

Eukaryotic cells have evolved multiple responses that allow endoplasmic reticulum (ER) homeostasis to be maintained even in the face of acute or chronic stresses. In this issue, Yu et al (2020) describe how site‐specific phosphorylation switches protein disulfide isomerase (PDI) from a folding enzyme to a holdase chaperone which regulates ER stress responses, thus highlighting PDI as a key player in ER homeostasis.
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control
A new study reveals that the chaperone activity of protein disulfide isomerase is reversibly modulated to protect cells against acute ER stress.
Cells interact via secreted and membrane proteins. In every eukaryotic cell, these are produced in the ER. It is thus essential for cell and organism homeostasis that protein production in the ER proceeds with high fidelity. This is made possible through a comprehensive ER protein folding machinery that supports protein folding, accelerates slow reactions in protein structure formation, and inhibits side reactions (Ellgaard et al, 2016).
In light of these critical functions, the ER has to have fail‐safe mechanisms that engage in the event that protein folding is compromised, be it due to acute stresses like sudden spikes in protein production or more persistent stressors like hypoxia or the presence of permanently misfolded proteins (Preissler & Ron, 2019). One of the mechanisms evolved to sense and safeguard the integrity of the ER is the unfolded protein response (UPR). UPR stress sensing in the mammalian ER is based on three sensors, IRE1, PERK, and ATF6 (Karagoz et al, 2019). The UPR mounts a transcriptional and translational response to re‐establish ER homeostasis by increasing ER protein folding and quality control capacity while reducing the flux of new proteins into the organelle. If cells fail to overcome ER stress, apoptosis is induced to protect the organism at the expense of sacrificing individual cells (Hetz & Papa, 2018). However, UPR measures generally work on the hour‐timescale, which may be insufficient to deal with acute stress scenarios. Indeed, cells appear to also be capable of inducing rapid responses to ER protein folding stress. This has been delineated for BiP, a major ER chaperone, which becomes inactivated by oligomerization and AMPylation. Upon ER stress, these processes are rapidly reversed, allowing BiP to quickly act as a chaperone when needed (Preissler & Ron, 2019). This provides a fast response independent of de novo protein synthesis.
Yu et al (2020) now show that protein disulfide isomerase (PDI) is another key protein in protecting cells against acute ER stress. PDI was the first folding enzyme to be identified in the ER (Goldberger et al, 1963) and even after more than 50 years of study it still holds surprises. Yu and colleagues take an approach from phenotype to molecular characterization; they show that deletion of the secretory pathway protein kinase Fam20C increases ER stress signaling by IRE1. Having identified Fam20C as a UPR modulator, the authors used mass spectrometry to identify a possible link between Fam20C and IRE1. This revealed PDI as a major stress‐regulated interaction partner of Fam20C. The authors show that multiple ER stressors induce rapid phosphorylation of a major fraction of PDI within minutes—before any translational effects of the UPR. Three serines within PDI are phosphorylated by Fam20C, and among those, the authors focus on phosphorylation of Ser357. The evolutionarily conserved Ser357 is positioned in a strategically very interesting position in PDI, the flexible linker that connects the last two of its four thioredoxin domains. Why interesting? Because PDI can act as a disulfide isomerase as well as a chaperone, and how regulation of these functions is coupled to the intrinsic flexibility of this multidomain protein has remained poorly understood (Freedman et al, 2017). Yu et al now provide new insights into this issue (Fig 1). Biochemical and in silico data suggest that PDI adopts an open conformation once Ser357 is phosphorylated, exposing an extended hydrophobic patch. This goes hand in hand with an increased chaperone activity and a decreased oxidoreductase function for PDI. PDI phosphorylation thus has pronounced effects on PDI function—and also on ER homeostasis as a whole. In cells, this functional switch appears to counteract protein misfolding in the ER, which the authors find to strictly depend on Ser357 within PDI. Loss of PDI (or this critical Ser) rendered cells more susceptible to lethal ER stress. And this phosphorylation switch appeared to have yet another function; it increased association of PDI with IRE1 in a cysteine‐independent, chaperone‐like manner. IRE1 binding was paralleled by a reduction in its XBP1 mRNA splicing activity. Thus, PDI is the link that closes the loop between Fam20C deletion and increased ER stress. Finally, the weight of this study comes to light when the authors move to animal models, generating mice where PDI lacks this key serine residue. Cellular responses after ER stress show that lack of PDI phosphorylation led to an exacerbated UPR response, including upregulation of apoptosis markers, as well as pro‐inflammatory pathways, resulting in aggravated liver damage.
Figure 1. Timecourse of ER stress responses and the role of PDI.
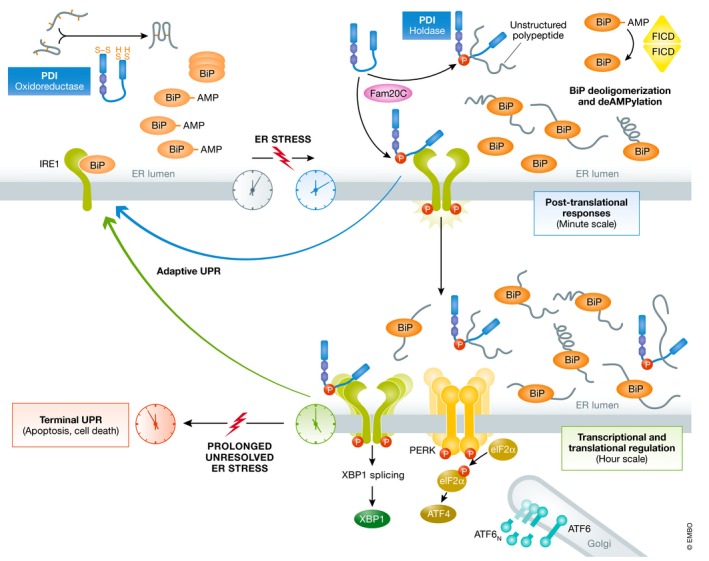
In non‐stress conditions, PDI (in blue) is acting as a pro‐folding enzyme, through the reduction, oxidation, and reshuffling of disulfide bonds. The UPR is kept inactivate e.g. by the stabilization of monomeric IRE1 (in green) through BiP binding (in orange); BiP itself is partially inactivated through oligomerization and AMPylation. Minutes after the begin of ER stress, Fam20C kinase (in pink) phosphorylates PDI at Ser357 promoting an open conformation which enhances PDIs holdase activity, thus enabling interaction with unfolded peptides and protection against aggregation in the ER lumen. In the same timescale, the pool of inactive BiP is activated by (i) deoligomerization, and (ii) deAMPylation carried out by FICD dimers. BiP displacement from IRE1 leads to its dimerization, trans‐autophosphorylation, and splicing of XBP1 transcripts. Phosphorylated PDI is able to bind IRE1, and this phosphorylation‐dependent interaction modulates the adaptive UPR. Prolonged, unsolvable ER stress leads to (hyper)activation of the different UPR branches and will ultimately induce apoptosis.
By using comprehensive approaches from molecular simulations to mouse models, Yu et al make a major contribution to our understanding of ER proteostasis. The authors reveal how different functions of one of the best‐understood ER folding enzymes/chaperones can be regulated, how they affect ER protein (mis)folding, and what the consequences on ER stress and cellular survival ex and in vivo are. PDI phosphorylation as a novel, fast means to control stress responses is in line with recent reports on other rapid ER stress responses. The study also further highlights the intimate connections between direct rapid cellular responses (PDI chaperone functions) and the regulation of slower adaptive measures cells take to maintain ER homeostasis (via the discovered PDI:IRE1 axis). It nicely fits into an emerging concept that under acute stress, the ER folding machinery first inhibits aggregation rather than supporting folding, as also exemplified by BiP re‐activation (Preissler & Ron, 2019), and inactivation of oxidative folding in the ER—if conditions become too oxidative (Sevier et al, 2007)—and now by PDI, which switches from promoting oxidative folding to protecting its clients from aggregation. Proteins involved in these rapid responses appear to often feed back into the UPR signaling branches. This likely allows cells a smoother transition from acute to chronic ER stress responses, without ever leaving a dangerous gap of strong protein misfolding that may irreversibly commit cells to apoptosis (Lam et al, 2020) when rescue was still possible.
Important findings always open up more questions. It will be very interesting to see if other Fam20C clients are also involved in ER stress responses, since abundance does not necessarily mean importance. A key question is also the regulation of Fam20C function and trafficking itself. The study additionally provides new perspectives on PDI regulation, which warrant further investigation, and further establishes the many ER PDIs not only as folding enzymes, but also as regulators of ER homeostasis (Oka et al, 2019). Lastly, the interplay between PDI and IRE1 adds a very important new aspect to the intricate regulation of this stress sensor (Karagoz et al, 2019). Future studies will now have to further refine the emerging picture of closely intertwined ER stress responses, which act on a very different timescale (Fig 1) and involve post‐translational, as well as transcriptional and translational responses to protect this key organelle.
The EMBO Journal (2020) 39: e104880
See also: https://doi.org/10.15252/embj.2019103841 (May 2020)
References
- Ellgaard L, McCaul N, Chatsisvili A, Braakman I (2016) Co‐ and post‐translational protein folding in the ER. Traffic 17: 615–638 [DOI] [PubMed] [Google Scholar]
- Freedman RB, Desmond JL, Byrne LJ, Heal JW, Howard MJ, Sanghera N, Walker KL, Wallis AK, Wells SA, Williamson RA et al (2017) ‘Something in the way she moves’: the functional significance of flexibility in the multiple roles of protein disulfide isomerase (PDI). Biochim Biophys Acta Proteins Proteom 1865: 1383–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger RF, Epstein CJ, Anfinsen CB (1963) Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem 238: 628–635 [PubMed] [Google Scholar]
- Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69: 169–181 [DOI] [PubMed] [Google Scholar]
- Karagoz GE, Acosta‐Alvear D, Walter P (2019) The unfolded protein response: detecting and responding to fluctuations in the protein‐folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol 11: a033886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam M, Marsters SA, Ashkenazi A, Walter P (2020) Misfolded proteins bind and activate death receptor 5 to induce apoptosis during unresolved endoplasmic reticulum stress. eLife 9: e52291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka OB, van Lith M, Rudolf J, Tungkum W, Pringle MA, Bulleid NJ (2019) ERp18 regulates activation of ATF6alpha during unfolded protein response. EMBO J 38: e100990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, Ron D (2019) Early events in the endoplasmic reticulum unfolded protein response. Cold Spring Harb Perspect Biol 11: a033894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevier CS, Qu H, Heldman N, Gross E, Fass D, Kaiser CA (2007) Modulation of cellular disulfide‐bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129: 333–344 [DOI] [PubMed] [Google Scholar]
- Yu J, Li T, Liu Y, Wang X, Zhang J, Wang X, Shi G, Lou J, Wang L, Wang C et al (2020) Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J 10.15252/embj.2019103841 [DOI] [PMC free article] [PubMed] [Google Scholar]