.uk/software/figtree/). Phylogenetic evidence for recombination was detected with Splits Tree v. 4.1 program (Huson and Bryant 2006). A statistical analysis was also carried out by pairwise homoplasy test (PHI) (Bruen et al. 2006) to verify the presence of recombination. The RDP Beta 5.3 version was employed to investigate the extent of recombination, recombination breakpoints, and the major/minor parents of the two characterized isolates. Default parameters and 0.05 highest acceptable Bonferroni-corrected p value of RDP Beta 5.3 version program (Martin et al. 2015) were considered for the analysis. Recombination events detected by at least three out of seven algorithms (RDP, BOOTSCAN, GENCONV, SISCAN, MAXCHI, CHIMAERA and 3SEQ) were taken to be relevant for avoiding false-positive results.
Results
WMV detection and sequencing
In 2013–2014, disease symptoms like mosaic and mottling caused by WMV were observed in watermelon (C. lanatus L.) fields located in the Alwar and Sikar districts, Rajasthan, India (Fig. 1a). To identify the etiology of the disease, 20 leaf samples of watermelon were collected and processed immediately for further analysis. Out of 20, only 8 symptomatic leaves yielded a ~ 800 bp DNA fragment by RT-PCR using potyvirus CP degenerated primer. No fragments were amplified from the asymptomatic samples (Fig. 1b). To further verify the existence of WMV, the complete genome of WMV was amplified by using seven sets of oligo primers, cloned and sequenced. The WMV genome consists of 10,030 nucleotides (nt) determined by the assembly of product by the DAMBE software. The sequences from all the positive samples showed more than 200 nt overlapping regions, indicating that they were from the same genome and not from the different components of a genomic mixture. Only two full length sequences of WMV clones RKG1 (KM597070, Sikar) and RKG2 (KM597071, Alwar) were submitted to NCBI and used for further bioinformatics analysis.
Genome complexity and genetic variability
The BLASTn analysis of the full-length nucleotide sequence against the NCBI database indicated that it had significant similarity to viruses belonging to the genus Potyvirus, sharing the closest relationship to WMV-FMF00-LL1 (France, EU660581) with 96% identity. Thus, based on the recent species demarcation threshold of potyviruses, both the virus isolates in this study were regarded as WMV isolates (Wylie et al. 2017). The results suggested that the putative virus was present in the sequenced samples and named as WMV-RKG1 and WMV-RKG2. The polyprotein of WMV isolates yielded all the 10 characteristic functional potyvirus proteins including the additional 11th protein P3N-PIPO. This viral protein is thought to be evolved through polymerase slippage mechanism required for efficient viral intercellular movement (White 2015).
The analysis involved 88 nucleotide sequences for which the estimated transition/transversion bias (R) was found to be 7.64. The substitution patterns and rates were estimated using the general time-reversible model (Nei and Kumar 2000). The discrete gamma (+G) distribution was used to model the evolutionary rate differences among sites (3 categories, [+G, parameter = 0.2611]). The rates of transitional substitutions ranged from 16.56 to 29.68, whereas transversional substitutions varied from 1.20 to 2.18. For estimating the ML values, a tree topology was studied by using computer simulation. The codon positions included were 1st + 2nd + 3rd + noncoding. All positions containing gaps and missing data were eliminated. There were a total 9814 positions present in the final dataset.
Analysis of genetic variability in virus populations belonging to different geographical areas can enhance our understanding of emergence, host range, epidemiology and endurance of viruses in the ever-changing environment, which can be beneficial in devising disease control strategies. The P1 genome segment of WMV was the leading protein in genetic diversity and had the highest number of mutations (966; 72.52%), which was followed by P3 (506; 48.6%), Hc-Pro (644, 46.87%) and CI (812; 42.35%) (Table 1). The greatest number of segregating sites was also observed in P1 (765; 57.43%) and P3 (427; 41.01%), followed by HC-Pro and CI genome segments. Our analysis established that the overall nucleotide diversity of WMV was 0.077 (7.7%), while P1 protein exhibited a diversity of 0.123 (12.3%). Low values were found for the 6K2, 6K1 and CP cistronic regions, while the others demonstrated relatively high diversity values. The mean nucleotide diversity between the two WMV isolates (RKG1/RKG2) in the present study was 0.021. The diversity of RKG ranged from 0.059 (EU660581; France) to 0.203 (KF274031; China) and those of RKG2 ranged from 0.042 (EU660581; France) to 0.203 (KF274031; China). The overall mean value for the polyprotein-coding region was 0.0849 ± 0.0017. We also tested the pairwise genetic differences at non-synonymous (dN) and synonymous (dS) nucleotide positions using the Pamilo–Bianchi–Li (PBL) method (Li 1993; Pamilo and Bianchi 1993). The dN/dS ratio can indicate the pattern of selective constraint in evolutionary relationships (Nei and Gojobori 1986). The ratios of Nib, CI, P1 and P3 cistronic regions were significantly higher than those of other cistronic regions.
Table 1.
Genetic variability analysis in different cistrons of the WMV population
| Region | Number of polymorphic (segregating) sites, S | Total number of mutations, Eta | Haplotype (gene) diversity, Hd | Nucleotide diversity, Pi | Average number of nucleotide differences between sequences (k) | Watterson’s estimate of the population mutation rate based on the total number of segregating sites (θ–w) | Watterson’s estimate of the population mutation rate based on the total number of mutations (θ–η) | dS | dN | dN/dS |
|---|---|---|---|---|---|---|---|---|---|---|
| Poly protein(10,215) | 4064 (39.78%) | 4941 (48.37%) | 1.00 | 0.077 | 759.70 | 0.081 | 0.09969 | 0.074 ± 0.002 | 0.087 ± 0.002 | 1.17 |
| CP (849) | 235 (27.67% | 266 = 31.33% | 0.995 | 0.047 | 39.53 | 0.055 | 0.06316 | 0.151 ± 0.015 | 0.010 ± 0.002 | 0.06 |
| Nia-VPg (570) | 197 = 34.56% | 223 = 39.12 | 0.990 | 0.075 | 42.53 | 0.069 | 0.078 | 0.256 ± 0.029 | 0.016 ± 0.004 | 0.06 |
| P3 (1041) | 427 = 41.01% | 506 = 48.60 | 0.998 | 0.067 | 68.62 | 0.083 | 0.098 | 0.211 ± 0.014 | 0.022 ± 0.002 | 0.10 |
| P1 (1332) | 765 = 57.43% | 966 = 72.52 | 0.999 | 0.123 | 161.79 | 0.115 | 0.123 | 0.318 ± 0.20 | 0.080 ± 0.006 | 0.25 |
| HC-pro (1374) | 539 = 39.32 | 644 = 46.87 | 0.999 | 0.075 | 102.56 | 0.078 | 0.093 | 0.275 ± 0.015 | 0.013 ± 0.001 | 0.04 |
| CI (1917) | 680 = 35.47 | 812 = 42.35 | 0.998 | 0.073 | 139.55 | 0.071 | 0.094 | 0.278 ± 0.14 | 0.081 ± 0.001 | 0.29 |
| NIB (1551) | 551 = 35.52 | 629 = 40.55 | 0.999 | 0.063 | 97.37 | 0.070 | 0.080 | 0.214 ± 0.012 | 0.107 ± 0.001 | 0.50 |
| NIA-Pro (733) | 255 = 34.78 | 296 = 40.38 | 0.994 | 0.073 | 52.77 | 0.069 | 0.081 | 0.200 ± 0.021 | 0.030 ± 0.005 | 0.15 |
Phylogenetic analysis
The complete genome sequences of 88 WMV isolates reported from different geographical areas were retrieved from the NCBI GenBank, including two WMV isolates of this study. The GTR + G + I substitution model was found to be more appropriate for our dataset by using the model selection tool implemented in TOPALi software. Subsequently, the aligned sequences were used to construct a maximum likelihood (ML) tree using MEGA X with 1000 bootstrap values. The WMVs were closely related with nucleotide identities of 91% (Chinese isolate; KC292915) to 96% (French isolate; EU660581) to the WMV isolates from different parts of the world. Phylogenetic analysis based on the complete sequence of WMV showed that all the isolates fell within three distinct genogroups, namely G1, GII and GIII (Desbiez et al. 2007) (Fig. 2).
Fig. 2.
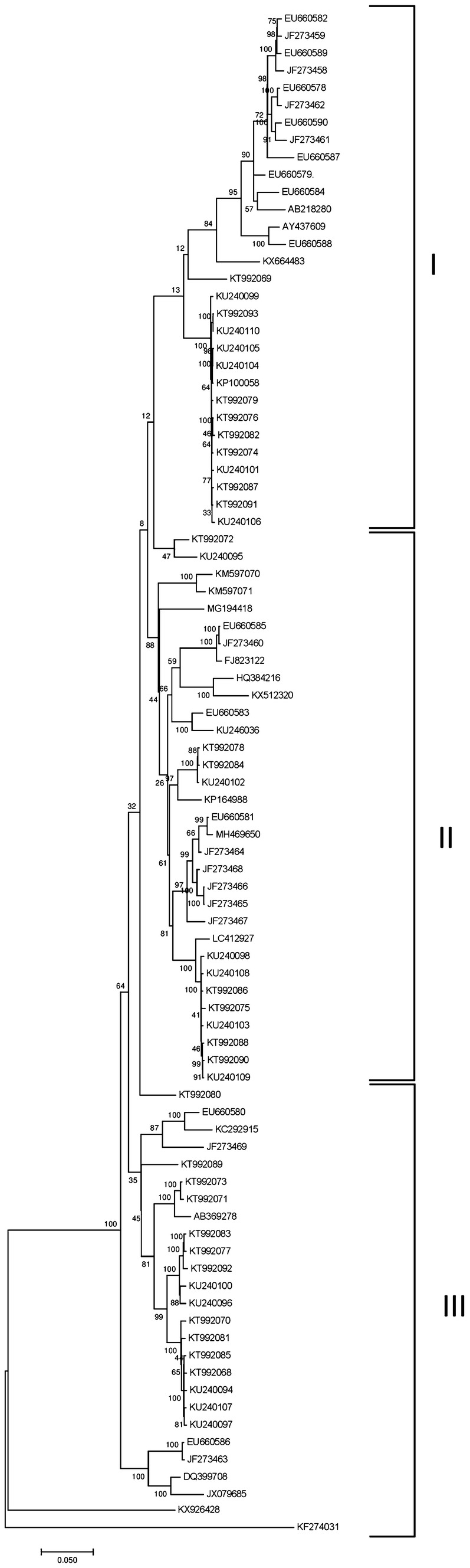
a The maximum likelihood (ML) tree of the aligned complete genome sequences of WMV isolates representing the common ancestry with other French WMV isolates. b The reticulate network in the split tree of WMV isolates showed the presence of potential recombination breakpoints
Recombination analysis
Since recombination and mutation are the two leading forces in the evolution and emergence of potyviruses (Verma et al. 2015), we scrutinized the incidence of recombination in the genomic sequences of earlier reported strains of WMV along with the two WMV isolates identified in the present study. Split decomposition analysis was performed to obtain the network structure of this phylogenetic incompatibility in the WMV population. The reticulate network in the split tree showed the presence of potential recombination events between the isolates. Furthermore, the output of CLUSTAL W alignment was exported to RDP, BOOTSCAN, GENCONV, SISCAN, MAXCHI, CHIMAERA and 3SEQ algorithms implemented in RDP Beta 5.3 version and subjected to recombination analysis. A total of 65 recombination breakpoints were obtained in all 88 WMV nucleotide sequences (Table S1). The sequences of WMV isolates (RKG1 and RKG2) exhibited seven potential recombination breakpoints in their genomes and served as the major parent for recombination in EU660579 and EU660582 (Table 2). A total of six recombination breakpoints were found to be common in both the WMV isolates.
Table 2.
Recombination breakpoints in WMV isolates calculated by different algorithms implemented in RDP.4.0
| S. no. | Begin | End | Recombinant sequence (s) | Minor parental sequence (s) | Major parental sequence (s) | Detection methods and their p values | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RDP | GENECONV | Bootscan | Maxchi | Chimaera | SiSscan | 3Seq | ||||||
| 1 | 564 | 1622 | KM597070/KM597071 | JF273468 | Unknown | 1.21E−34 | 3.89E−35 | NS | 1.38E−13 | 4.13E−12 | 1.62E−28 | 8.95E−08 |
| 2 | 96 | 864 | KM597070/KM597071 | Unknown | EU660581 | 3.83E−31 | 1.29E−34 | NS | 2.93E−05 | 1.13E−09 | 2.46E−21 | 1.06E−11 |
| 3 | 5663 | 6918 | KM597070 | JF273464 | KT992074 | 1.27E−15 | 2.15E−15 | NS | 5.50E−11 | 9.85E−09 | 1.12E−16 | 3.73E−03 |
| 4 | 5549 | 6198 | KM597071 | EU660581 | Unknown | 4.75E−10 | 4.64E−14 | 9.81E−11 | 1.53E−08 | 1.28E−08 | 7.02E−12 | 2.98E−09 |
| 5 | 8331 | 8918 | KM597070 | JF273462 | Unknown | 6.79E−05 | 6.34E−07 | 7.75E−04 | 3.21E−05 | 1.97E−04 | 4.75E−08 | NS |
| 6 | 4825 | 5075 | KM597070/KM597071 | EU660580 | KT992086 | 2.13E−04 | 6.54E−03 | 3.24E−02 | NS | 8.84E−03 | 1.48E−04 | 1.51E−02 |
| 7 | 3146 | 3958 | KM597070 KM597071 | EU660580 | KU240099 | 2.00E−07 | 9.95E−05 | 2.66E−07 | 5.97E−04 | 1.14E−04 | 9.98E−07 | 3.22E−04 |
| 8 | 2600 | 3088 | KM597070 KM597071 | EU660580 | DQ399708 | 1.57E−07 | 7.80E−06 | 2.67E−05 | 2.21E−04 | 2.92E−05 | 1.85E−08 | 7.37E−03 |
Discussion
WMV is one of the most prevalent potyvirus causing severe damage to cucurbits worldwide including India. This study provided a snapshot of the genetic variability, phylogeny and recombination of WMV isolates collected from symptomatic watermelon plants grown in the Alwar and Sikar districts of Rajasthan, India. The nucleotide sequences of both the isolates showed the characteristic size and genomic organization of a typical potyvirus (Perotto et al. 2016). The sequences of WMV-RKG1 and WMV-RKG2 shared the highest nucleotide similarity (96%) with the WMV sequences reported from France (EU660581). Interestingly, the phylogenetic analyses of the two WMV isolates (RKG1 and RKG2) clustered with French isolates to group II (Fig. 2), suggesting the possible immigration of WMV isolates from France to India. These results indicate that the genomes of the Indian WMV isolates and French isolates have arisen from recombination supporting the previous data on WMV genetic diversification and evolutionary history (Garcia-Arenal et al. 2001).
Our elucidation indicated that the genetic variations were not distributed evenly among the different regions of the WMV genome. The P1 and P3 cistronic regions demonstrated higher diversity than other coding regions. Protein P1 of potyvirus modulates replication and host defense, whereas protein P3 determines host range and symptomatology (Suehiro et al. 2004; Pasin et al. 2014). Our result suggests that the P1 and P3 genomic segments are under lesser evolutionary constraints than the other cistrons, and these regions were hence determined to be the mutational hotspots of WMV.
The high degree of genetic variability in the P1 region may be due to the recombination or mutation resulting from the different selection pressures encountered by the various WMV strains from different geographical regions. These results are consistent with those from the analysis of the other potyviruses such as onion yellow dwarf virus (OYDV) (Verma et al. 2015), potato virus Y (Visser et al. 2012; Karasev et al. 2011), SMV (Seo et al. 2009), turnip mosaic virus (Ohshima et al. 2002, 2007) and zucchini yellow mosaic virus (ZYMV) (Lecoq et al. 2009). Additionally, the coding sequences were aligned using the MEGA X to investigate the pattern of nucleotide substitution per site (dN and dS). The substitutions at the synonymous sites (dS) were significantly higher than those at the non-synonymous sites (dN) in every cistronic region (Table 1). It is estimated that the change in the amino acid sequence can be either synonymous (i.e., no effect on the protein product of a gene) or nonsynonymous (i.e., do not change the function of the protein in which they appear) (Plotkin and Kudla 2010).
The ratio (dN/dS ratio) between the nucleotide substitutions at the non-synonymous (dN) and synonymous sites (dS) asserted the extent of variations in the nucleic acid that led to differences in the encoded protein as well as the degree of negative selection in the genes and the degree of functional constraint for the maintenance of the translated protein sequences (Cuevas et al. 2012; Hughes 2009). For most coding genes of WMV, the dNS/dS ratio was quite low, which is consistent with the negative selection against protein change (Nei 1987). The higher dN/dS ratio of the Nib, CI, P1 and P3 regions suggests that these are under lesser evolutionary constraints. The higher transitions/transversions bias (R = 7.64) was consistent with the principle that transitions are more conservatives than transversions. Estimation of transitions/transversions rate bias is significantly important for understanding the evolution of the genomes as well as reconstruction of phylogeny (Batista et al. 2011; Yang and Yoder 1999).
Recombination is vital for acquiring sequence diversity and complex genotyping system for viruses, and the process can also act as one of the sources of evolution (Lian et al. 2013; Posada et al. 2002). Earlier investigations demonstrated that the HC-to-CI region is a hotspot for recombination in WMV isolates (Desbeiz et al. 2007, 2011). In the present study, recombination events were identified in all the genomic segments of WMV, with a higher frequency in the HC-to-CI, P1 and P3 regions (Table S1). Our WMV isolates (RKG and RKG2) possessed seven common recombination breakpoints through preponderance in the HC-to-CI region. RKG1 and RKG2 strains also exhibited putative recombination breakpoints in the P1, NIa-Pro and Nib-CP regions (Table 2). The presence of recombination breakpoints in the WMV isolates under this study revealed the existence of assorted pressures during the natural selection of the isolates.
Analysis of molecular variability and the recombination origin of WMV based on P1, CP and CI genes have been already discussed and explained in many countries such as France, Iran, Spain, Italy and Pakistan (Ali et al. 2006; Desbeiz et al. 2009; Finetti-Sialer et al. 2012; Sharifi et al. 2008; Desbiez and Lecoq 2008; Moreno et al. 2004). These finding suggest a significant role of recombination during the genetic variants of WMV isolates around the world. Conventional strategies for the management of virus infections in plants fails to control the rapidly evolving and emerging plant viruses. Under these circumstances, genome engineering strategies that depend on genetic variability analysis have been recently introduced as promising tools to control the plant virus infections (Borrelli et al. 2018).
Thus, understanding the molecular evolutionary pattern and phylogeny of WMV, as well as finding novel host emergence and molecular mechanisms, will enable researchers to explore new management strategies for plants that are constantly challenged by potyviruses.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1: Recombination Break points in watermelon mosaic virus (WMV) geographical isolates calculated by different algorithms implemented in RDP.4.0 (DOCX 32 kb)
Acknowledgements
The authors are thankful to the Department of Biotechnology, GOI, India (BT/PR14902/BRB/10/889/2010), for financial support. The authors are also thankful to Dr. Govind P. Rao, Principal Scientist, Indian Agricultural Research Institute, Pusa Campus, New Delhi, India, for his critical review and suggestions during the manuscript preparation.
Author contributions
RKV, MM and AM did the experimental work and RKG did the experiment design and manuscript editing.
Compliance with ethical standards
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical statement
This article does not contain any studies with human participants or animals performed by any of the authors.
Contributor Information
Rakesh Kumar Verma, Email: rkwat4@yahoo.com.
Megha Mishra, Email: meghamishra1228@gmail.com.
Avinash Marwal, Email: marwal_avinash@yahoo.co.in.
R. K. Gaur, Email: gaurrajarshi@hotmail.com
References
- Ali A, Natsuaki T, Okuda S. The complete nucleotide sequence of a Pakistani isolate of watermelon mosaic virus provides further insights into the taxonomic status in the bean common mosaic virus subgroup. Virus Genes. 2006;32:307–311. doi: 10.1007/s11262-005-6915-z. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Aragonés V, Pérez-de-Castro A, Cordero T, Cebolla-Cornejo J, López C, Picó B, Daròs J-A. A Watermelon mosaic virus clone tagged with the yellow visual maker phytoene synthase facilitates scoring infectivity in melon breeding programs. Eur J Plant Pathol. 2019;153:317–323. [Google Scholar]
- Arsovski AA, Pradinuk J, Guo XQ, Wang S, Adams KL. Evolution of cis-regulatory elements and regulatory networks in duplicated genes of Arabidopsis. Plant Pathol. 2015;169(4):2982–2991. doi: 10.1104/pp.15.00717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista MVA, Ferreira TAE, Freitas AC, Balbino VQ. An entropy-based approach for the identification of phylogenetically informative genomic regions of Papillomavirus. Infect Genet Evol. 2011;11:2026–2033. doi: 10.1016/j.meegid.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–2681. doi: 10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli VMG, Brambilla V, Rogowsky P, Marocco A, Lanubile A. The Enhancement of Plant Disease Resistance Using CRISPR/Cas9 Technology. Front Plant Sci. 2018;9:1245. doi: 10.3389/fpls.2018.01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas JM, Delaunay A, Rupar M, Jacquot E, Elena SF. Molecular evolution and phylogeography of potato virus Y based on the CP gene. J Gen Virol. 2012;93:2496–2501. doi: 10.1099/vir.0.044347-0. [DOI] [PubMed] [Google Scholar]
- Desbiez C, Costa C, Wipf-Scheibel C, Girard M, Lecoq H. Serological and molecular variability of watermelon mosaic virus (genus Potyvirus) Arch Virol. 2007;152:775–781. doi: 10.1007/s00705-006-0899-4. [DOI] [PubMed] [Google Scholar]
- Desbiez C, Lecoq H. The nucleotide sequence of Watermelon mosaic virus (WMV, Potyvirus) reveals interspecific recombination between two related potyviruses in the 5 part of the genome. Arch Virol. 2004;149:1. doi: 10.1007/s00705-004-0340-9. [DOI] [PubMed] [Google Scholar]
- Desbiez C, Lecoq H. Evidence for multiple intraspecific recombinants in natural populations of Watermelon mosaic virus (WMV, Potyvirus) Arch Virol. 2008;153:1749–1754. doi: 10.1007/s00705-008-0170-2. [DOI] [PubMed] [Google Scholar]
- Desbiez C, Joannon B, Wipf-Scheibel C, Chandeysson C, Lecoq H. Emergence of new strains of Watermelon mosaic virus in South-eastern France: evidence for limited spread but rapid local population shift. Virus Res. 2009;141:201–208. doi: 10.1016/j.virusres.2008.08.018. [DOI] [PubMed] [Google Scholar]
- Desbiez C, Joannon B, Wipf-Scheibel C, Chandeysson C, Lecoq H. Recombination in natural populations of watermelon mosaic virus: new agronomic threat or damp squib. J Gen Virol. 2011;92:1939–1948. doi: 10.1099/vir.0.031401-0. [DOI] [PubMed] [Google Scholar]
- Ellis AC, Dudenbostel T, Crowe-White K (2019) Watermelon juice: a novel functional food to increase circulating lycopene in older adult women. Plant Foods Hum Nutr. [DOI] [PubMed]
- Finetti-Sialer MM, Mascia T, Cillo F, Vovlas C, Gallitelli D. Biological and molecular characterization of a recombinant isolate of Watermelon mosaic virus associated with a watermelon necrotic disease in Italy. Eur J Plant Pathol. 2012;132:317–322. [Google Scholar]
- Garcia-Arenal F, Fraile A, Malpica JM. Variability and genetic structure of plant virus populations. Annu Rev Phytopathol. 2001;39:157–186. doi: 10.1146/annurev.phyto.39.1.157. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Small effective population sizes and rare nonsynonymous variants in potyviruses. Virology. 2009;393:127–134. doi: 10.1016/j.virol.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Karasev AV, Hu X, Brown CJ, Kerlan C, Nikolaeva OV, Crosslin JM, Gray SM. Genetic diversity of the ordinary strain of Potato virus Y (PVY) and origin of recombinant PVY strains. Phytopathology. 2011;101:778–785. doi: 10.1094/PHYTO-10-10-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar PS, Kulkarni VS. An economic analysis of production management of watermelon in Haveri (Karnataka) and Ananthapur Districts (Andhra Pradesh): a comparative analysis. Int J Curr Microbiol. 2018;7:2945–2957. [Google Scholar]
- Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecoq H, Wipf-Schibel C, Chandeysson C, Le Van A, Fabre F, Desbiez C (2009) Molecular epidemiology of Zucchini yellow mosaic virus in France: an historical overview. Virus Res 141:190–200 [DOI] [PubMed]
- Li WH. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Lian S, Lee JS, Cho WK, Yu J, Kim MK, Choi HS, Kim KH. Phylogenetic and recombination analysis of tomato spotted wilt virus. PLoS ONE. 2013;8:e63380. doi: 10.1371/journal.pone.0063380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):3. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno IM, Malpica JM, Diaz-Pendón JA, Moriones E, Fraile A, Garcia-Arenal F. Variability and genetic structure of the population of watermelon mosaic virus infecting melon in Spain. Virology. 2004;318:451–460. doi: 10.1016/j.virol.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Naz A, Butt MS, Sultan MT, Qayyum MMN, Niaz RS. Watermelon lycopene and allied health claims. EXCLI J. 2014;13:650–660. [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3(5):418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987. p. 512. [Google Scholar]
- Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press, Oxford; 2000. [Google Scholar]
- Ohshima K, Yamaguchi Y, Hirota R, Hamamoto T, Tomimura K, Tan Z, Sano T, Azuhata F, Walsh JA, Fletcher J, Chen J, Gera A, Gibbs A (2002) Molecular evolution of Turnip mosaic virus: evidence of host adaptation, genetic recombination and geographical spread. J General Virol 83:1511–1521 [DOI] [PubMed]
- Ohshima K, Tomitaka Y, Wood JT, Minematsu Y, Kajiyama H, Tomimura K, Gibbs AJ. Patterns of recombination in turnip mosaic virus genomic sequences indicate hotspots of recombination. J Gen Virol. 2007;88:298–315. doi: 10.1099/vir.0.82335-0. [DOI] [PubMed] [Google Scholar]
- Ouibrahim L, Mazier M, Estevan J, Pagny G, Decroocq V, Desbiez C, Moretti A, Gallois JL, Caranta C. Cloning of the Arabidopsis rwm1 gene for resistance to Watermelon mosaic virus points to a new function for natural virus resistance genes. Plant J. 2014;79:705–716. doi: 10.1111/tpj.12586. [DOI] [PubMed] [Google Scholar]
- Pamilo P, Bianchi NO. Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol. 1993;10(2):271–281. doi: 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- Perotto MC, Celli MG, Pozzi EA, Luciani CE, Conci VC. Occurrence and characterization of a severe isolate of Watermelon mosaic virus from Argentina. Eur J Plant Pathol. 2016;146:213–218. [Google Scholar]
- Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D, Crandall KA, Holmes EC. Recombination in evolutionary genomics. Annu Rev Genet. 2002;36:75–97. doi: 10.1146/annurev.genet.36.040202.111115. [DOI] [PubMed] [Google Scholar]
- Purcifull DE, Hiebert E, Edwardson J (1984) Watermelon mosaic virus. No. 293. In: Description of Plant Virus, CMI/ABB, Surrey, England, p 7
- Revers F, García JA (2015) Molecular biology of potyviruses. In: Advances in virus research, Elsevier, pp 101–199 [DOI] [PubMed]
- Rodamilans B, Valli A, Mingot A, San León D, Baulcombe D, López-Moya JJ, García JA. RNA polymerase slippage as a mechanism for the production of frameshift gene products in plant viruses of the potyviridae family. J Virol. 2015;89:6965–6967. doi: 10.1128/JVI.00337-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Ferrer-Mata A, Sánchez-Delbarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Santosa AI, Al-Shahwan IM, Abdalla OA, Al-Saleh MA, Amer MA. Characterization of a watermelon mosaic virus isolate inducing a severe disease in watermelon in Saudi Arabia. J Agric Sci Tech A. 2018;8:220–229. [Google Scholar]
- Seo JK, Ohshima K, Lee HG, Son M, Choi HS, Lee SH, Sohn SH, Kim KH. Molecular variability and genetic structure of the population of Soybean mosaic virus based on the analysis of complete genome sequences. Virology. 2009;393:91–103. doi: 10.1016/j.virol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Sharifi M, Massumi H, Heydarnejad J, Hosseini PA, Shaabanian M, Rahimian H. Analysis of the biological and molecular variability of Watermelon mosaic virus isolates from Iran. Virus Genes. 2008;37:304–313. doi: 10.1007/s11262-008-0271-8. [DOI] [PubMed] [Google Scholar]
- Verma RK, Mishra R, Gaur RK. Potato virus Y genetic variability: a review. In: Gaur RK, Petrov NM, Patil BL, Stoyanova MI, editors. Plant viruses: evolution and management. Singapore: Springer Singapore; 2016. pp. 205–214. [Google Scholar]
- Verma RK, Mishra R, Petrov NM, Stoyanova M, Stoev A, Bakardjieva N, Gaur RK. Molecular characterization and recombination analysis of an Indian isolate of Onion yellow dwarf virus. Eur J Plant Pathol. 2015;143:437–445. [Google Scholar]
- Visser JC, Bellstedt DU, Pirie MD. The recent recombinant evolution of a major crop pathogen, Potato virus Y. PLoS ONE. 2012;7:e50631. doi: 10.1371/journal.pone.0050631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KA (2015) The polymerase slips and PIPO exists. EMBO Rep 16(8):885–886. 10.15252/embr.201540871 [DOI] [PMC free article] [PubMed]
- Wylie SJ, Adams M, Chalam C, Kreuze J, López-Moya JJ, Ohshima K, Praveen S, Rabenstein F, Stenger D, Wang A, Zerbini FM. ICTV virus taxonomy profile: Potyviridae. J Gen Virol. 2017;98:352–354. doi: 10.1099/jgv.0.000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol. 2013;30:1720–1728. doi: 10.1093/molbev/mst064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Tomimura K, Gera A, Chen J, Hamamoto T, Sano T, Ohshima K, Walsh JA, Fletcher J, Hirota R, Tan Z, Gibbs A, Azuhata F. Molecular evolution of Turnip mosaic virus: evidence of host adaptation, genetic recombination and geographical spread. J Gen Virol. 2002;83:1511–1521. doi: 10.1099/0022-1317-83-6-1511. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yoder AD. Estimation of the transition/transversion rate bias and species sampling. J Mol Evol. 1999;48:274–283. doi: 10.1007/pl00006470. [DOI] [PubMed] [Google Scholar]
- Zhang D, Hamauzu Y. Phenolic compounds and their antioxidant properties in different tissues of carrots (Daucus carota L.) J Food Agric Environ. 2004;2:95–100. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Recombination Break points in watermelon mosaic virus (WMV) geographical isolates calculated by different algorithms implemented in RDP.4.0 (DOCX 32 kb)