

Abstract
Objective
We aimed to use next‐generation sequencing (NGS) for the early diagnosis of primary immunodeficiency diseases (PIDs) and define its effects on medical management for an infant cohort in early life.
Methods
A single‐centre study was conducted from November 2015 to April 2018. Infants less than 3 months old with infections or abnormal white blood cell counts were enrolled in the study. Gene variants were analysed by NGS, and once a mutation was found in a PID‐associated gene, the immune functions associated with this mutation were detected. The diagnosis rate of PIDs in the cohort was the main outcome. The patients received corresponding management and follow‐up treatments.
Results
Among 2392 patients who were genetically tested with NGS, 51 infants were diagnosed with PIDs. Seven types of PIDs were detected, and the most common (25/51, 49%) were combined immunodeficiencies with associated or syndromic features. Thirty‐five patients (68.6%) were cured or had improved outcomes after being diagnosed with PID. The NGS cost was US$280 per case.
Conclusions
This study not only highlighted the potential of NGS to rapidly deliver molecular diagnoses of PIDs but also indicated that the prevalence of PIDs is underestimated. With broader use, this approach has the potential to alter clinical strategies.
Keywords: clinical utility, infants, next‐generation sequencing, primary immunodeficiency diseases
This study highlights advantages of next‐generation sequencing for primary immunodeficiency diseases (PID) screening in early life infants. Based on the diagnosis, early precision therapy improved the prognosis of individuals with PIDs.
Introduction
Primary immunodeficiency diseases (PIDs) are disorders associated with defects in the immune system. More than 400 different types of monogenic inherited PIDs are currently recognised; these PIDs affect different immune cells (including T cells, B cells and granulocytes) and the complement system. 1 , 2 , 3 Early diagnosis of PIDs is extremely difficult because these diseases do not have specific characteristic symptoms but instead have diverse clinical manifestations, particularly in early life. A lack of a diagnosis can have considerable adverse effects on patients and their families; such effects can include failure to identify potential treatments, recognise the risk of genetic transmission during subsequent pregnancies, and provide anticipatory guidance and prognostic information.
With progress in gene sequencing technologies, increasing numbers of PIDs are being diagnosed. The emerging dominance of next‐generation sequencing (NGS) has driven a rapid increase in the number of recognised PIDs. In the past 2 years, 64 different genetic defects have been recognised. 1 , 2 , 4 Whole‐exome sequencing (WES) can enable rapid diagnosis and has been adopted by several laboratories as the method of choice for diagnosing various PIDs. 5 , 6 , 7 , 8 , 9 For genetically undiagnosed patients with PIDs, NGS has the ability to contribute significantly to the identification of molecular mechanisms. 10
The genetic and phenotypic heterogeneity of PIDs makes timely and accurate diagnosis challenging, hindering the prompt treatment of patients with PIDs. Here, we report the high diagnostic yield of NGS for PIDs in early life infants.
Results
Clinical characteristics
From November 2015 to April 2018, 2392 patients were enrolled, including 1353 boys and 1039 girls. The enrolment and outcomes of the subjects participating in the study are shown in Figure 1. The ages of the patients ranged from 6 to 87 days. In all, 1680 (70.23%) patients required intensive care unit (ICU) support; 1773 (74.12%) patients had been diagnosed with infectious diseases, including sepsis, severe pneumonia and purulent meningitis; and 619 (25.88%) patients had no defined infectious disease, but had abnormal routine blood tests, including 102 with white blood cells < 4000 cells µL−1, 433 with white blood cells > 20 000 cells µL−1 and 84 had platelets < 100 × 109 L−1. The baseline characteristics of the patients are shown in Table 1. Forty‐five patients with PIDs had particular infections, 12 patients had haematological abnormalities, and eight patients had both a particular infection and haematological abnormalities (Table 2).
Figure 1.
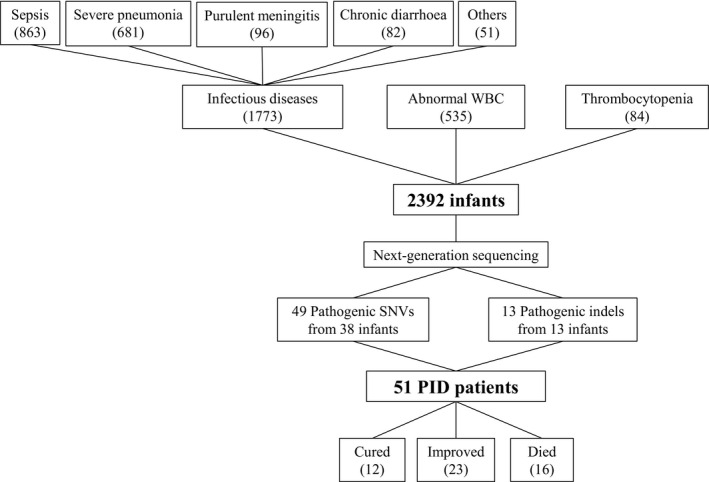
Enrolment and the outcomes of subjects participating in the study. PID, primary immunodeficiency diseases; WBC, white blood cell.
Table 1.
Baseline characteristics of enrolled patients
| Characteristics | N (%) |
|---|---|
| Sex | |
| Female | 1039 (43.44%) |
| Male | 1353 (56.56%) |
| Severity of diseases | |
| Requiring ICU support | 1680 (70.23%) |
| Non‐requiring ICU support | 712 (29.77%) |
| Infectious diseases | |
| Types of diseases | |
| Sepsis | 863 (36.08%) |
| Severe pneumonia | 681 (28.47%) |
| Purulent meningitis | 96 (4.01%) |
| Chronic diarrhoea | 82 (3.43%) |
| Other infectious disease | 51 (2.13%) |
| Pathogens | |
| Bacteria | 1035 (58.38%) |
| Fungal | 12 (0.68%) |
| Virus | 303 (17.09%) |
| Others | 15 (0.85%) |
| Unclear | 408 (23.01%) |
| Abnormal WBC without infection | 535 (22.37%) |
| Thrombocytopenia | 84 (3.51%) |
ICU, intensive care unit; WBC, white blood cell.
Table 2.
The number of patients with particular infections or haematological abnormalities in patients with defined genetic defects
| Disease‐causing gene | The number of total patients | The number of patients with particular infections | The number of patients with haematological abnormalities |
|---|---|---|---|
| 22q11 del | 13 | 12 | 2 |
| CHD7 | 3 | 3 | 0 |
| WAS | 4 | 3 | 4 |
| ATM | 3 | 3 | 0 |
| ELANE | 3 | 2 | 3 |
| IL10RA | 3 | 3 | 0 |
| BTK | 2 | 2 | 0 |
| CYBB | 2 | 2 | 0 |
| IL12RB1 | 2 | 2 | 0 |
| SH2D1A | 2 | 1 | 1 |
| CD40LG | 1 | 1 | 0 |
| CSF3R | 1 | 1 | 0 |
| CYBA | 1 | 1 | 0 |
| IKBKG | 1 | 1 | 0 |
| IL2RG | 1 | 1 | 0 |
| ITGB2 | 1 | 1 | 1 |
| LIG4 | 1 | 1 | 0 |
| NOD2 | 1 | 0 | 0 |
| RAG1 | 1 | 1 | 0 |
| STAT1 | 1 | 1 | 0 |
| STAT3 | 1 | 0 | 0 |
| STXBP2 | 1 | 1 | 0 |
| TNFRSF13B | 1 | 1 | 0 |
| VPS13B | 1 | 1 | 1 |
Identified disease‐causing variants
Approximately 20 000 single nucleotide variants (SNVs) and 10 copy number variations (CNVs) were detected from each patient’s original sequencing data. Variants were automatically classified as potential causal variants based on the following considerations derived from the American College of Medical Genetics and Genomics (ACMG) guidelines 11 , 12 : (1) the association between the variation‐affected gene and the individual’s phenotypes; (2) the inheritance model of relevant genes; (3) the zygosity of variants; and (4) whether a compound heterozygous event had occurred. Variations that satisfied these criteria were diagnosed as pathogenic variations.
In general, approximately 150 SNVs were classified as candidate variants for each patient. Lists of variants were then further manually prioritised based on phenotypic information and subjected to Sanger sequencing. All candidate disease‐causing variants were additionally confirmed via Sanger sequencing. Moreover, CNV analyses were performed for whom no causal SNVs or indels could be identified; 0–2 potential CNVs were detected in each patient. All candidate disease‐causing CNVs were confirmed using internally controlled multiplex PCR amplification. In addition, clinical data or functional test results are consistent with the phenotype of variants. Finally, 49 SNVs from 38 infants and 13 of the suspected exonic deletions from 13 infants were confirmed to be disease‐causing variants. A summary of the confirmed disease‐causing variants is shown in Supplementary table 1.
Diagnostic efficiency
Fifty‐one individuals (2.1%, 51/2392) had disease‐related PID gene mutations detected using NGS (Supplementary table 1). According to the PID classification, 4 seven types of immunodeficiency diseases were found: four cases were immunodeficiencies affecting cellular and humoral immunity; 25 cases were combined immunodeficiencies with associated or syndromic features; three cases were predominantly antibody deficiencies; six cases involved diseases of immune dysregulation; nine cases were congenital defects of phagocyte number or function; three cases were defects in intrinsic and innate immunity; and one case was an autoinflammatory disorder (Figure 2). No case consisted of complement deficiencies or phenocopies of inborn errors of immunity in the cohort. Forty‐seven (47/1680, 2.80%) patients with PID were identified in patients requiring ICU support, and 4 (4/712, 0.56%) were identified in patients not requiring ICU support, and there was a significant difference (P < 0.01).
Figure 2.
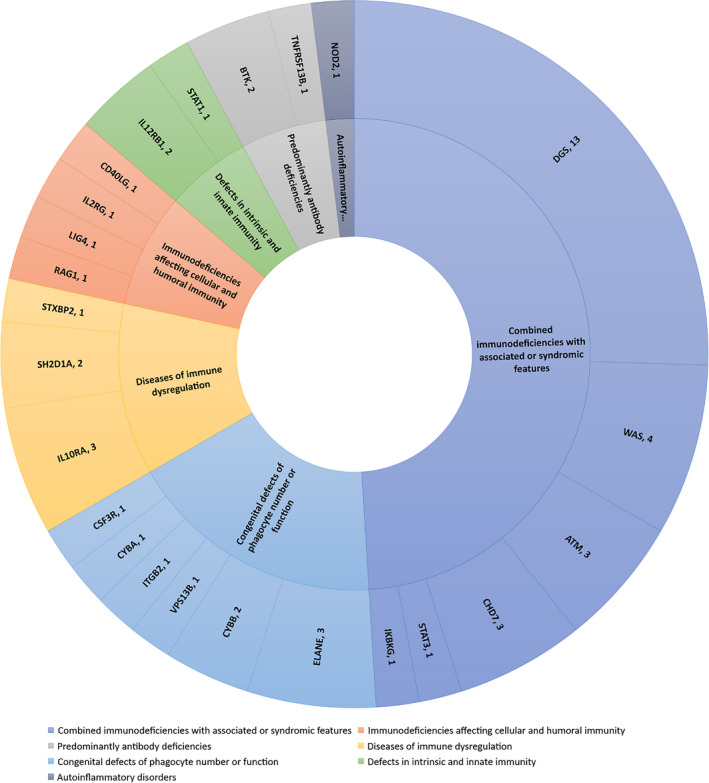
Primary immunodeficiency diseases types, disease‐causing genes and the number of cases identified in the infant cohort in this study.
Among the diagnosed PIDs, the most common type of disease was combined immunodeficiencies with associated or syndromic features, which accounted for almost one‐half (49%, 25/51) of all diagnosed PIDs. DiGeorge syndrome was particularly common. Thirteen patients were diagnosed with DiGeorge Syndrome, seven patients had congenital cardiac disease, and two had hypocalcaemia. The top 10 specific disease‐causing genes are shown in Figure 3.
Figure 3.
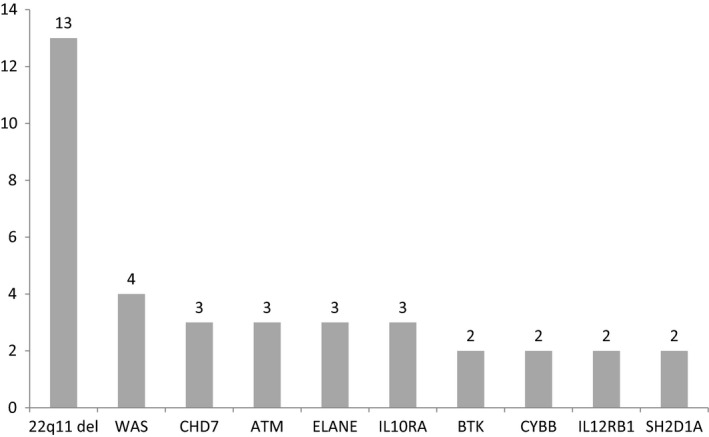
Top 10 primary immunodeficiency diseases‐causing genes and the number of cases in the infant cohort in this study.
T‐cell receptor excision circles/kappa‐deleting recombination excision circles (TRECs/KRECs) detection
T‐cell receptor excision circles/kappa‐deleting recombination excision circles (TRECs/KRECs) detection is widely used for newborn screening, and we used the detection for all of the above‐mentioned 51 patients. Lymphocytes from the positive patient were subjected to lymphocyte subset detection by flow cytometry. The results showed that 12 patients had decreased T cells (< 1000 µL−1) and two patients had decreased B cells (< 300 µL−1). The patients with decreased T cells included five with DiGeorge Syndrome, one with CHD7 mutation, one with ATM mutation, one with CD40LG mutation, one with IKBKG mutation, one with IL2RG mutation, one with LIG4 mutation and one with RAG1 mutation. The two patients with decreased B cells had BTK mutation. This result indicates that only 14 patients could be identified with PIDs by TRECs/KRECs detection in the infant cohort.
Clinical management and treatment implications
The positive diagnostic results produced widespread effects on clinical management, resulting in the offering of appropriate treatment or changes in treatment and prompting the provision of appropriate genetic counselling. In this study, 51 patients (2.1%) were diagnosed with PIDs. After diagnosis, all patients received the corresponding treatment. Thirty‐five patients were cured or improved, including 30 patients who had specific therapy (Supplementary table 1).
The diseases of patients with IL2RG, LIG4 and RAG1 gene mutations were confirmed by lymphocyte subsets and immunoglobulin detection. These patients had extremely low CD3+ T‐cell counts. The diagnosis of severe combined immunodeficiency (SCID) was clear. SCID has high mortality if not diagnosed in a timely manner. Haematopoietic stem cell transplantation (HSCT) is the way to cure this disease. The three patients with SCID received HSCT, and all were cured. A CD40LG mutation results in hyper‐IgM syndrome, one kind of CID. The one patient with the CD40LG mutation had CMV and BCG infections when they were diagnosed. The patient received HSCT after the infection was controlled, and he was cured.
Twenty‐five patients were diagnosed with combined immunodeficiencies with associated or syndromic features, including 13 patients with DiGeorge syndrome, three patients with CHARGE syndrome, four patients with Wiskott–Aldrich syndrome (WAS), three patients with ataxia‐telangiectasia, one patient with hyper‐IgE syndrome, and one patient with an IKBKG mutation. All four patients with WAS had platelets < 100 × 109 L−1 and received HSCT, two were cured, and two died. The three patients with ataxia‐telangiectasia received regular intravenous immunoglobulin (IVIG) therapy. The other eighteen patients received symptomatic treatment. Patients with DiGeorge syndrome or CHARGE syndrome still had occasional infections.
For the three patients with predominantly antibody deficiencies, two patients had BTK mutation, and one had TNFRSF13B mutation. Both patients with the BTK mutation had good prognoses with regular IVIG therapy. The patient with the TNFRSF13B mutation had severe pneumonia at 2 months old. Therefore, we included him in the study and detected the gene variants. Because he had normal immunoglobulin at that time, we followed this patient. When he was 8 months old, he had a Candida albicans infection and hypogammaglobulinaemia. And then, he received regular IVIG therapy.
Among the six patients with immune dysregulation diseases, three patients had IL10RA mutation, two patients had SH2D1A mutation, and one patient had STXBP2 mutation. All three patients with IL10RA mutation had chronic diarrhoea when they were diagnosed and received HSCT, and two of these patients were cured. Among the two patients with SH2D1A mutation and one patient with STXBP2 mutation, two patients died of severe EBV infection during the neonatal period, and one patient was cured using HSCT.
In addition, three patients had ELANE mutation, one patient had CSF3R mutation, and one patient had VPS13B mutation. The neutrophil count was regularly monitored. One patient with severe neutropenia as a result of ELANE mutation received HSCT and was cured. Three patients were diagnosed with chronic granulomatous disease (CGD) because of CYBB or CYBA mutation; one patient received HSCT, and the other two patients received antifungal drugs and interferon‐γ therapy. The patient with ITGB2 mutation was cured by HSCT.
Two patients with IL12RB1 mutation received interferon‐γ therapy. They are living well. After one patient with STAT1 mutation was found, we performed functional analysis. The mutation was confirmed to be a gain‐of‐function mutation. Then, the patient received antifungal drugs. For the patients with autoinflammatory disorders, including one patient with NOD2 mutation, symptomatic treatment was administered.
NGS costs
The cost of NGS was approximately US$280 per sample. It is clear that NGS is a more rapid alternative to Sanger sequencing with respect to reagent costs for all genes. In addition, much greater labour is required for classical Sanger sequencing than for NGS.
Discussion
Primary immunodeficiency diseases constitute a group of highly heterogeneous genetic disorders. Approximately 3110 genes may potentially cause PIDs based on their biological functions and human gene connectome analysis. 13 However, the prevalence of various forms of PIDs is likely underestimated. 14 There are currently more than 400 forms of PIDs defined by the International Union of Immunological Societies (IUIS). 1 , 2 The main reason that PIDs cannot be identified earlier is a lack of specific symptoms during early infancy.
Gene discovery and an understanding of the molecular bases of PIDs are essential starting points for determining a molecular diagnosis and providing genetic counselling 15 and may provide clues to the development of new therapeutic approaches for different diseases. In recent years, several studies have investigated the clinical efficacy of gene sequencing for different PIDs. 16 , 17 , 18 , 19 , 20 , 21 , 22 These studies focused on patients with specific PID phenotypes.
In our study, hospitalised infants without clear PID phenotypes were investigated. We identified disease‐causing genes in 51 out of 2392 patients during early life. This proportion is an encouraging diagnostic rate of approximately 2.1% from an infant cohort. Our results indicated that the prevalence of PID is underestimated. Interestingly, there is a difference in the rate of PID identification in children of different severities. More severe diseases result in a higher rate of PID identification. This result indicated that we should consider the possibility of PID for patients with severe infection.
The phenotyping of PID patients often requires detailed, functional immunological assays to determine specific defects in cellular functions. 23 Many patients may be misdiagnosed because of a lack of specific symptoms. Neonatal TRECs/KRECs screening is widely used for early PID diagnosis. 24 , 25 , 26 This is very useful for the identification of immunodeficiencies that affect the numbers of T and B cells. However, almost half of the patients in our cohort had combined immunodeficiencies with associated or syndromic features. Only one quarter had decreased T cells or B cells. Routine TRECs/KRECs screening could miss a PID diagnosis. Our results showed the clinical importance of genetic investigation in infants with infection during early life.
Definitive diagnosis is beneficial for patients and their families in various ways, including allowing for identification of potential treatments, recognition of the risk of transmission in subsequent pregnancies, and provision of anticipatory guidance and prognosis. In our study, approximately 60% of patients (30/51) received specific treatments after being diagnosed with PID and consequently had better prognoses. If they had not been promptly diagnosed, most of these patients would have died, especially the SCID patients. The turnaround time of NGS was 2 weeks. Some PIDs can be identified quickly by functional testing. For example, we can identify some patients with SCID by flow cytometry. However, some patients do not have specific characteristic immune phenotypes. For these patients, NGS is needed. Even for patients with specific immune phenotypes, the genetic diagnosis is still useful. As such, our genetic test might reasonably be offered as a first‐line genetic assessment for early life infants who have infections in clinics. This study highlights advantages of NGS for PID screening in early life infants. In particular, the cost of sequencing is only US$280 per sample. This expense is conducive to the popularisation of the tested approach, especially in developing countries.
In conclusion, this study shows the clinical applications and benefits of genetic testing in the clinical management of early life infants. Such testing is extremely useful for the early identification of PIDs. This study not only highlighted the potential of NGS to rapidly provide molecular diagnoses of PIDs but also indicated that the prevalence of PIDs is underestimated. With broader use, this approach has the potential to alter clinical strategies in early life infants.
Methods
This study was approved by the ethics committee of the Children’s Hospital of Fudan University. Written informed consent was obtained from the parents of all of the patients. The methods were conducted in accordance with the approved guidelines.
Patients and sample preparation
To explore the use of NGS in the early identification of PIDs, patients who met the following criteria were included in the study from November 2015 to April 2018. Inclusion criteria were as follows: (1) age <3 months and (2) hospitalised for infectious diseases, regardless of the extent and location of the infection or abnormal routine blood test (white blood cells < 4000 µL−1 or > 20 000 µL−1; or platelets < 100 × 109 L−1).
After the collection of 2 mL of EDTA‐anticoagulated blood, DNA was extracted from peripheral blood mononuclear cells, which were isolated using lymphocyte separation medium (Beyotime Biotechnology, Shanghai, China).
NGS and variation analysis
Fragments of patients’ genomic DNA were enriched for panel sequencing using the Agilent ClearSeq Inherited Disease panel kit. The panel included all known PID‐associated genes 27 at that time. The methods for NGS and variation analysis have been reported previously. 28 Enriched DNA samples were indexed and sequenced on a HiSeq2000 sequencer (Illumina, San Diego, CA, USA) using standard protocols in a Clinical Laboratory Improvement Amendments (CLIA)‐compliant sequencing laboratory at WuXi NextCODE (CLIA ID 99D2064856). Sequencing provided at least 50 million 125‐bp paired‐end reads for each sample.
Reads were mapped to the human reference genome hg19 using Burrows‐Wheeler Aligner (BWA). The average coverage sequencing depth for the official targets was at least 180× and was higher than 20× for > 99% of the target region. Variants were called in accordance with the gold standard of the GATK Best Practices. Deleterious mutations and novel variants detected using NGS were confirmed via Sanger sequencing. A coverage‐based algorithm, CANOES, was used to detect large exonic deletions and duplications. Based on the validation results of the array comparative genomic hybridisation (CGH) experiments, ratios < 0.65 and > 1.35 were scored as deletions and duplications, respectively. All positive calls were further investigated and confirmed via Sanger sequencing or quantitative polymerase chain reaction (qPCR). The turnaround time of the entire experimental process was 2 weeks.
Management of patients diagnosed with PIDs
Once a mutation in a PID‐associated gene was found in patients, functional testing associated with this mutation will be finished. The patients were diagnosed based on clinical symptoms, genetic data and functional testing. Then, the patients received corresponding management and follow‐up treatments.
Conflict of interest
The authors declare no competing financial interests.
Author Contributions
Jinqiao Sun: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Writing‐original draft. Lin Yang: Data curation; Formal analysis; Investigation; Methodology; Writing‐original draft. Yulan Lu: Formal analysis; Investigation; Methodology; Software. Huijun Wang: Data curation; Investigation; Methodology. Xiaomin Peng: Data curation; Formal analysis; Methodology. Xinran Dong: Data curation; Formal analysis; Methodology. Guoqiang Cheng: Conceptualization; Investigation. Yun Cao: Conceptualization; Methodology. Bingbing Wu: Conceptualization; Investigation; Methodology; Software; Writing‐review & editing. Xiaochuan Wang: Conceptualization; Data curation; Investigation; Methodology; Writing‐review & editing. Wenhao Zhou: Conceptualization; Data curation; Funding acquisition; Investigation; Methodology; Writing‐review & editing.
Supporting information
Supplementary Material
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81471484 and 81501289), the National Key Research and Development Program of China (2016YFC0905100), the Shanghai Shen Kang Hospital Development Center (SHDC12017110) and the Shanghai Science and Technology Commission (19411969900).
Contributor Information
Bingbing Wu, Email: bingbingwu2010@163.com.
Xiaochuan Wang, Email: xchwang@shmu.edu.cn.
Wenhao Zhou, Email: zhouwenhao@fudan.edu.cn.
References
- 1. Bousfiha A, Jeddane L, Picard C et al Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol 2020; 40: 66–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tangye SG, Al‐Herz W, Bousfiha A et al Human inborn errors of immunity: 2019 update on the classification from the international union of immunological societies expert committee. J Clin Immunol 2020; 40: 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pan‐Hammarström Q, Abolhassani H, Hammarström L. Defects in plasma cell differentiation are associated with primary immunodeficiency in human subjects. J Allergy Clin Immunol 2018; 141: 1217–1219. [DOI] [PubMed] [Google Scholar]
- 4. Picard C, Bobby Gaspar H, Al‐Herz W et al International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol 2018; 38: 96–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chou J, Ohsumi TK, Geha RS. Use of whole exome and genome sequencing in the identification of genetic causes of primary immunodeficiencies. Curr Opin Allergy Clin Immunol 2012; 12: 623–628. [DOI] [PubMed] [Google Scholar]
- 6. Nijman IJ, van Montfrans JM, Hoogstraat M et al Targeted next‐generation sequencing: a novel diagnostic tool for primary immunodeficiencies. J Allergy Clin Immunol 2014; 133: 529–534. [DOI] [PubMed] [Google Scholar]
- 7. Ghosh S, Krux F, Binder V et al Array‐based sequence capture and next‐generation sequencing for the identification of primary immunodeficiencies. Scand J Immunol 2012; 75: 350–354. [DOI] [PubMed] [Google Scholar]
- 8. Stoddard JL, Niemela JE, Fleisher TA et al Targeted NGS: a costeffective approach to molecular diagnosis of PIDs. Front Immunol 2014; 5: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moens LN, Falk‐Sorqvist E, Asplund AC et al Diagnostics of primary immunodeficiency diseases: a sequencing capture approach. PLoS One 2014; 9: e114901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yska HAF, Elsink K, Kuijpers TW et al Diagnostic yield of next generation sequencing in genetically undiagnosed patients with primary immunodeficiencies: a systematic review. J Clin Immunol 2019; 39: 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wright CF, Fitzgerald TW, Jones WD et al Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome‐wide research data. Lancet 2015; 385: 1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang Y, Muzny DM, Reid JG et al Clinical whole‐exome sequencing for the diagnosis of mendelian disorders. The New Engl J Med 2013; 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Itan Y, Casanova JL. Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Front Immunol 2015; 6: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bousfiha AA, Jeddane L, Ailal F et al Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol 2013; 33: 1–7. [DOI] [PubMed] [Google Scholar]
- 15. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol 2010; 125: S182–S194. [DOI] [PubMed] [Google Scholar]
- 16. Al‐Mousa H, Abouelhoda M, Monies DM et al Unbiased targeted next‐generation sequencing molecular approach for primary immunodeficiency diseases. J Allergy Clin Immunol 2016; 137: 1780–1787. [DOI] [PubMed] [Google Scholar]
- 17. Yu H, Zhang VW, Stray‐Pedersen A et al Rapid molecular diagnostics of severe primary immunodeficiency determined by using targeted next‐generation sequencing. J Allergy Clin Immunol 2016; 138: 1142–1151. [DOI] [PubMed] [Google Scholar]
- 18. Rae W, Ward D, Mattocks C et al Clinical efficacy of a next‐generation sequencing gene panel for primary immunodeficiency diagnostics. Clin Genet 2018; 93: 647–655. [DOI] [PubMed] [Google Scholar]
- 19. Maffucci P, Filion CA, Boisson B et al Genetic diagnosis using whole exome sequencing in common variable immunodeficiency. Front Immunol 2016; 7: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fang M, Abolhassani H, Lim CK et al Next generation sequencing data analysis in primary immunodeficiency disorders ‐ future directions. J Clin Immunol 2016; 36: 68–75. [DOI] [PubMed] [Google Scholar]
- 21. Abolhassani H, Aghamohammadi A, Fang M et al Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med 2019; 21: 243–251. [DOI] [PubMed] [Google Scholar]
- 22. Rudilla F, Franco‐Jarava C, Martínez‐Gallo M et al Expanding the clinical and genetic spectra of primary immunodeficiency‐related disorders with clinical exome sequencing: expected and unexpected findings. Front Immunol 2019; 10: 2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hsieh EW, Hernandez JD. Novel tools for primary immunodeficiency diagnosis: making a case for deep profiling. Curr Opin Allergy Clin Immunol 2016; 16: 549–556. [DOI] [PubMed] [Google Scholar]
- 24. Dorsey MJ, Puck JM. Newborn screening for severe combined immunodeficiency in the united states: lessons learned. Immunol Allergy Clin North Am 2019; 39: 1–11. [DOI] [PubMed] [Google Scholar]
- 25. Amatuni GS, Currier RJ, Church JA et al Screening for severe combined immunodeficiency and T‐cell lymphopenia in California, 2010–2017. Pediatrics 2019; 143: e20182300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hammarström L. Primary immunodeficiencies screening: neonatal screening for T/B cell disorders ‐ a triplex PCR method for quantitation of TRECs and KRECs in newborns. Clin Exp Immunol 2014; 178: 14–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Picard C, Al‐Herz W, Bousfiha A et al Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol 2015; 35: 696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang L, Kong Y, Dong X et al Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genet Med 2019; 21: 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material